Simultaneous Quantification of Protein Binding Kinetics in Whole Cells with Surface Plasmon Resonance Imaging and Edge Deformation Tracking



Abstract
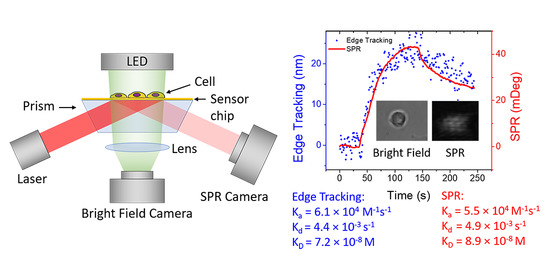
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Sensor Chip Preparation
2.3. Instrumentation
2.4. Experimental Protocol
2.5. Data Processing
3. Results and Discussion
3.1. Overview of the Integrated Imaging System
3.2. Single Cell Binding Kinetics Measured by Simultaneous Edge Tracking and SPR
3.3. Variability Validation
3.4. Correlation Analysis of the Observed Kinetics Constant
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Tamayo Tenorio, A.; Boom, R.M.; van der Goot, A.J. Understanding leaf membrane protein extraction to develop a food-grade process. Food Chem. 2017, 217, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Boussambe, G.N.M.; Guillet, P.; Mahler, F.; Marconnet, A.; Vargas, C.; Cornut, D.; Soulie, M.; Ebel, C.; Le Roy, A.; Jawhari, A.; et al. Fluorinated diglucose detergents for membrane-protein extraction. Methods 2018, 147, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.L.; Kemmer, G.C.; Pomorski, T.G. Membrane protein reconstitution into giant unilamellar vesicles: A review on current techniques. Eur. Biophys. J. 2017, 46, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Castell, O.K.; Dijkman, P.M.; Wiseman, D.N.; Goddard, A.D. Single molecule fluorescence for membrane proteins. Methods 2018, 147, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, S.; Yin, L.; Yang, Y.; Guan, Y.; Wang, W.; Xu, H.; Tao, N. Quantification of Epidermal Growth Factor Receptor Expression Level and Binding Kinetics on Cell Surfaces by Surface Plasmon Resonance Imaging. Anal. Chem. 2015, 87, 9960–9965. [Google Scholar] [CrossRef] [PubMed]
- Lepage, D.; Dubowski, J.J. Miniaturized quantum semiconductor surface plasmon resonance platform for detection of biological molecules. Biosensors 2013, 3, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jing, W.; Hunt, A.; Yu, H.; Yang, Y.; Wang, S.; Chen, H.Y.; Tao, N. Label-Free Quantification of Small-Molecule Binding to Membrane Proteins on Single Cells by Tracking Nanometer-Scale Cellular Membrane Deformation. ACS Nano 2018, 12, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Shan, X.; Zhang, F.; Wang, S.; Chen, H.Y.; Tao, N. Kinetics of small molecule interactions with membrane proteins in single cells measured with mechanical amplification. Sci. Adv. 2015, 1, e1500633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guan, Y.; Yang, Y.; Hunt, A.; Wang, S.; Chen, H.Y.; Tao, N. Optical Tracking of Nanometer-Scale Cellular Membrane Deformation Associated with Single Vesicle Release. ACS Sens. 2019, 4, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.L.; Papalia, G.A.; Flynn, P.J.; Furneisen, J.; Quinn, J.; Klein, J.S.; Katsamba, P.S.; Waddell, M.B.; Scott, M.; Thompson, J.; et al. A global benchmark study using affinity-based biosensors. Anal. Biochem. 2009, 386, 194–216. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.L.; Alvarez, M.; Fonseca, F.L.; Casadevall, A. Binding of the wheat germ lectin to Cryptococcus neoformans suggests an association of chitinlike structures with yeast budding and capsular glucuronoxylomannan. Eukaryot Cell 2008, 7, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Monsigny, M.; Roche, A.C.; Sene, C.; Maget-Dana, R.; Delmotte, F. Sugar-lectin interactions: How does wheat-germ agglutinin bind sialoglycoconjugates? Eur. J. Biochem. 1980, 104, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Boldt, D.H. Interaction of wheat germ agglutinin with human peripheral blood mononuclear cells. Binding kinetics and flow microfluorometric analysis. Mol. Immunol. 1980, 17, 47–55. [Google Scholar] [CrossRef]
- Bhavanandan, V.P.; Katlic, A.W. The interaction of wheat germ agglutinin with sialoglycoproteins. The role of sialic acid. J. Biol. Chem. 1979, 254, 4000–4008. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Edge Tracking ka (M−1s−1) | Edge Tracking kd (s−1) | Edge Tracking KD (M) | SPR ka (M−1s−1) | SPR kd (s−1) | SPR KD (M) | |
|---|---|---|---|---|---|---|
| Cell1 | 6.1 × 104 | 4.4 × 10−3 | 7.21 × 10−8 | 5.5 × 104 | 4.9 × 10−3 | 8.91 × 10−8 |
| Cell2 | 6.4 × 104 | 4.6 × 10−3 | 7.19 × 10−8 | 5.9 × 104 | 4.8 × 10−3 | 8.14 × 10−8 |
| Cell3 | 5.5 × 104 | 3.5 × 10−3 | 6.36 × 10−8 | 4.8 × 104 | 4.5 × 10−3 | 9.37 × 10−8 |
| Cell4 | 5.3 × 104 | 5.5 × 10−3 | 10.4 × 10−8 | 7.6 × 104 | 5.3 × 10−3 | 6.97 × 10−8 |
| Cell5 | 6.1 × 104 | 6.5 × 10−3 | 10.7 × 10−8 | 7.7 × 104 | 5.8 × 10−3 | 7.53 × 10−8 |
| Cell6 | 6.4 × 104 | 2.6 × 10−3 | 4.06 × 10−8 | 5.3 × 104 | 4.1 × 10−3 | 7.74 × 10−8 |
| Cell7 | 5.4 × 104 | 4.3 × 10−3 | 7.96 × 10−8 | 6.8 × 104 | 4.9 × 10−3 | 7.21 × 10−8 |
| Mean | (5.9 ± 0.47) × 104 | (4.5 ± 1.27) × 10−3 | (7.69 ± 2.29) × 10−8 | (6.2 ± 1.15) × 104 | (4.9 ± 0.54) × 10−3 | (7.98 ± 0.89) × 10−8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, W.; Hunt, A.; Tao, N.; Zhang, F.; Wang, S. Simultaneous Quantification of Protein Binding Kinetics in Whole Cells with Surface Plasmon Resonance Imaging and Edge Deformation Tracking. Membranes 2020, 10, 247. https://doi.org/10.3390/membranes10090247
Jing W, Hunt A, Tao N, Zhang F, Wang S. Simultaneous Quantification of Protein Binding Kinetics in Whole Cells with Surface Plasmon Resonance Imaging and Edge Deformation Tracking. Membranes. 2020; 10(9):247. https://doi.org/10.3390/membranes10090247
Chicago/Turabian StyleJing, Wenwen, Ashley Hunt, Nongjian Tao, Fenni Zhang, and Shaopeng Wang. 2020. "Simultaneous Quantification of Protein Binding Kinetics in Whole Cells with Surface Plasmon Resonance Imaging and Edge Deformation Tracking" Membranes 10, no. 9: 247. https://doi.org/10.3390/membranes10090247
APA StyleJing, W., Hunt, A., Tao, N., Zhang, F., & Wang, S. (2020). Simultaneous Quantification of Protein Binding Kinetics in Whole Cells with Surface Plasmon Resonance Imaging and Edge Deformation Tracking. Membranes, 10(9), 247. https://doi.org/10.3390/membranes10090247