
Phosphorylation of mRNA-Binding Proteins Puf1 and Puf2 by TORC2-Activated Protein Kinase Ypk1 Alleviates Their Repressive Effects


Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Yeast Strains and Growth Conditions
2.2. Plasmids and Recombinant DNA Methods
2.3. Purification of GST-Fusion Proteins
2.4. Production and Purification of Ypk1-as from S. cerevisiae
2.5. Protein Kinase Assay
2.6. Total Protein Extraction
2.7. Immunoblotting
2.8. Fluorescence Microscopy
2.9. RT-qPCR
2.10. In Vivo Dual Fluorescence Reporter Assay
3. Results
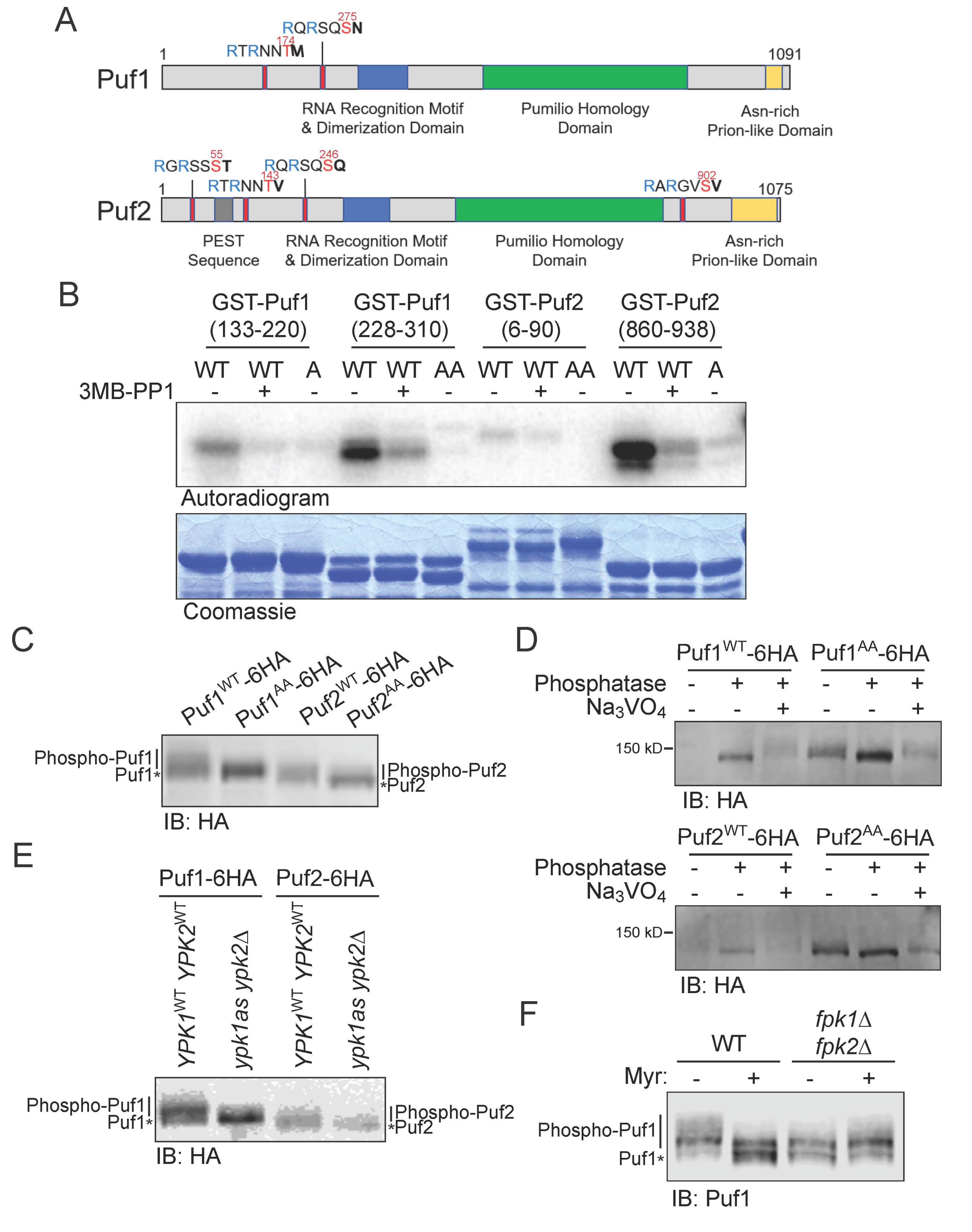
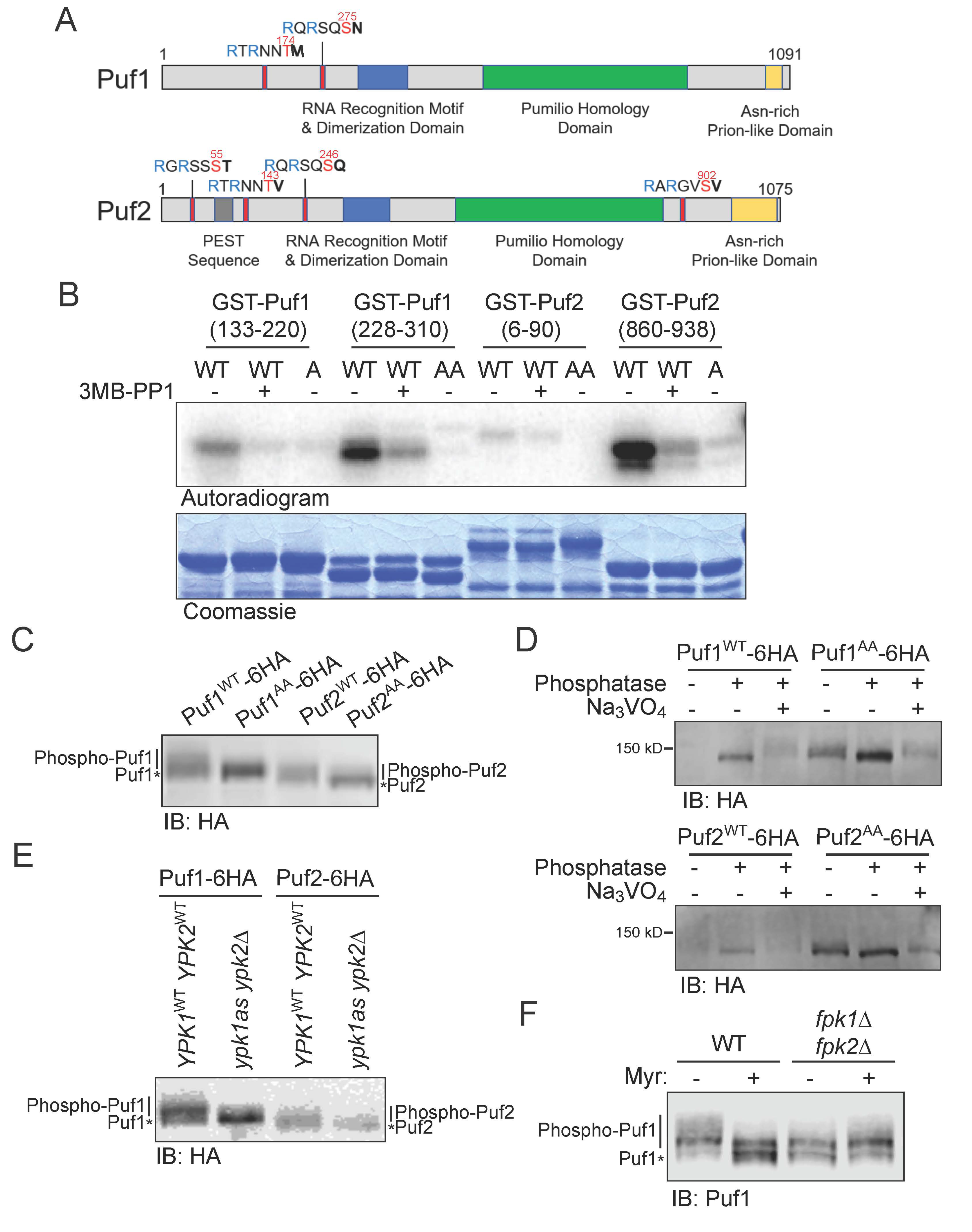
3.1. Puf1 and Puf2 Are Substrates of Protein Kinase Ypk1
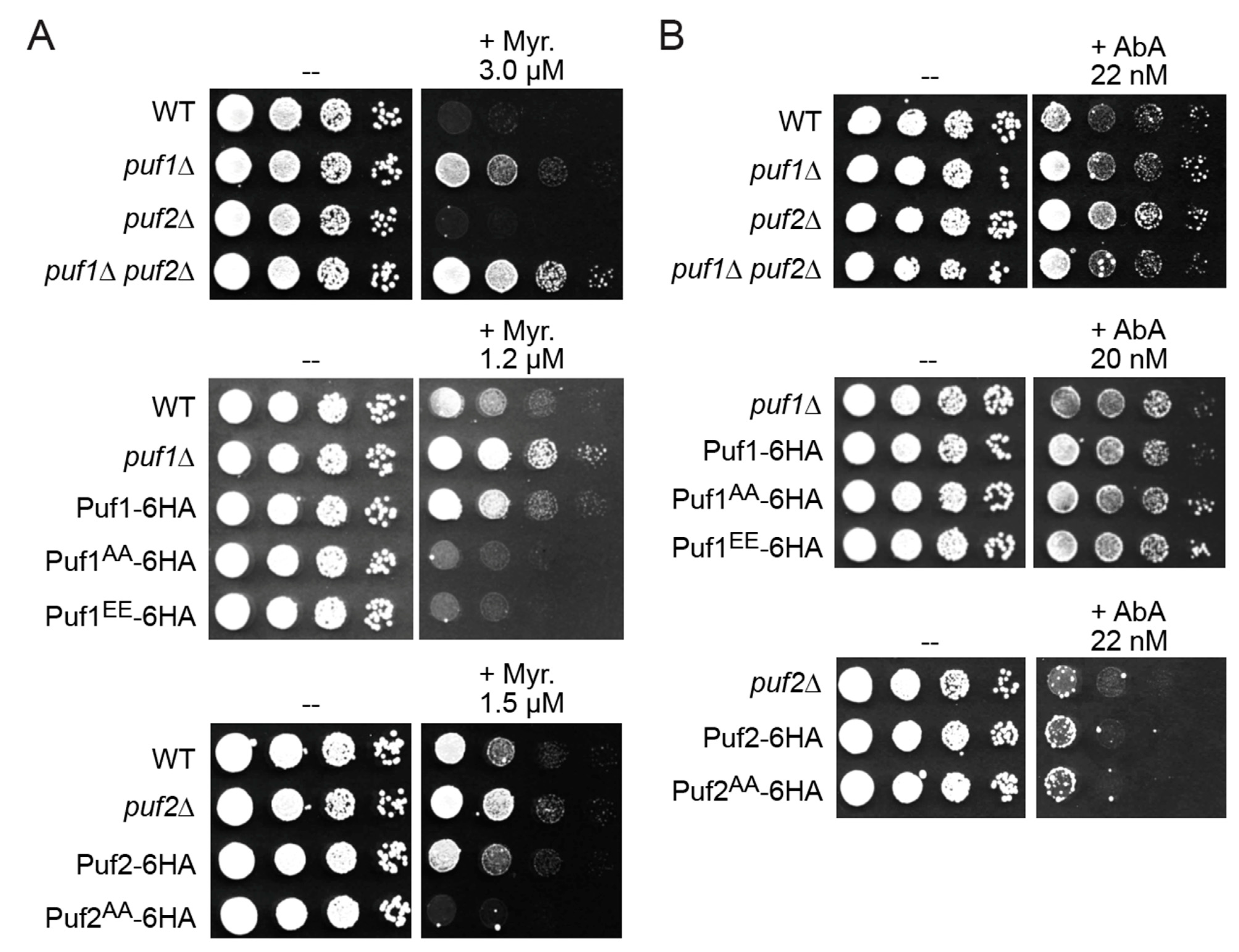
3.2. Phosphorylation by Ypk1 Down-Regulates the Function of Puf1 and Puf2
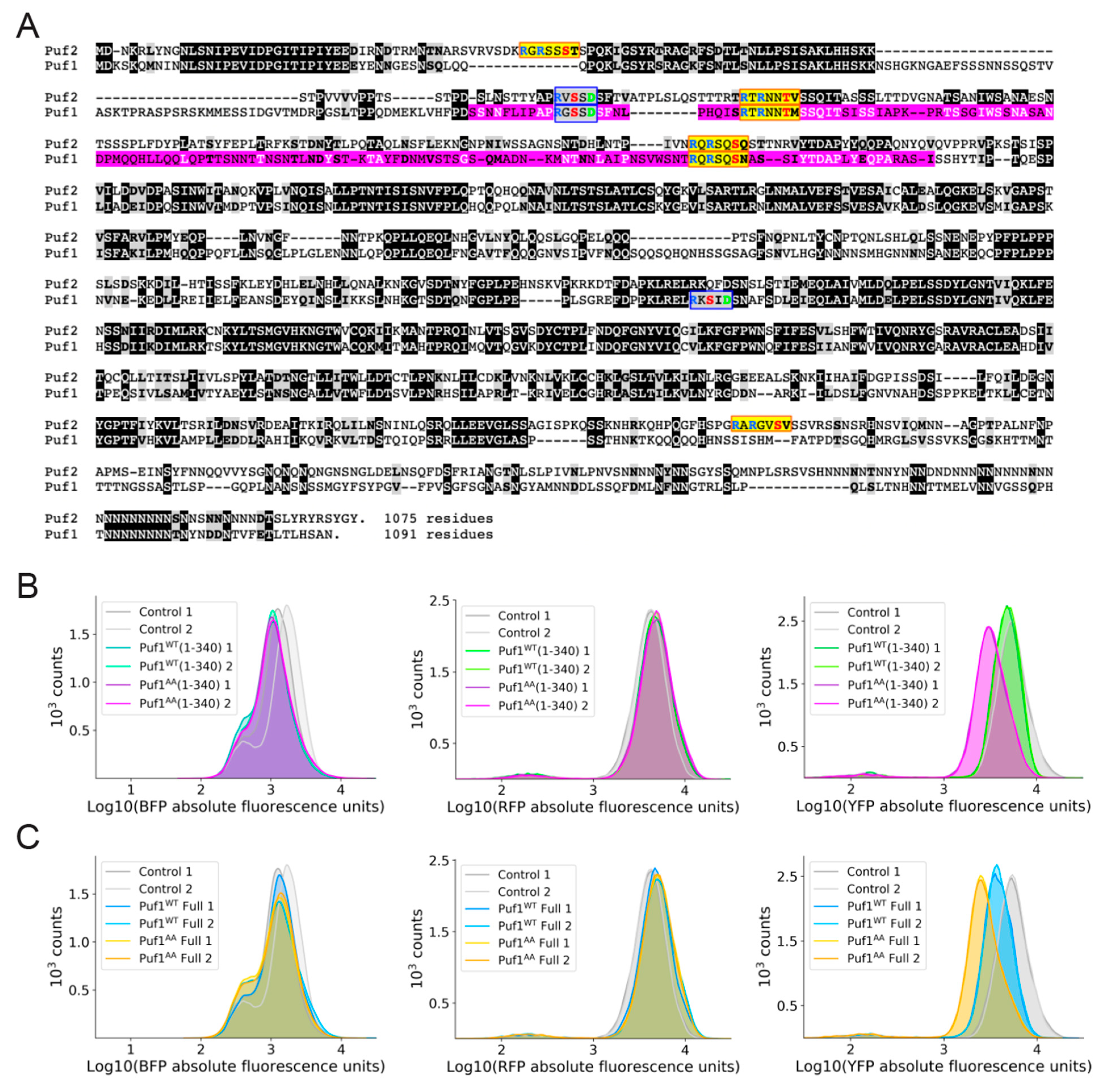
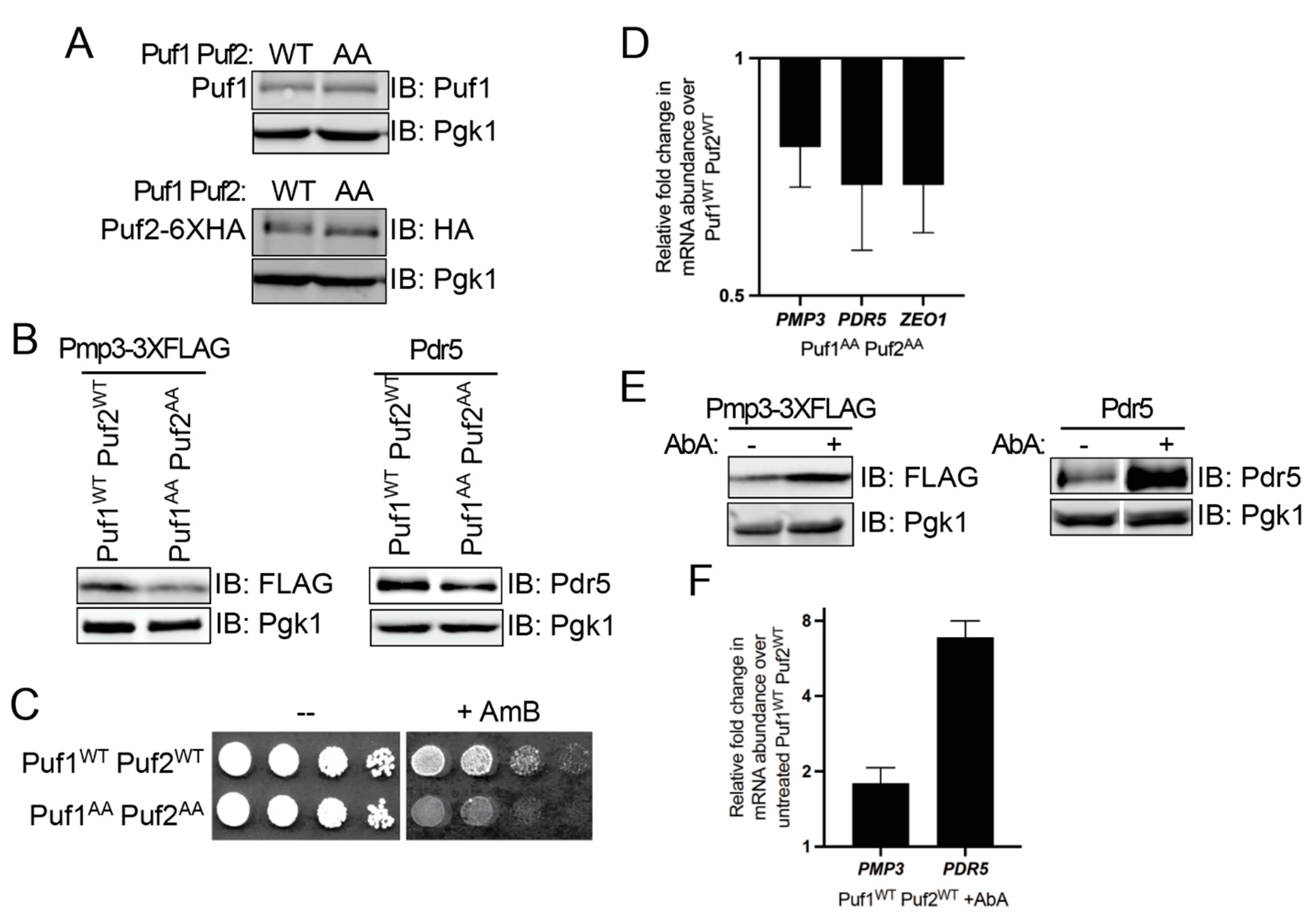
3.3. Ypk1 Phosphorylation Promotes Protein Production from Puf1- and Puf2-Bound mRNAs
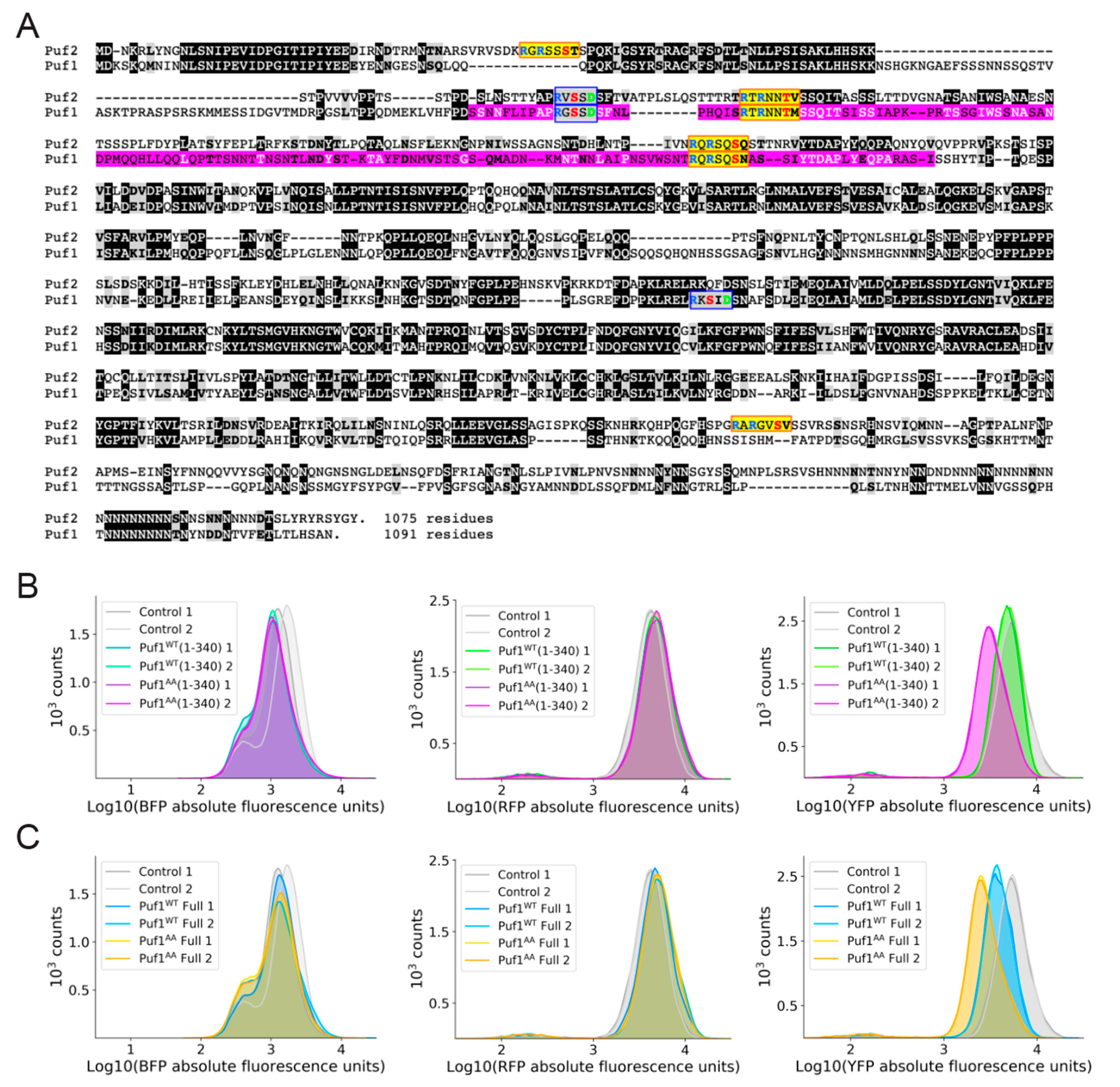
3.4. Additional Mechanistic Insight from Application of a Heterologous Protein-RNA Tethering and Fluorescent Protein Reporter Assay
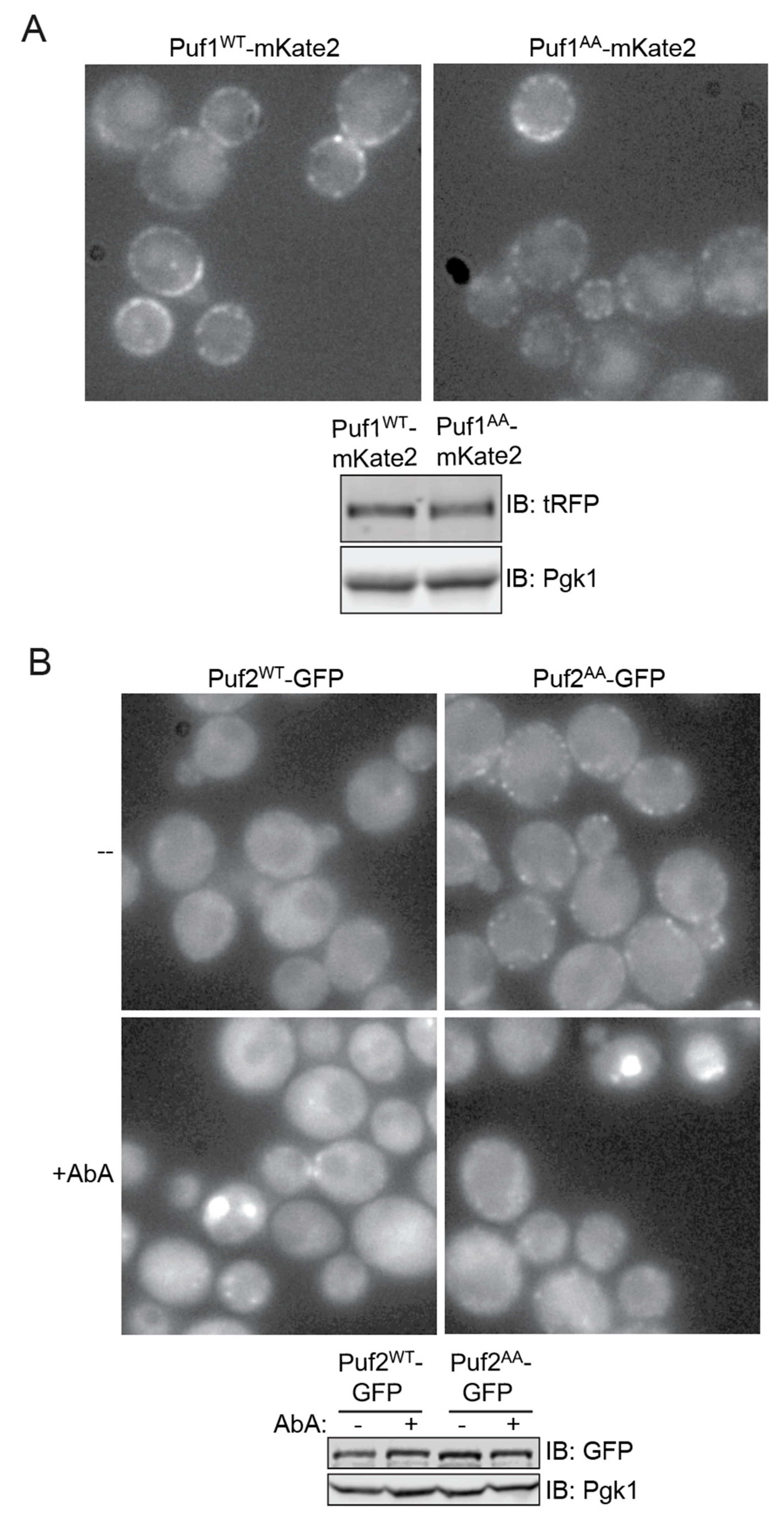
3.5. Differential Subcellular Localization of Puf1 and Puf2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.R.; Mukherjee, N.; Keene, J.D. Systematic analysis of posttranscriptional gene expression. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; McKnight, S.L. A solid-state conceptualization of information transfer from gene to message to protein. Annu. Rev. Biochem. 2018, 87, 351–390. [Google Scholar] [CrossRef]
- Kelaini, S.; Chan, C.; Cornelius, V.A.; Margariti, A. RNA-binding proteins hold key roles in function, dysfunction, and disease. Biology 2021, 10, 366. [Google Scholar] [CrossRef]
- Weis, K. Dead or alive: DEAD-box ATPases as regulators of ribonucleoprotein complex condensation. Biol. Chem. 2021, 402, 653–661. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Das, S.; Das, B. Localization elements and zip codes in the intracellular transport and localization of messenger RNAs in Saccharomyces cerevisiae. Wiley Interdiscip. Rev. RNA 2020, 11, e1591. [Google Scholar] [CrossRef]
- Wickens, M.; Bernstein, D.S.; Kimble, J.; Parker, R. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet. 2002, 18, 150–157. [Google Scholar] [CrossRef]
- Quenault, T.; Lithgow, T.; Traven, A. PUF proteins: Repression, activation and mRNA localization. Trends Cell Biol. 2011, 21, 104–112. [Google Scholar] [CrossRef]
- Wang, M.; Ogé, L.; Perez-Garcia, M.D.; Hamama, L.; Sakr, S. The PUF protein family: Overview on PUF RNA targets, biological functions, and post transcriptional regulation. Int. J. Mol. Sci. 2018, 19, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamore, P.D.; Williamson, J.R.; Lehmann, R. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 1997, 3, 1421–1433. [Google Scholar]
