
Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor

 ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Ivacaftor Binding Affinity of Murine P-gp
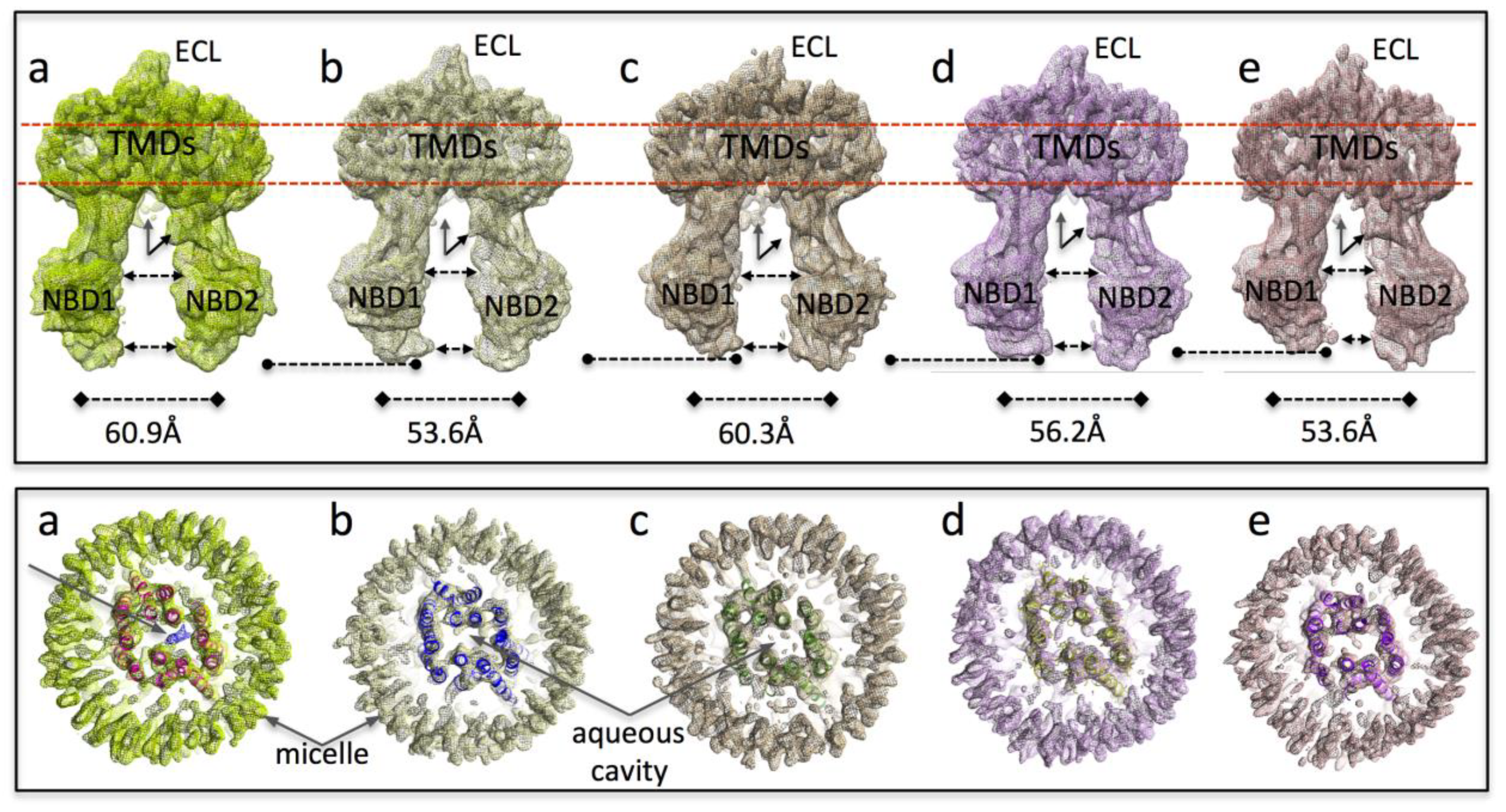
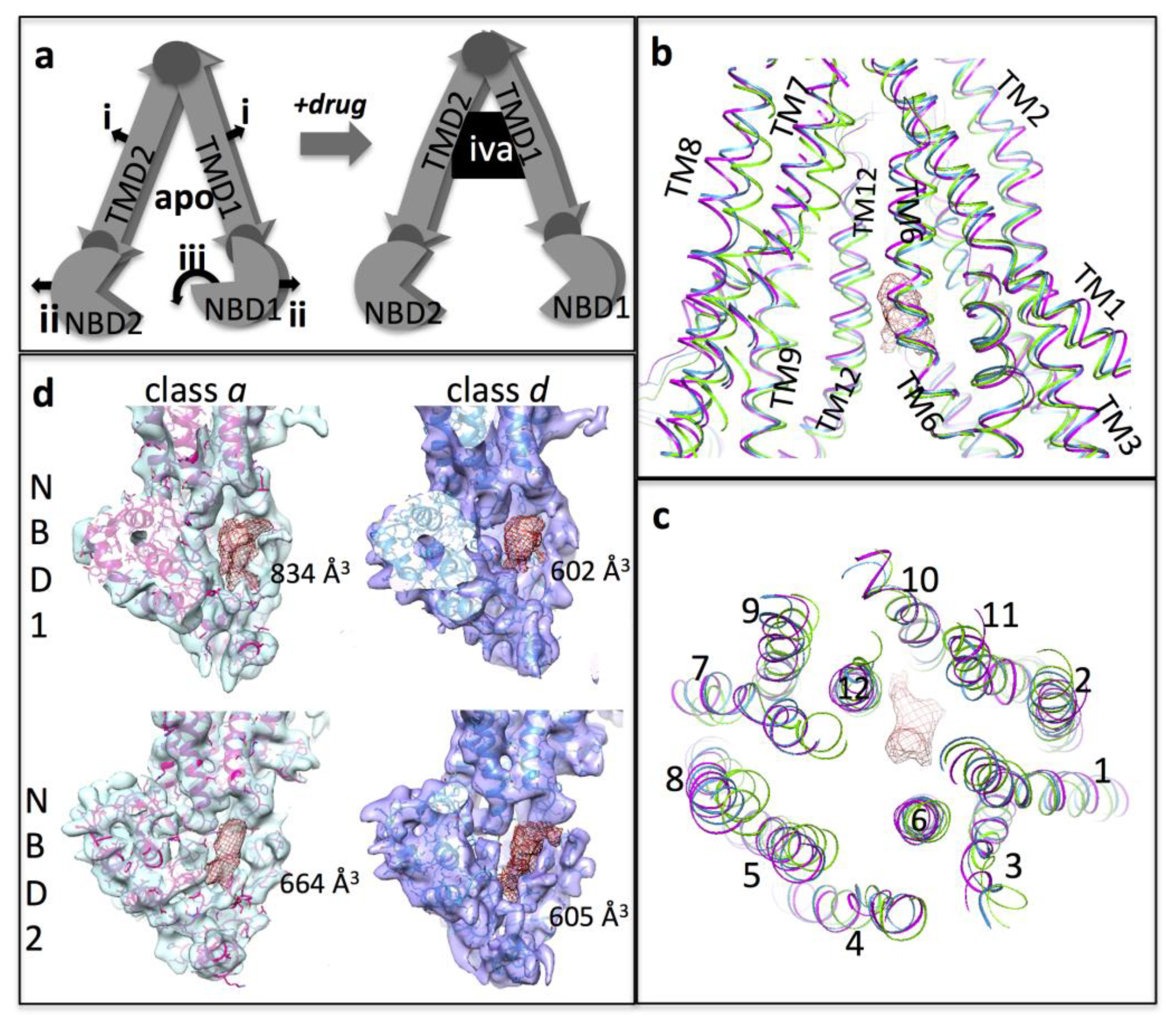
3.2. Cryo-EM of P-gp in the Presence of Ivacaftor
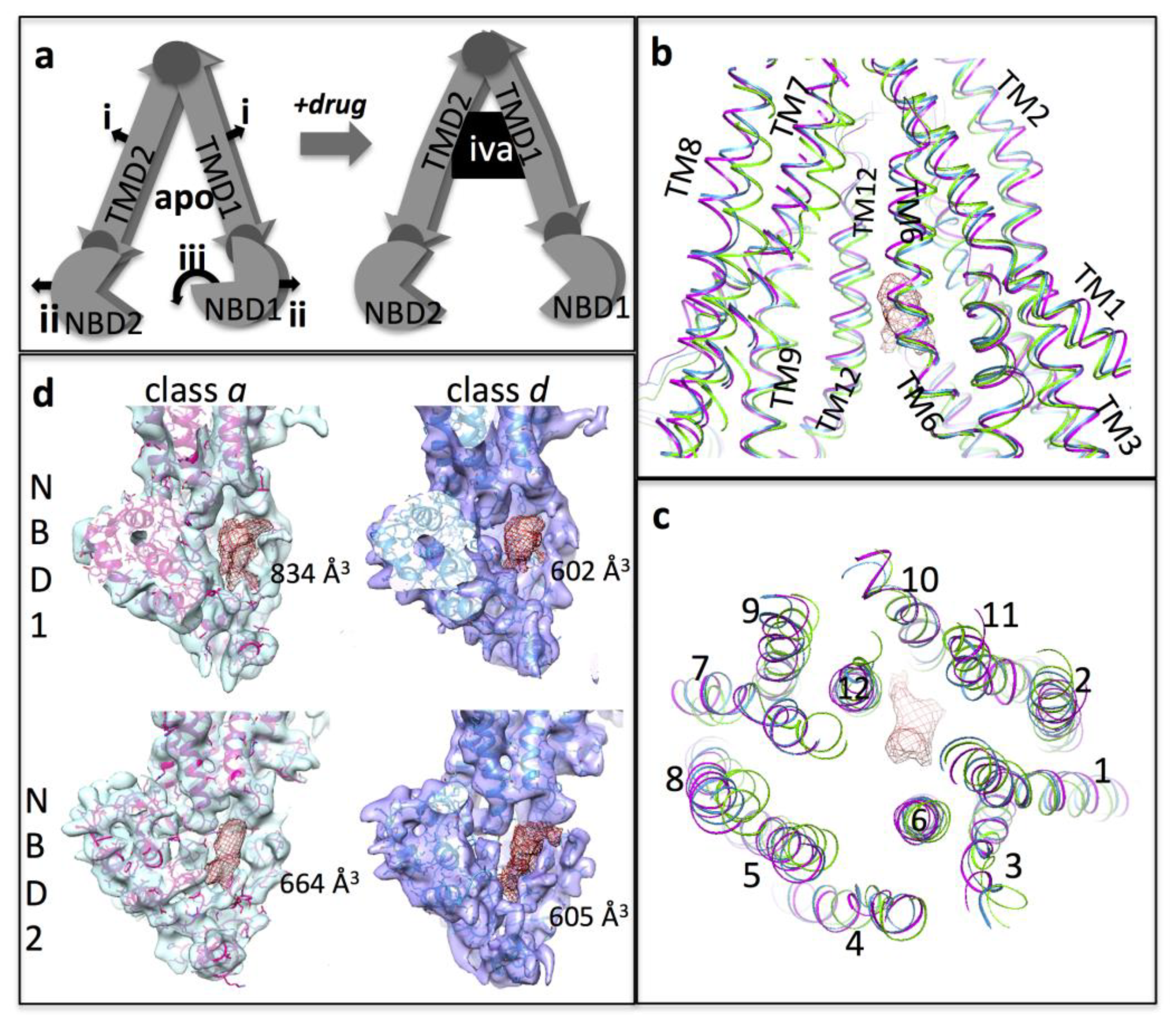
3.3. Interpretation of the Differences between the 3D Maps
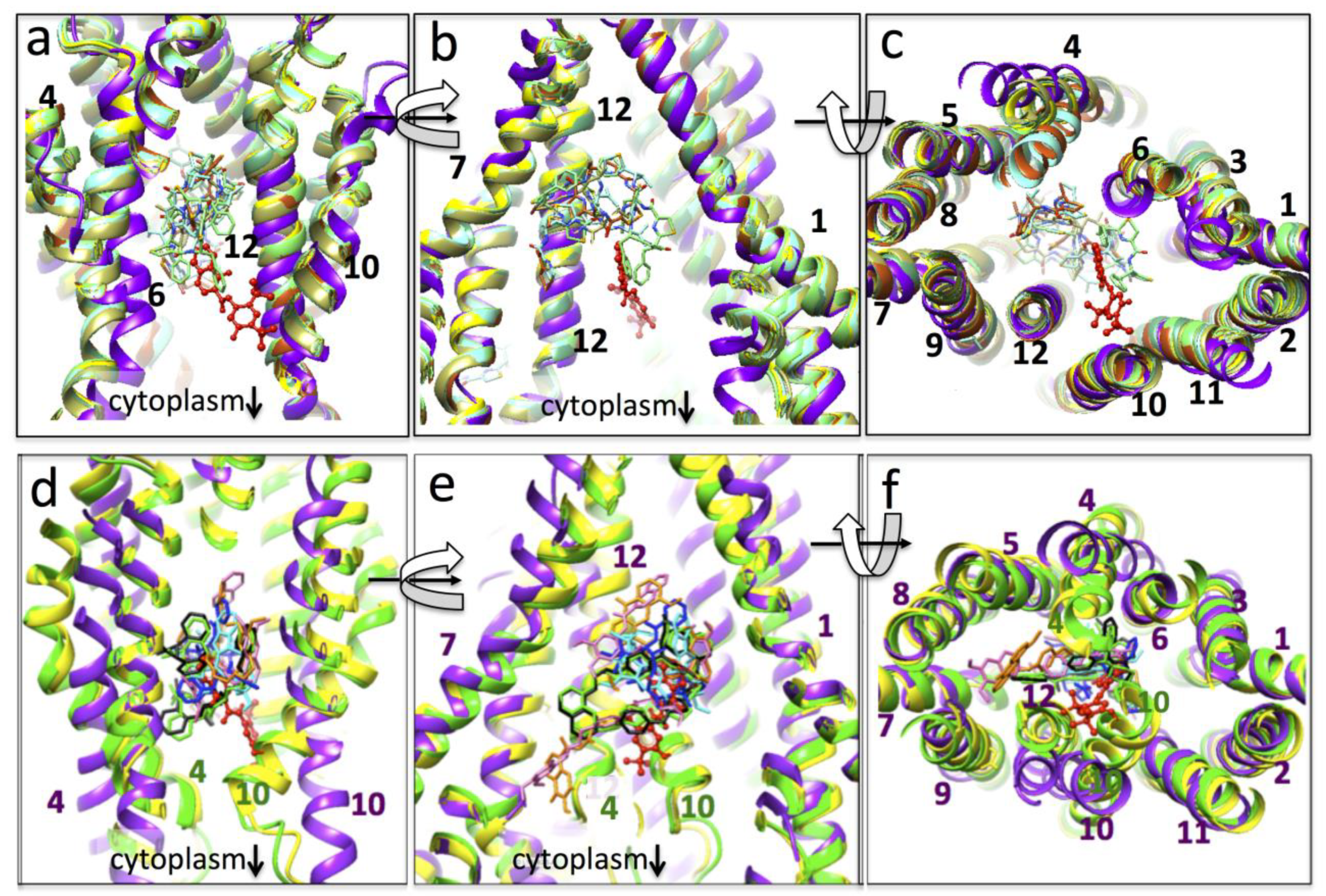
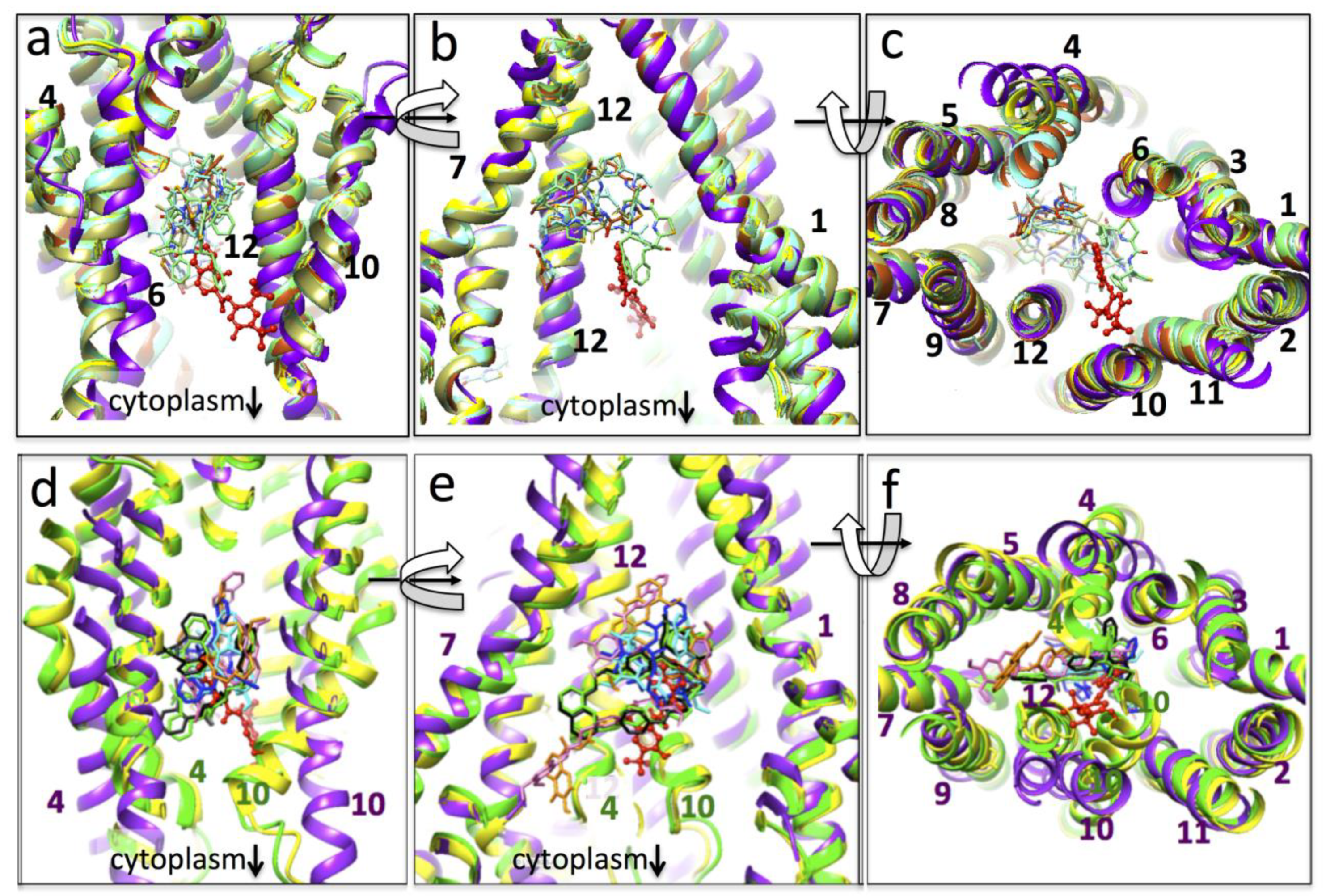
3.4. Conformational Changes Associated with the Additional Density
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Callaghan, R.; Ford, R.C.; Kerr, I.D. The translocation mechanism of P-glycoprotein. FEBS Lett. 2006, 580, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Callaghan, R.; Linton, K.J.; Rosenberg, M.F.; Ford, R.C. Structure of the multidrug resistance P-glycoprotein. Semin. Cancer Biol. 1997, 8, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.F.; Callaghan, R.; Ford, R.C.; Higgins, C.F. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J. Biol. Chem. 1997, 272, 10685–10694. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, M.F.; Velarde, G.; Ford, R.C.; Martin, C.; Berridge, G.; Kerr, I.D.; Callaghan, R.; Schmidlin, A.; Wooding, C.; Linton, K.J.; et al. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. Embo J. 2001, 20, 5615–5625. [Google Scholar] [CrossRef] [Green Version]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Alam, A.; Kung, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef] [Green Version]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Ford, R.C.; Marshall-Sabey, D.; Schuetz, J. Linker Domains: Why ABC Transporters ‘Live in Fragments no Longer’. Trends Biochem. Sci. 2020, 45, 137–148. [Google Scholar] [CrossRef]
- Ford, R.C.; Beis, K. Learning the ABCs one at a time: Structure and mechanism of ABC transporters. Biochem. Soc. Trans. 2019, 47, 23–36. [Google Scholar] [CrossRef]
- Ford, R.C.; Hellmich, U.A. What monomeric nucleotide binding domains can teach us about dimeric ABC proteins. FEBS Lett. 2020, 594, 3857–3875. [Google Scholar] [CrossRef]
- Hofmann, S.; Januliene, D.; Mehdipour, A.R.; Thomas, C.; Stefan, E.; Bruchert, S.; Kuhn, B.T.; Geertsma, E.R.; Hummer, G.; Tampe, R.; et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 2019, 571, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [Green Version]
- Le, C.A.; Harvey, D.S.; Aller, S.G. Structural definition of polyspecific compensatory ligand recognition by P-glycoprotein. IUCrJ 2020, 7, 663–672. [Google Scholar] [CrossRef]
- Nicklisch, S.C.; Rees, S.D.; McGrath, A.P.; Gokirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001. [Google Scholar] [CrossRef] [Green Version]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.K.