Water Splitting and Transport of Ions in Electromembrane System with Bilayer Ion-Exchange Membrane


Abstract
1. Introduction
2. Experimental
2.1. Objects of the Study
2.1.1. Preparation of Bilayer Membranes with a Thin Cation-Exchange Film (BM-a)
2.1.2. Preparation of Bilayer Membranes with a Thin Anion-Exchange Film (BM-c-A80)
2.1.3. Bilayer Membranes with Water Splitting Catalysts (BM-ac)
2.1.4. Electrodeposition of Transition Metal Hydroxides on the Bipolar Boundary
2.1.5. Deposition of Iron (III) Hydroxide Sol
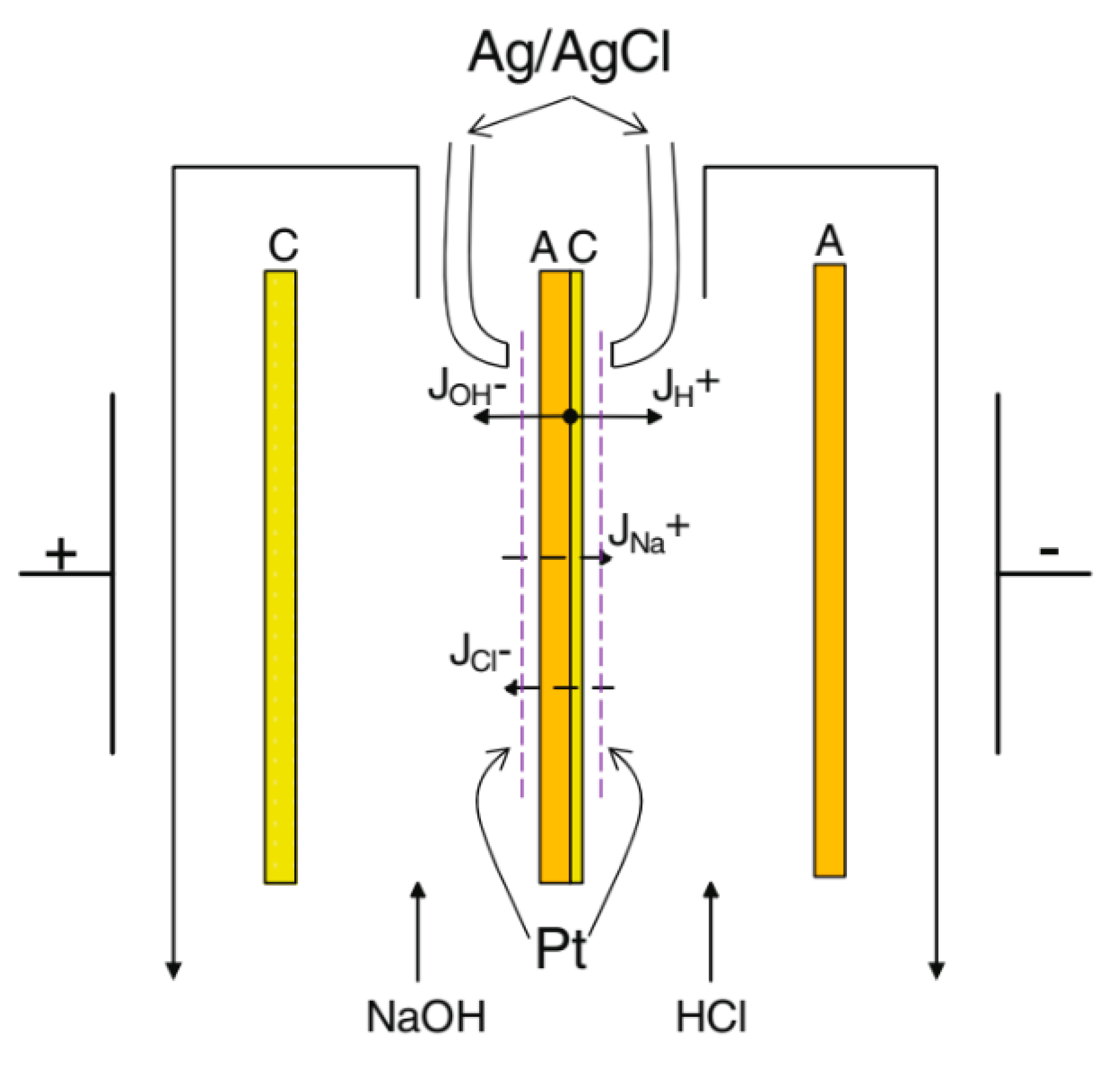