- Zhang, B.; Gallegos, M.; Puoti, A.; Durkin, E.; Fields, S.; Kimble, J.; Wickens, M.P. A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line. Nature 1997, 390, 477–484. [Google Scholar] [CrossRef]
- Muir, A.; Ramachandran, S.; Roelants, F.M.; Timmons, G.; Thorner, J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLife 2014, 3, e03779. [Google Scholar] [CrossRef] [PubMed]
- Machin, N.A.; Lee, J.M.; Barnes, G. Microtubule stability in budding yeast: Characterization and dosage suppression of a benomyl-dependent tubulin mutant. Mol. Biol. Cell 1995, 6, 1241–1259. [Google Scholar] [CrossRef] [Green Version]
- Gerber, A.P.; Herschlag, D.; Brown, P.O. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004, 2, E79. [Google Scholar] [CrossRef]
- Tkach, J.M.; Yimit, A.; Lee, A.Y.; Riffle, M.; Costanzo, M.; Jaschob, D.; Hendry, J.A.; Ou, J.; Moffat, J.; Boone, C.; et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 2012, 14, 966–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.F.; Koh, Y.Y.; VanVeller, B.; Raines, R.T.; Wickens, M. Target selection by natural and redesigned PUF proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 15868–15873. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP complex is required for cellular sphingolipid homeostasis. eLife 2015, 4, e08712. [Google Scholar] [CrossRef]
- Wadsworth, J.M.; Clarke, D.J.; McMahon, S.A.; Lowther, J.P.; Beattie, A.E.; Langridge-Smith, P.R.R.; Broughton, H.B.; Dunn, T.M.; Naismith, J.H.; Campopiano, D.J. The chemical basis of serine palmitoyltransferase inhibition by myriocin. J. Am. Chem. Soc. 2013, 135, 14276–14285. [Google Scholar] [CrossRef] [Green Version]
- Roelants, F.M.; Breslow, D.K.; Muir, A.; Weissman, J.S.; Thorner, J. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 19222–19227. [Google Scholar] [CrossRef] [Green Version]
- Klug, L.; Daum, G. Yeast lipid metabolism at a glance. FEMS Yeast Res. 2014, 14, 369–388. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, T.; Parmryd, I. Interleaflet coupling, pinning, and leaflet asymmetry-major players in plasma membrane nanodomain formation. Front. Cell Dev. Biol. 2017, 4, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez Marshall, M.N.; Emmerstorfer-Augustin, A.; Leskoske, K.L.; Zhang, L.H.; Li, B.; Thorner, J. Analysis of the roles of phosphatidylinositol-4,5-bisphosphate and individual subunits in assembly, localization, and function of Saccharomyces cerevisiae target of rapamycin complex 2. Mol. Biol. Cell 2019, 30, 1555–1574. [Google Scholar] [CrossRef]
- Leskoske, K.L.; Roelants, F.M.; Martinez Marshall, M.N.; Hill, J.M.; Thorner, J. The stress-sensing TORC2 complex activates yeast AGC-family protein kinase Ypk1 at multiple novel sites. Genetics 2017, 207, 179–195. [Google Scholar] [CrossRef]