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Frank, G.A.; Shukla, S.; Rao, P.; Borgnia, M.J.; Bartesaghi, A.; Merk, A.; Mobin, A.; Esser, L.; Earl, L.A.; Gottesman, M.M.; et al. Cryo-EM Analysis of the Conformational Landscape of Human P-glycoprotein (ABCB1) During its Catalytic Cycle. Mol. Pharmacol. 2016, 90, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thonghin, N.; Collins, R.F.; Barbieri, A.; Shafi, T.; Siebert, A.; Ford, R.C. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 A resolution. BMC Struct. Biol. 2018, 18, 17. [Google Scholar] [CrossRef]
- Lingam, S.; Thonghin, N.; Ford, R.C. Investigation of the effects of the CFTR potentiator ivacaftor on human P-glycoprotein (ABCB1). Sci. Rep. 2017, 7, 17481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, S.M.; Luo, X.; Dubey, N.; Li, C.; Chavan, A.B.; Gilmartin, G.S.; Higgins, M.; Mahnke, L. Clinical drug-drug interaction assessment of ivacaftor as a potential inhibitor of cytochrome P450 and P-glycoprotein. J. Clin. Pharmacol. 2015, 55, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Liddy, A.M.; McLaughlin, G.; Schmitz, S.; D’Arcy, D.M.; Barry, M.G. The pharmacokinetic interaction between ivacaftor and ritonavir in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2235–2241. [Google Scholar] [CrossRef]
- Wang, H.W.; Fan, X. Challenges and opportunities in cryo-EM with phase plate. Curr. Opin. Struct. Biol. 2019, 58, 175–182. [Google Scholar] [CrossRef]
