
Descriptors of Secondary Active Transporter Function and How They Relate to Partial Reactions in the Transport Cycle


 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Methods
3. Results
3.1. The Minimal Model of Secondary Active Transporter
3.2. Kinetic Models can Predict Experimental Outcomes
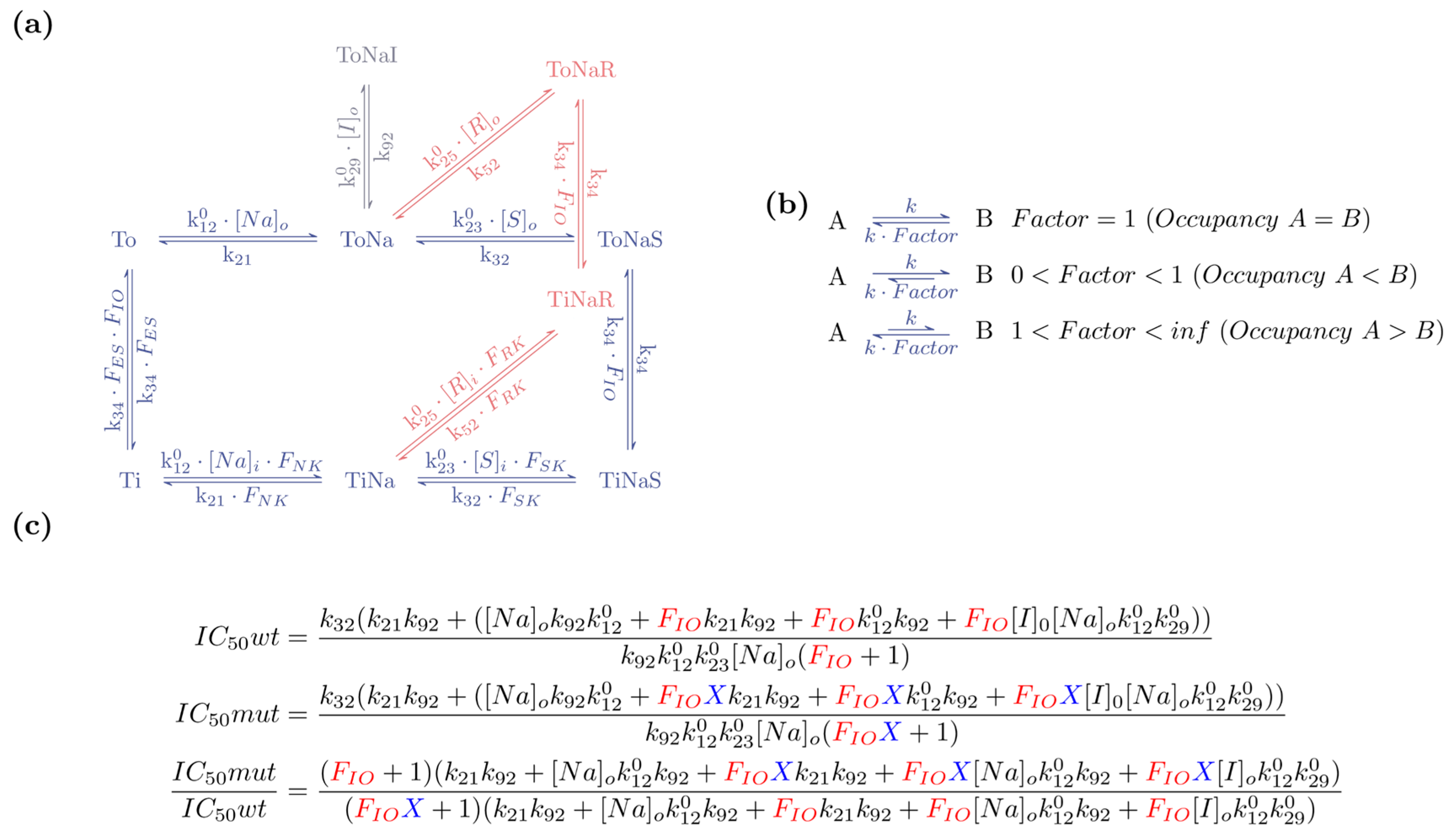
3.3. Explicit Mathematical Expressions Defining Descriptors
3.4. Experimental Outcomes of Descriptors Offer Diagnosis to the Type of Functional Defect Caused by a Mutation
3.5. Anomalous Substrate Release
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A. Derivation of the Explicit Terms for State Occupancies and Net-Fluxes though Cycles at Steady State

References
- Hediger, M.A.; Clémençon, B.; Burrier, R.E.; Bruford, E.A. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol. Asp. Med. 2013, 34, 95–107. [Google Scholar] [CrossRef]
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J. Pharm. Sci. 2020, 15, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef]
- Jardetzky, O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Ung, P.M.; Schlessinger, A. SLC Transporters: Structure, Function, and Drug Discovery. Medchemcomm 2016, 7, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- César-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef]
- Bhat, S.; El-Kasaby, A.; Freissmuth, M.; Sucic, S. Functional and Biochemical Consequences of Disease Variants in Neurotransmitter Transporters: A Special Emphasis on Folding and Trafficking Deficits. Pharmacol. Ther. 2020, 222, 107785. [Google Scholar] [CrossRef]
- Schmidt, H.; Jirstrand, M. Systems Biology Toolbox for MATLAB: A computational platform for research in systems biology. Bioinformatics 2006, 22, 514–515. [Google Scholar] [CrossRef]
- Humphreys, C.J.; Wall, S.C.; Rudnick, G. Ligand binding to the serotonin transporter: Equilibria, kinetics, and ion dependence. Biochemistry 1994, 33, 9118–9125. [Google Scholar] [CrossRef]
- Roux, M.J.; Supplisson, S. Neuronal and glial glycine transporters have different stoichiometries. Neuron 2000, 25, 373–383. [Google Scholar] [CrossRef]
- Hasenhuetl, P.S.; Bhat, S.; Mayer, F.P.; Sitte, H.H.; Freissmuth, M.; Sandtner, W. A kinetic account for amphetamine-induced monoamine release. J. Gen. Physiol. 2018, 150, 431–451. [Google Scholar] [CrossRef]
- Erdem, F.A.; Ilic, M.; Koppensteiner, P.; Gołacki, J.; Lubec, G.; Freissmuth, M.; Sandtner, W. A comparison of the transport kinetics of glycine transporter 1 and glycine transporter 2. J. Gen. Physiol. 2019, 151, 1035–1050. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, B.; Loo, D.D.; Wright, E.M. Relationships between Na+/glucose cotransporter (SGLT1) currents and fluxes. J. Membr. Biol. 1998, 162, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, V.; Schicker, K.; Freissmuth, M.; Sandtner, W. Kinetic Models of Secondary Active Transporters. Int. J. Mol. Sci. 2019, 20, 5365. [Google Scholar] [CrossRef]