- Roelants, F.M.; Leskoske, K.L.; Martinez Marshall, M.N.; Locke, M.N.; Thorner, J. The TORC2-dependent signaling network in the yeast Saccharomyces cerevisiae. Biomolecules 2017, 7, 66. [Google Scholar] [CrossRef]
- Riggi, M.; Kusmider, B.; Loewith, R. The flipside of the TOR coin—TORC2 and plasma membrane homeostasis at a glance. J. Cell Sci. 2020, 133, jcs242040. [Google Scholar] [CrossRef]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192–205.e3. [Google Scholar] [CrossRef] [Green Version]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Roelants, F.M.; Torrance, P.D.; Bezman, N.; Thorner, J. Pkh1 and Pkh2 differentially phosphorylate and activate Ypk1 and Ykr2 and define protein kinase modules required for maintenance of cell wall integrity. Mol. Biol. Cell 2002, 13, 3005–3028. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Jeschke, G.R.; Roelants, F.M.; Thorner, J.; Turk, B.E. Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol. Cell Biol. 2012, 32, 4705–4717. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Roelants, F.M.; Timmons, G.; Leskoske, K.L.; Thorner, J. Down-regulation of TORC2-Ypk1 signaling promotes MAPK-independent survival under hyperosmotic stress. eLife 2015, 4, e09336. [Google Scholar] [CrossRef]
- Roelants, F.M.; Baltz, A.G.; Trott, A.E.; Fereres, S.; Thorner, J. A protein kinase network regulates the function of aminophospholipid flippases. Proc. Natl. Acad. Sci. USA 2010, 107, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Roelants, F.M.; Chauhan, N.; Muir, A.; Davis, J.C.; Menon, A.K.; Levine, T.P.; Thorner, J. TOR complex 2-regulated protein kinase Ypk1 controls sterol distribution by inhibiting StARkin domain-containing proteins located at plasma membrane-endoplasmic reticulum contact sites. Mol. Biol. Cell 2018, 29, 2128–2136. [Google Scholar] [CrossRef]
- Topolska, M.; Roelants, F.M.; Si, E.P.; Thorner, J. TORC2-dependent Ypk1-mediated phosphorylation of Lam2/Ltc4 disrupts its association with the β-propeller protein Laf1 at endoplasmic reticulum-plasma membrane contact sites in the yeast Saccharomyces cerevisiae. Biomolecules 2020, 10, 1598. [Google Scholar] [CrossRef]
- Alvaro, C.G.; Aindow, A.; Thorner, J. Differential phosphorylation provides a switch to control how α-arrestin Rod1 down-regulates mating pheromone response in Saccharomyces cerevisiae. Genetics 2016, 203, 299–317. [Google Scholar] [CrossRef] [Green Version]
- Roelants, F.M.; Leskoske, K.L.; Pedersen, R.T.; Muir, A.; Liu, J.M.; Finnigan, G.C.; Thorner, J. TOR Complex 2-regulated protein kinase Fpk1 stimulates endocytosis via inhibition of Ark1/Prk1-related protein kinase Akl1 in Saccharomyces cerevisiae. Mol. Cell Biol. 2017, 37, e00627-16. [Google Scholar] [CrossRef] [Green Version]
- Bourgoint, C.; Rispal, D.; Berti, M.; Filipuzzi, I.; Helliwell, S.B.; Prouteau, M.; Loewith, R. Target of rapamycin complex 2-dependent phosphorylation of the coat protein Pan1 by Akl1 controls endocytosis dynamics in. J. Biol. Chem. 2018, 293, 12043–12053. [Google Scholar] [CrossRef] [Green Version]