- Hughes, T.E.T.; Lodowski, D.T.; Huynh, K.W.; Yazici, A.; Del Rosario, J.; Kapoor, A.; Basak, S.; Samanta, A.; Han, X.; Chakrapani, S.; et al. Structural basis of TRPV5 channel inhibition by econazole revealed by cryo-EM. Nat. Struct. Mol. Biol. 2018, 25, 53–60. [Google Scholar] [CrossRef]
- Twomey, E.C.; Yelshanskaya, M.V.; Vassilevski, A.A.; Sobolevsky, A.I. Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron 2018, 99, 956–968.e954. [Google Scholar] [CrossRef] [Green Version]
- Yelshanskaya, M.V.; Li, M.; Sobolevsky, A.I. Structure of an agonist-bound ionotropic glutamate receptor. Science 2014, 345, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Martin, G.M.; Yoshioka, C.; Rex, E.A.; Fay, J.F.; Xie, Q.; Whorton, M.R.; Chen, J.Z.; Shyng, S.L. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife 2017, 6, e24149. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jeong, E.; Jeong, J.H.; Kim, Y.; Cho, Y. Structural Basis for Activation of the Heterodimeric GABAB Receptor. J. Mol. Biol. 2020, 432, 5966–5984. [Google Scholar] [CrossRef] [PubMed]
- Polovinkin, L.; Hassaine, G.; Perot, J.; Neumann, E.; Jensen, A.A.; Lefebvre, S.N.; Corringer, P.J.; Neyton, J.; Chipot, C.; Dehez, F.; et al. Conformational transitions of the serotonin 5-HT3 receptor. Nature 2018, 563, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gui, M.; Wang, Z.F.; Gorgulla, C.; Yu, J.J.; Wu, H.; Sun, Z.J.; Klenk, C.; Merklinger, L.; Morstein, L.; et al. Cryo-EM structure of an activated GPCR-G protein complex in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 258–267. [Google Scholar] [CrossRef]