- Zhang, Z.; Tao, Z.; Gameiro, A.; Barcelona, S.; Braams, S.; Rauen, T.; Grewer, C. Transport direction determines the kinetics of substrate transport by the glutamate transporter EAAC1. Proc. Natl. Acad. Sci. USA 2007, 104, 18025–18030. [Google Scholar] [CrossRef]
- Schicker, K.; Uzelac, Z.; Gesmonde, J.; Bulling, S.; Stockner, T.; Freissmuth, M.; Boehm, S.; Rudnick, G.; Sitte, H.H.; Sandtner, W. Unifying concept of serotonin transporter-associated currents. J. Biol. Chem. 2012, 287, 438–445. [Google Scholar] [CrossRef] [PubMed]
- King, E.L.; Altman, C. A Schematic Method of Deriving the Rate Laws for Enzyme-Catalyzed Reactions. J. Phys. Chem. 1956, 60, 1375–1378. [Google Scholar] [CrossRef]
- Hill, T.L. Free Energy Transduction and Biochemical Cycle Kinetics; Dover Publications: Mineola, NY, USA, 2004; ISBN 9780486441948. [Google Scholar]
- Kahlig, K.M.; Binda, F.; Khoshbouei, H.; Blakely, R.D.; McMahon, D.G.; Javitch, J.A.; Galli, A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc. Natl. Acad. Sci. USA 2005, 102, 3495–3500. [Google Scholar] [CrossRef] [PubMed]
- Sandtner, W.; Schmid, D.; Schicker, K.; Gerstbrein, K.; Koenig, X.; Mayer, F.P.; Boehm, S.; Freissmuth, M.; Sitte, H.H. A quantitative model of amphetamine action on the 5-HT transporter. Br. J. Pharmacol. 2014, 171, 1007–1018. [Google Scholar] [CrossRef]
- Mazei-Robison, M.S.; Bowton, E.; Holy, M.; Schmudermaier, M.; Freissmuth, M.; Sitte, H.H.; Galli, A.; Blakely, R.D. Anomalous dopamine release associated with a human dopamine transporter coding variant. J. Neurosci. 2008, 28, 7040–7046. [Google Scholar] [CrossRef]
- Bowton, E.; Saunders, C.; Erreger, K.; Sakrikar, D.; Matthies, H.J.; Sen, N.; Jessen, T.; Colbran, R.J.; Caron, M.G.; Javitch, J.A.; et al. Dysregulation of dopamine transporters via dopamine D2 autoreceptors triggers anomalous dopamine efflux associated with attention-deficit hyperactivity disorder. J. Neurosci. 2010, 30, 6048–6057. [Google Scholar] [CrossRef]
- Herborg, F.; Andreassen, T.F.; Berlin, F.; Loland, C.J.; Gether, U. Neuropsychiatric disease-associated genetic variants of the dopamine transporter display heterogeneous molecular phenotypes. J. Biol. Chem. 2018, 293, 7250–7262. [Google Scholar] [CrossRef] [PubMed]
- Erreger, K.; Grewer, C.; Javitch, J.A.; Galli, A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J. Neurosci. 2008, 28, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, P.J.; Campbell, N.G.; Sharma, S.; Erreger, K.; Herborg Hansen, F.; Saunders, C.; Belovich, A.N.; Sahai, M.A.; Cook, E.H.; NIH ARRA Autism Sequencing Consortium; et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 2013, 18, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, P.J.; Shekar, A.; Belovich, A.N.; Christianson, N.B.; Campbell, N.G.; Sutcliffe, J.S.; Galli, A.; Matthies, H.J.; Erreger, K. Zn(2+) reverses functional deficits in a de novo dopamine transporter variant associated with autism spectrum disorder. Mol. Autism 2015, 6, 8. [Google Scholar] [CrossRef][Green Version]