- Locke, M.N.; Thorner, J. Rab5 GTPases are required for optimal TORC2 function. J. Cell Biol. 2019, 218, 961–976. [Google Scholar] [CrossRef]
- Riggi, M.; Bourgoint, C.; Macchione, M.; Matile, S.; Loewith, R.; Roux, A. TORC2 controls endocytosis through plasma membrane tension. J. Cell Biol. 2019, 218, 2265–2276. [Google Scholar] [CrossRef] [Green Version]
- Amberg, D.C.; Burke, D.J.; Strathern, J.N. Methods in Yeast Genetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2005. [Google Scholar]
- Leskoske, K.L.; Roelants, F.M.; Emmerstorfer-Augustin, A.; Augustin, C.M.; Si, E.P.; Hill, J.M.; Thorner, J. Phosphorylation by the stress-activated MAPK Slt2 down-regulates the yeast TOR complex 2. Genes Dev. 2018, 32, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012; Volume 1 and 2. [Google Scholar]
- Reynaud, K.K. Surveying the Global Landscape of Post-Transcriptional Regulation. Ph.D. Thesis, University of California, Berkeley, Berkeley, CA, USA, 2020. [Google Scholar]
- Finnigan, G.C.; Thorner, J. mCAL: A new approach for versatile multiplex action of Cas9 using one sgRNA and loci flanked by a programmed target sequence. G3 Genes Genomes Genet. 2016, 6, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Egner, R.; Mahé, Y.; Pandjaitan, R.; Kuchler, K. Endocytosis and vacuolar degradation of the plasma membrane-localized Pdr5 ATP-binding cassette multidrug transporter in Saccharomyces cerevisiae. Mol. Cell Biol. 1995, 15, 5879–5887. [Google Scholar] [CrossRef] [Green Version]
- Baum, P.; Thorner, J.; Honig, L. Identification of tubulin from the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1978, 75, 4962–4966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynaud, K.; Brothers, M.; Ly, M.; Ingolia, N.T. Dynamic post-transcriptional regulation by Mrn1 links cell wall homeostasis to mitochondrial structure and function. PLoS Genet. 2021, 17, e1009521. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villén, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Lanz, M.C.; Yugandhar, K.; Gupta, S.; Sanford, E.J.; Faça, V.M.; Vega, S.; Joiner, A.M.N.; Fromme, J.C.; Yu, H.; Smolka, M.B. In-depth and 3-dimensional exploration of the budding yeast phosphoproteome. EMBO Rep. 2021, 22, e51121. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R.; Wickner, R.B. Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Acc. Chem. Res. 2013, 46, 1487–1496. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, D.; Piccolis, M.; Chiaruttini, N.; Riezman, I.; Riezman, H.; Roux, A.; Walther, T.C.; Loewith, R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 2012, 14, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Niles, B.J.; Powers, T. Plasma membrane proteins Slm1 and Slm2 mediate activation of the AGC kinase Ypk1 by TORC2 and sphingolipids in S. cerevisiae. Cell Cycle 2012, 11, 3745–3749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, J.; Kim, P.M.; Lam, H.Y.; Piccirillo, S.; Zhou, X.; Jeschke, G.R.; Sheridan, D.L.; Parker, S.A.; Desai, V.; Jwa, M.; et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 2010, 3, ra12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, C.P.; Smolka, M.B.; Payne, S.H.; Bafna, V.; Eng, J.; Zhou, H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell Proteom. 2008, 7, 1389–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulard, A.; Cremonesi, A.; Moes, S.; Schütz, F.; Jenö, P.; Hall, M.N. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol. Biol. Cell 2010, 21, 3475–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGilvray, M.E.; Shishkova, E.; Place, M.; Wagner, E.R.; Coon, J.J.; Gasch, A.P. Phosphoproteome response to dithiothreitol reveals unique versus shared features of Saccharomyces cerevisiae stress responses. J. Proteome Res. 2020, 19, 3405–3417. [Google Scholar] [CrossRef]
- Heidler, S.A.; Radding, J.A. The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 1995, 39, 2765–2769. [Google Scholar] [CrossRef] [Green Version]
- Posada, J.; Cooper, J.A. Requirements for phosphorylation of MAP kinase during meiosis in Xenopus oocytes. Science 1992, 255, 212–215. [Google Scholar] [CrossRef]
- Navarre, C.; Goffeau, A. Membrane hyperpolarization and salt sensitivity induced by deletion of PMP3, a highly conserved small protein of yeast plasma membrane. EMBO J. 2000, 19, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Ulbricht, R.J.; Olivas, W.M. Puf1p acts in combination with other yeast Puf proteins to control mRNA stability. RNA 2008, 14, 246–262. [Google Scholar] [CrossRef] [Green Version]
- Yosefzon, Y.; Koh, Y.Y.; Chritton, J.J.; Lande, A.; Leibovich, L.; Barziv, L.; Petzold, C.; Yakhini, Z.; Mandel-Gutfreund, Y.; Wickens, M.; et al. Divergent RNA binding specificity of yeast Puf2p. RNA 2011, 17, 1479–1488. [Google Scholar] [CrossRef] [Green Version]
- Stovicek, V.; Holkenbrink, C.; Borodina, I. CRISPR/Cas system for yeast genome engineering: Advances and applications. FEMS Yeast Res. 2017, 17, fox030. [Google Scholar] [CrossRef] [PubMed]
- Rainha, J.; Rodrigues, J.L.; Rodrigues, L.R. CRISPR-Cas9: A powerful tool to efficiently engineer Saccharomyces cerevisiae. Life 2020, 11, 13. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, K.; Zhang, J.; Li, Y.; Wang, H.; Cui, D.; Tang, J.; Liu, Y.; Shi, X.; Li, W.; et al. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell Rep. 2013, 3, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Bari, V.K.; Sharma, S.; Alfatah, M.; Mondal, A.K.; Ganesan, K. Plasma membrane proteolipid 3 protein modulates amphotericin B resistance through sphingolipid biosynthetic pathway. Sci. Rep. 2015, 5, 9685. [Google Scholar] [CrossRef] [Green Version]