- Nys, M.; Wijckmans, E.; Farinha, A.; Yoluk, O.; Andersson, M.; Brams, M.; Spurny, R.; Peigneur, S.; Tytgat, J.; Lindahl, E.; et al. Allosteric binding site in a Cys-loop receptor ligand-binding domain unveiled in the crystal structure of ELIC in complex with chlorpromazine. Proc. Natl. Acad. Sci. USA 2016, 113, E6696–E6703. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Du, J.; Goehring, A.; Gouaux, E. Cryo-EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science 2017, 355, eaal3729. [Google Scholar] [CrossRef] [Green Version]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, Y.; Li, X. Structure of human Dispatched-1 provides insights into Hedgehog ligand biogenesis. Life Sci. Alliance 2020, 3, e202000776. [Google Scholar] [CrossRef] [PubMed]
- Krintel, C.; Dorosz, J.; Larsen, A.H.; Thorsen, T.S.; Venskutonyte, R.; Mirza, O.; Gajhede, M.; Boesen, T.; Kastrup, J.S. Binding of a negative allosteric modulator and competitive antagonist can occur simultaneously at the ionotropic glutamate receptor GluA2. FEBS J. 2021, 288, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Swartz, D.J.; Protasevich, I.I.; Brouillette, C.G.; Harrell, P.M.; Hildebrandt, E.; Gasser, B.; Mattanovich, D.; Ward, A.; Chang, G.; et al. A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS ONE 2011, 6, e22577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudet, L.; Urbatsch, I.L.; Gros, P. High-level expression of mouse Mdr3 P-glycoprotein in yeast Pichia pastoris and characterization of ATPase activity. Methods Enzymol. 