- Grewer, C.; Gameiro, A.; Mager, T.; Fendler, K. Electrophysiological characterization of membrane transport proteins. Annu. Rev. Biophys. 2013, 42, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Hasenhuetl, P.S.; Freissmuth, M.; Sandtner, W. Electrogenic Binding of Intracellular Cations Defines a Kinetic Decision Point in the Transport Cycle of the Human Serotonin Transporter. J. Biol. Chem. 2016, 291, 25864–25876. [Google Scholar] [CrossRef] [PubMed]
- Chiba, P.; Freissmuth, M.; Stockner, T. Defining the blanks--pharmacochaperoning of SLC6 transporters and ABC transporters. Pharmacol. Res. 2014, 83, 63–73. [Google Scholar] [CrossRef]
- Li, Y.; Hasenhuetl, P.S.; Schicker, K.; Sitte, H.H.; Freissmuth, M.; Sandtner, W. Dual Action of Zn2+ on the Transport Cycle of the Dopamine Transporter. J. Biol. Chem. 2015, 290, 31069–31076. [Google Scholar] [CrossRef]
- Niello, M.; Gradisch, R.; Loland, C.J.; Stockner, T.; Sitte, H.H. Allosteric Modulation of Neurotransmitter Transporters as a Therapeutic Strategy. Trends Pharmacol. Sci. 2020, 41, 446–463. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. An automatic method for deriving steady-state rate equations. Biochem. J. 1977, 165, 55–59. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original | Reparameterized |
|---|---|
| k43 | k34 × FIO |
| k18 | k34 × FES |
| k81 | k34 × FIO × FES |
| k56 | k34 |
| k65 | k34 × FIO |
| k076 | k025 × FRK |
| k52 | k34 × FRK |
| k087 | k012 × FNK |
| k78 | K21 × FNK |
| k074 | k023 × FSK |
| k47 | k32 × FSK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schicker, K.; Bhat, S.; Farr, C.; Burtscher, V.; Horner, A.; Freissmuth, M.; Sandtner, W. Descriptors of Secondary Active Transporter Function and How They Relate to Partial Reactions in the Transport Cycle. Membranes 2021, 11, 178. https://doi.org/10.3390/membranes11030178
Schicker K, Bhat S, Farr C, Burtscher V, Horner A, Freissmuth M, Sandtner W. Descriptors of Secondary Active Transporter Function and How They Relate to Partial Reactions in the Transport Cycle. Membranes. 2021; 11(3):178. https://doi.org/10.3390/membranes11030178
Chicago/Turabian StyleSchicker, Klaus, Shreyas Bhat, Clemens Farr, Verena Burtscher, Andreas Horner, Michael Freissmuth, and Walter Sandtner. 2021. "Descriptors of Secondary Active Transporter Function and How They Relate to Partial Reactions in the Transport Cycle" Membranes 11, no. 3: 178. https://doi.org/10.3390/membranes11030178
APA StyleSchicker, K., Bhat, S., Farr, C., Burtscher, V., Horner, A., Freissmuth, M., & Sandtner, W. (2021). Descriptors of Secondary Active Transporter Function and How They Relate to Partial Reactions in the Transport Cycle. Membranes, 11(3), 178. https://doi.org/10.3390/membranes11030178