- Balzi, E.; Wang, M.; Leterme, S.; Van Dyck, L.; Goffeau, A. PDR5, a novel yeast multidrug resistance conferring transporter controlled by the transcription regulator PDR1. J. Biol. Chem. 1994, 269, 2206–2214. [Google Scholar] [CrossRef]
- Haramati, O.; Brodov, A.; Yelin, I.; Atir-Lande, A.; Samra, N.; Arava, Y. Identification and characterization of roles for Puf1 and Puf2 proteins in the yeast response to high calcium. Sci. Rep. 2017, 7, 3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivas, W.; Parker, R. The Puf3 protein is a transcript-specific regulator of mRNA degradation in yeast. EMBO J. 2000, 19, 6602–6611. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.S.; Houshmandi, S.S.; Lopez Leban, F.; Olivas, W.M. Recruitment of the Puf3 protein to its mRNA target for regulation of mRNA decay in yeast. RNA 2004, 10, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Green, R.; Lesage, G.; Sdicu, A.M.; Ménard, P.; Bussey, H. A synthetic analysis of the Saccharomyces cerevisiae stress sensor Mid2p, and identification of a Mid2p-interacting protein, Zeo1p, that modulates the PKC1-MPK1 cell integrity pathway. Microbiology 2003, 149, 2487–2499. [Google Scholar] [CrossRef]
- Corbet, G.A.; Parker, R. RNP granule formation: Lessons from P-bodies and stress granules. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 203–215. [Google Scholar] [CrossRef]
- Tauber, D.; Tauber, G.; Parker, R. Mechanisms and regulation of RNA condensation in RNP grranule formation. Trends Biochem. Sci. 2020, 45, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2021, 22, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Dutcher, R.C.; Porter, D.F.; Arava, Y.; Wickens, M.; Hall, T.M.T. Distinct RNA-binding modules in a single PUF protein cooperate to determine RNA specificity. Nucleic Acids Res. 2019, 47, 8770–8784. [Google Scholar] [CrossRef]
- Hogan, G.J.; Brown, P.O.; Herschlag, D. Evolutionary conservation and diversification of Puf RNA binding proteins and their mRNA targets. PLoS Biol. 2015, 13, e1002307. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Olivas, W.M. Roles of Puf proteins in mRNA degradation and translation. Wiley Interdiscip. Rev. RNA 2011, 2, 471–492. [Google Scholar] [CrossRef]
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef]
- Roy, B.; Jacobson, A. The intimate relationships of mRNA decay and translation. Trends Genet. 2013, 29, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Rocha, P.S. Plant abiotic stress-related RCI2/PMP3s: Multigenes for multiple roles. Planta 2016, 243, 1–12. [Google Scholar] [CrossRef]
- Kwok, A.C.M.; Zhang, F.; Ma, Z.; Chan, W.S.; Yu, V.C.; Tsang, J.S.H.; Wong, J.T.Y. Functional responses between PMP3 small membrane proteins and membrane potential. Environ. Microbiol. 2020, 22, 3066–3080. [Google Scholar] [CrossRef]