1998, 292, 397–413. [Google Scholar] [CrossRef]
- Thonghin, N.; Kargas, V.; Clews, J.; Ford, R.C. Cryo-electron microscopy of membrane proteins. Methods 2018, 147, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Kohlstaedt, M.; von der Hocht, I.; Hilbers, F.; Thielmann, Y.; Michel, H. Development of a Thermofluor assay for stability determination of membrane proteins using the Na(+)/H(+) antiporter NhaA and cytochrome c oxidase. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1112–1122. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Clews, J.; Kargas, V.; Wang, X.; Ford, R.C. The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell Mol. Life Sci. 2017, 74, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Danev, R.; Buijsse, B.; Khoshouei, M.; Plitzko, J.M.; Baumeister, W. Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 15635–15640. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Mooney, P.; Zheng, S.; Booth, C.R.; Braunfeld, M.B.; Gubbens, S.; Agard, D.A.; Cheng, Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 2013, 10, 584–590. [Google Scholar] [CrossRef] [Green Version]
- Grant, T.; Rohou, A.; Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. Elife 2018, 7, e35383. [Google Scholar] [CrossRef]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Kucukelbir, A.; Sigworth, F.J.; Tagare, H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 2014, 11, 63–65. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swint-Kruse, L.; Brown, C.S. Resmap: Automated representation of macromolecular interfaces as two-dimensional networks. Bioinformatics 2005, 21, 3327–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; McMullan, G.; Faruqi, A.R.; Murshudov, G.N.; Short, J.M.; Scheres, S.H.; Henderson, R. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 2013, 135, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D.; et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Aller, S.G. Allosteric Role of Substrate Occupancy Toward the Alignment of P-glycoprotein Nucleotide Binding Domains. Sci. Rep. 2018, 8, 14643. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.B.; Fox, K.; Lam, P.; Ling, V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem. 1999, 259, 841–850. [Google Scholar] [CrossRef]
- Pajeva, I.K.; Hanl, M.; Wiese, M. Protein contacts and ligand binding in the inward-facing model of human P-glycoprotein. ChemMedChem 2013, 8, 748–762. [Google Scholar] [CrossRef]
- Bocci, G.; Moreau, A.; Vayer, P.; Denizot, C.; Fardel, O.; Parmentier, Y. New insights in the in vitro characterisation and molecular modelling of the P-glycoprotein inhibitory promiscuity. Eur. J. Pharm. Sci. 2018, 121, 85–94. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter. | Map a | Map b | Map c | Map d | Map e |