- Roelants, F.M.; Su, B.M.; von Wulffen, J.; Ramachandran, S.; Sartorel, E.; Trott, A.E.; Thorner, J. Protein kinase Gin4 negatively regulates flippase function and controls plasma membrane asymmetry. J. Cell Biol. 2015, 208, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.D.; Tu, B.P. Glucose-regulated phosphorylation of the PUF protein Puf3 regulates the translational fate of its bound mRNAs and association with RNA granules. Cell Rep. 2015, 11, 1638–1650. [Google Scholar] [CrossRef] [Green Version]
- Waśkiewicz-Staniorowska, B.; Skała, J.; Jasiński, M.; Grenson, M.; Goffeau, A.; Ułaszewski, S. Functional analysis of three adjacent open reading frames from the right arm of yeast chromosome XVI. Yeast 1998, 14, 1027–1039. [Google Scholar] [CrossRef]
- Deng, Y.; Singer, R.H.; Gu, W. Translation of ASH1 mRNA is repressed by Puf6p-Fun12p/eIF5B interaction and released by CK2 phosphorylation. Genes Dev. 2008, 22, 1037–1050. [Google Scholar] [CrossRef] [Green Version]
- Kedde, M.; van Kouwenhove, M.; Zwart, W.; Oude Vrielink, J.A.; Elkon, R.; Agami, R. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010, 12, 1014–1020. [Google Scholar] [CrossRef]
- Lee, C.D.; Tu, B.P. Metabolic influences on RNA biology and translation. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 176–184. [Google Scholar] [CrossRef]
- Frederick, M.I.; Heinemann, I.U. Regulation of RNA stability at the 3′ end. Biol. Chem. 2020, 402, 425–431. [Google Scholar] [CrossRef]
- Velázquez-Cruz, A.; Baños-Jaime, B.; Díaz-Quintana, A.; De la Rosa, M.A.; Díaz-Moreno, I. Post-translational control of RNA binding proteins and disease-related dysregulation. Front. Mol. Biosci. 2021, 8, 658852. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scheme | Genotype | Source or Reference |
|---|---|---|
| BY4741 | MATahis3∆1 leu2∆0 ura3∆0 met15∆0 | Research Genetics, Inc. |
| BY4742 | MATα his3∆1 leu2∆0 ura3∆0 lys2∆0 | Research Genetics, Inc. |
| yAM135-A | BY4741 Ypk1(L424A)::ura3- ypk2∆::KanMX4 | [13] |
| yHG2 | BY4742 puf1∆::KanMX puf2∆::KanMX | Research Genetics, Inc. |
| yHG4 | BY4742 puf2∆::KanMX | Research Genetics, Inc. |
| yHG5 | BY4742 puf1∆::KanMX | This study |
| yHG8 | BY4742 Puf1-mKate2::SpHIS5 | This study |
| ySP1 | BY4742 Puf1(T174A S275A)-mKate2::SpHIS5 | This study |
| ySP8 | BY4742 Puf2-6HA-eGFP::SpHIS5-LEU2 | This study |
| ySP9 | BY4742 Puf2(S53A S54A S55A T56A T143A S246A S902A)-6HA-eGFP::SpHIS5-LEU2 | This study |
| yHG15 | BY4742 Puf1-6HA::LEU2 | This study |
| yHG16 | BY4742 Puf1(T174A S273A S275A)-6HA::LEU2 | This study |
| yHG22 | BY4742 Puf1(T174E S273E S275E)-6HA::LEU2 | This study |
| yHG17 | BY4742 Puf2-6HA::LEU2 | This study |
| yHG18 | BY4742 Puf2(S53A S54A S55A T56A T143A S246A S902A)-6HA::LEU2 | This study |
| YFR694 | BY4741 Puf1-6HA::LEU2 lys2∆0 MET15 | This study |
| YFR695 | BY4741 Puf1-6HA::LEU2 Ypk1(L424A)::URA3 ypk2∆::KanMX4 | This study |
| YFR699 | BY4741 Puf2-6HA::LEU2 | This study |
| YFR700 | BY4741 Puf2-6HA::LEU2 Ypk1(L424A)::URA3 ypk2∆::KanMX4 | This study |
| ySP18 | BY4742 Puf1::LEU2 Puf2-6HA::LEU2 Pmp3-3XFLAG met15∆0 | This study |
| ySP19 | BY4742 Puf1(T174A S273A S275A)::LEU2 Puf2(S53A S54A S55A T56A T143A S246A S902A)-6HA::LEU2 Pmp3-3XFLAG | This study |
| YFR205 | BY4741 fpk1∆::KanMX fpk2∆::KanMX lys2∆0 | [33] |
| YFR739-A | BY4742 Puf1::LEU2 Puf2-6HA::LEU2 met15∆0 | This study |
| YFR738 | BY4742 Puf1(T174A S273A S275A)::LEU2 Puf2(S53A S54A S55A T56A T143A S246A S902A)-6HA::LEU2 | This study |
| Plasmid | Description | Source/Reference |
|---|---|---|
| pAX50 | pBG1805 GAL1prom-Ypk1(L424A), URA3 | [13] |
| pGEX4T-1 | GST tag, bacterial expression vector, AmpR | GE Healthcare, Inc. |
| pHG10 | pGEX4T-1 Puf1(133-220) | This study |
| pHG11 | pGEX4T-1 Puf1(228-310) | This study |
| pHG12 | pGEX4T-1 Puf2(6-90) | This study |
| pHG13 | pGEX4T-1 Puf2(860-938) | This study |
| pHG18 | pGEX4T-1 Puf1(133-220) (T174A) | This study |
| pHG19 | pGEX4T-1 Puf1(228-310) (S273A S275A) | This study |
| pHG20 | pGEX4T-1 Puf2(6-90) (S53A S54A S55A T56A) | This study |
| pHG21 | pGEX4T-1 Puf2(860-938) (S902A) | This study |
| pKS137 | CEN, PGK1prom-λN-1XFLAG-BFP::SpHIS5 | [44] |
| pHG32 | pKS137-Puf1-λN-1XFLAG-BFP | This study |
| pHG33 | pKS137-Puf1(T174A S275A)-λN-1XFLAG-BFP | This study |
| pHG35 | pKS137-Puf1(1-340)-λN-1XFLAG-BFP | This study |
| pHG36 | pKS137-Puf1(1-340) T174A S275A-λN-1XFLAG-BFP | This study |
| pNTI473 | pPGK1::mCherry::(PP7)3 | [44] |
| pGF-V789 | pRS316-GALprom-Cas9 | [45] |
| pSP9 | pRS423-sgRNAPMP3 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galez, H.A.; Roelants, F.M.; Palm, S.M.; Reynaud, K.K.; Ingolia, N.T.; Thorner, J. Phosphorylation of mRNA-Binding Proteins Puf1 and Puf2 by TORC2-Activated Protein Kinase Ypk1 Alleviates Their Repressive Effects. Membranes 2021, 11, 500. https://doi.org/10.3390/membranes11070500
Galez HA, Roelants FM, Palm SM, Reynaud KK, Ingolia NT, Thorner J. Phosphorylation of mRNA-Binding Proteins Puf1 and Puf2 by TORC2-Activated Protein Kinase Ypk1 Alleviates Their Repressive Effects. Membranes. 2021; 11(7):500. https://doi.org/10.3390/membranes11070500
Chicago/Turabian StyleGalez, Henri A., Françoise M. Roelants, Sarah M. Palm, Kendra K. Reynaud, Nicholas T. Ingolia, and Jeremy Thorner. 2021. "Phosphorylation of mRNA-Binding Proteins Puf1 and Puf2 by TORC2-Activated Protein Kinase Ypk1 Alleviates Their Repressive Effects" Membranes 11, no. 7: 500. https://doi.org/10.3390/membranes11070500
APA StyleGalez, H. A., Roelants, F. M., Palm, S. M., Reynaud, K. K., Ingolia, N. T., & Thorner, J. (2021). Phosphorylation of mRNA-Binding Proteins Puf1 and Puf2 by TORC2-Activated Protein Kinase Ypk1 Alleviates Their Repressive Effects. Membranes, 11(7), 500. https://doi.org/10.3390/membranes11070500