|---|---|---|---|---|---|
| Global Resolution (Å, FSC = 0.143) from unmasked half maps (CisTEM) | 5.4 | 4.3 | 4.3 | 4.2 | 4.2 |
| Global Resolution (Å, FSC = 0.143) from map to model fit (Phenix) | 4.6 | 4.4 | 4.5 | 4.4 | 4.4 |
| Resolution (Å) from Resmap with masked half maps (Mean, Mode) | 5.0, 4.0 | 4.5, 4.0 | 4.6, 4.0 | 4.5, 4.0 | 4.4, 4.0 |
| CC map to model fit (mean from 1182 residues, Phenix) | 0.73 | 0.67 | 0.63 | 0.69 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbieri, A.; Thonghin, N.; Shafi, T.; Prince, S.M.; Collins, R.F.; Ford, R.C. Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes 2021, 11, 923. https://doi.org/10.3390/membranes11120923
Barbieri A, Thonghin N, Shafi T, Prince SM, Collins RF, Ford RC. Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes. 2021; 11(12):923. https://doi.org/10.3390/membranes11120923
Chicago/Turabian StyleBarbieri, Alessandro, Nopnithi Thonghin, Talha Shafi, Stephen M. Prince, Richard F. Collins, and Robert C. Ford. 2021. "Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor" Membranes 11, no. 12: 923. https://doi.org/10.3390/membranes11120923
APA StyleBarbieri, A., Thonghin, N., Shafi, T., Prince, S. M., Collins, R. F., & Ford, R. C. (2021). Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes, 11(12), 923. https://doi.org/10.3390/membranes11120923