2.2. Electrochemical Measurements
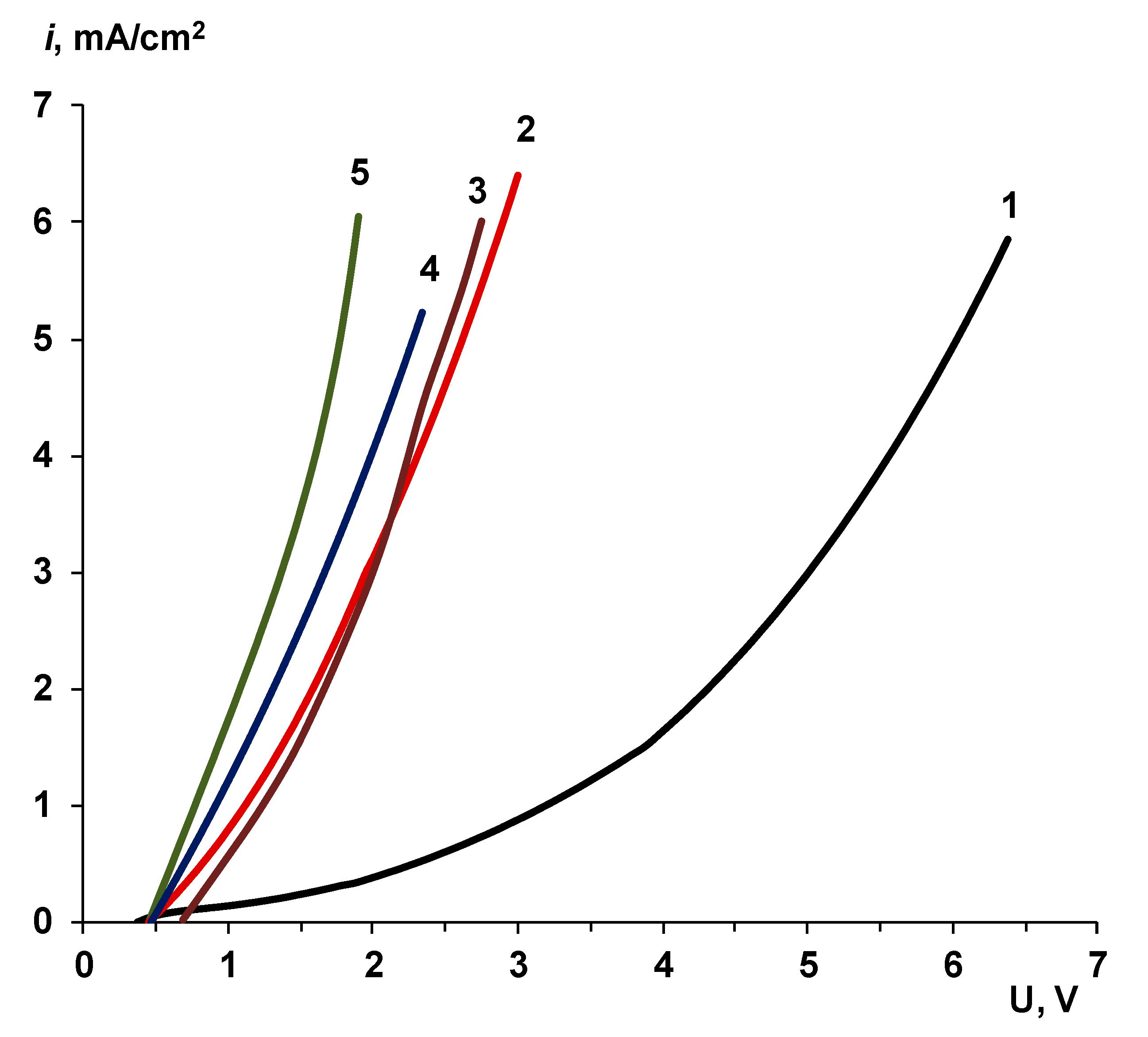
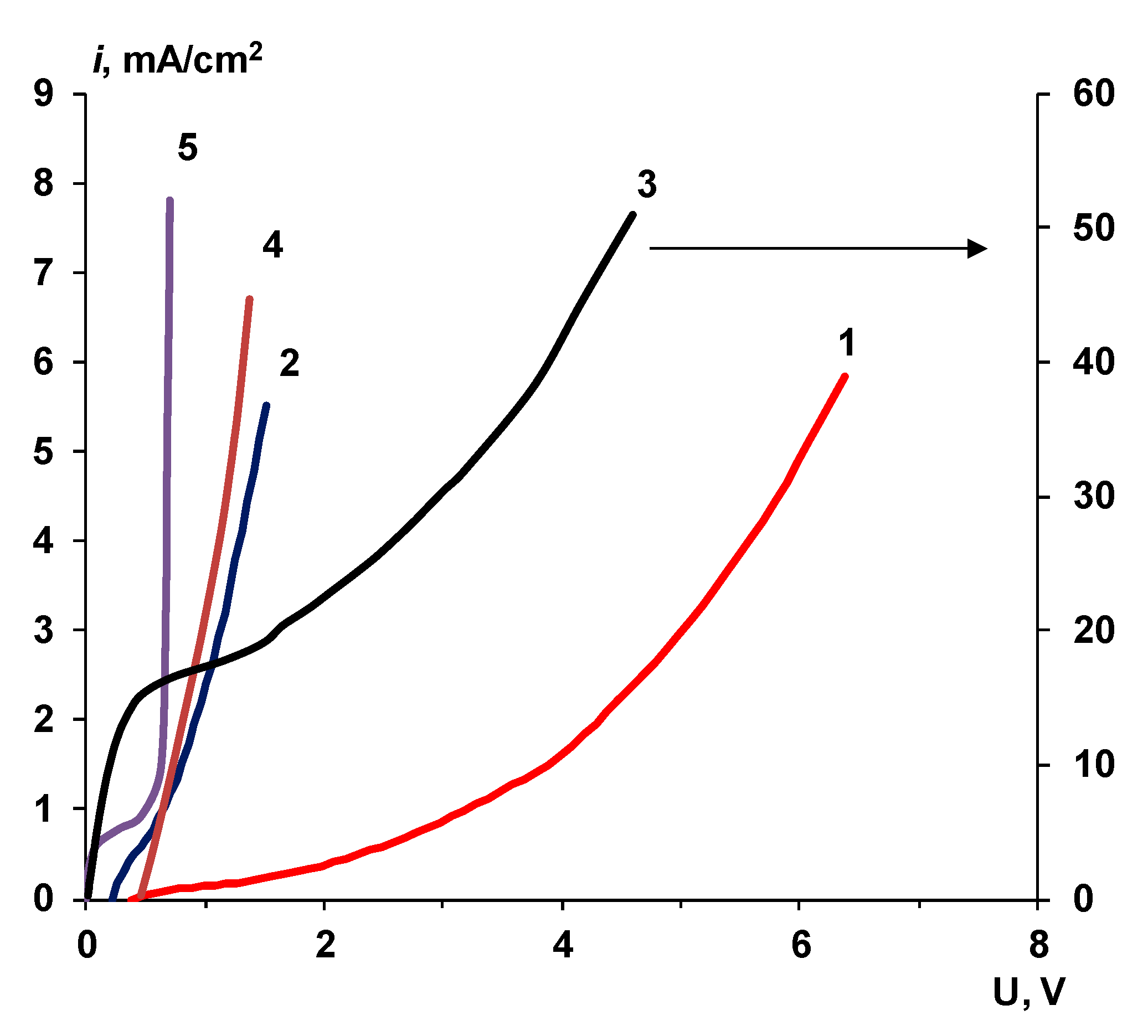
2.2.1. Voltammetry
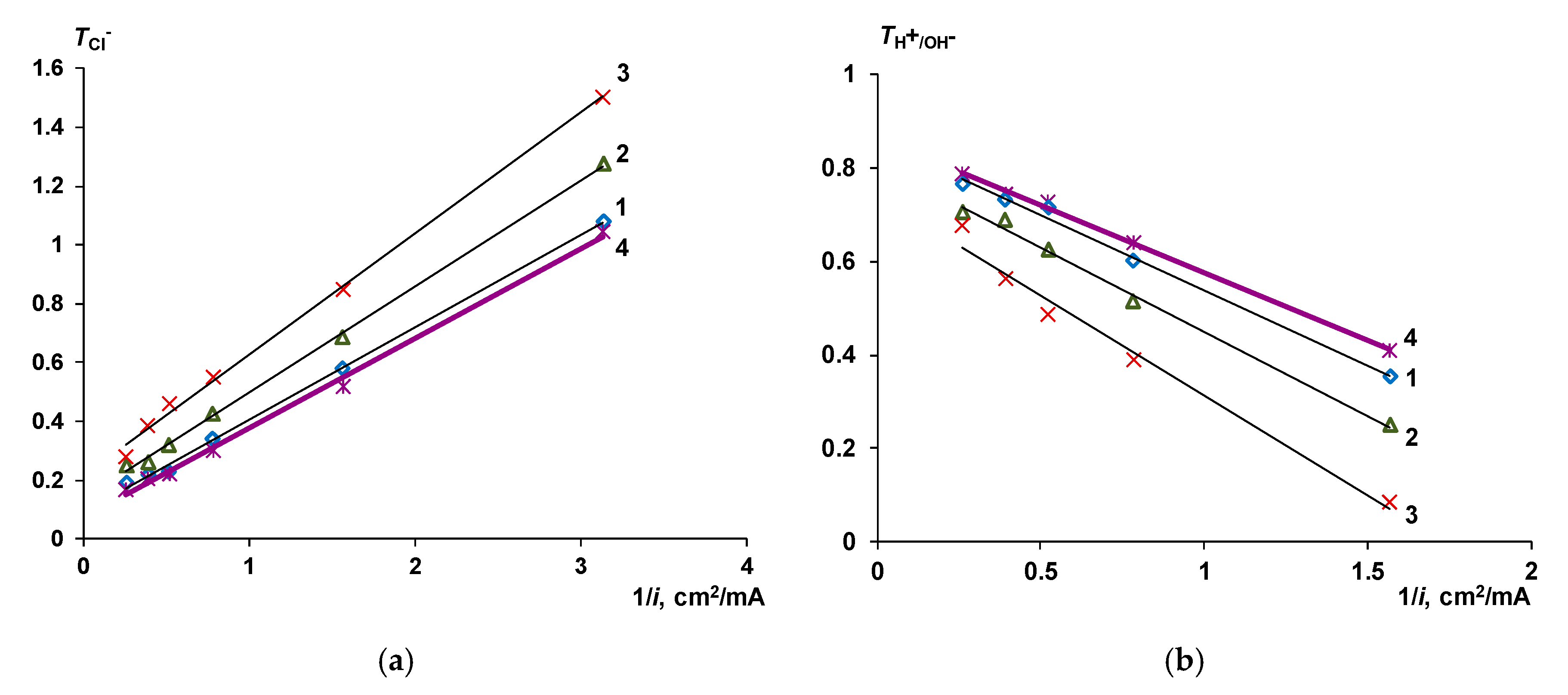
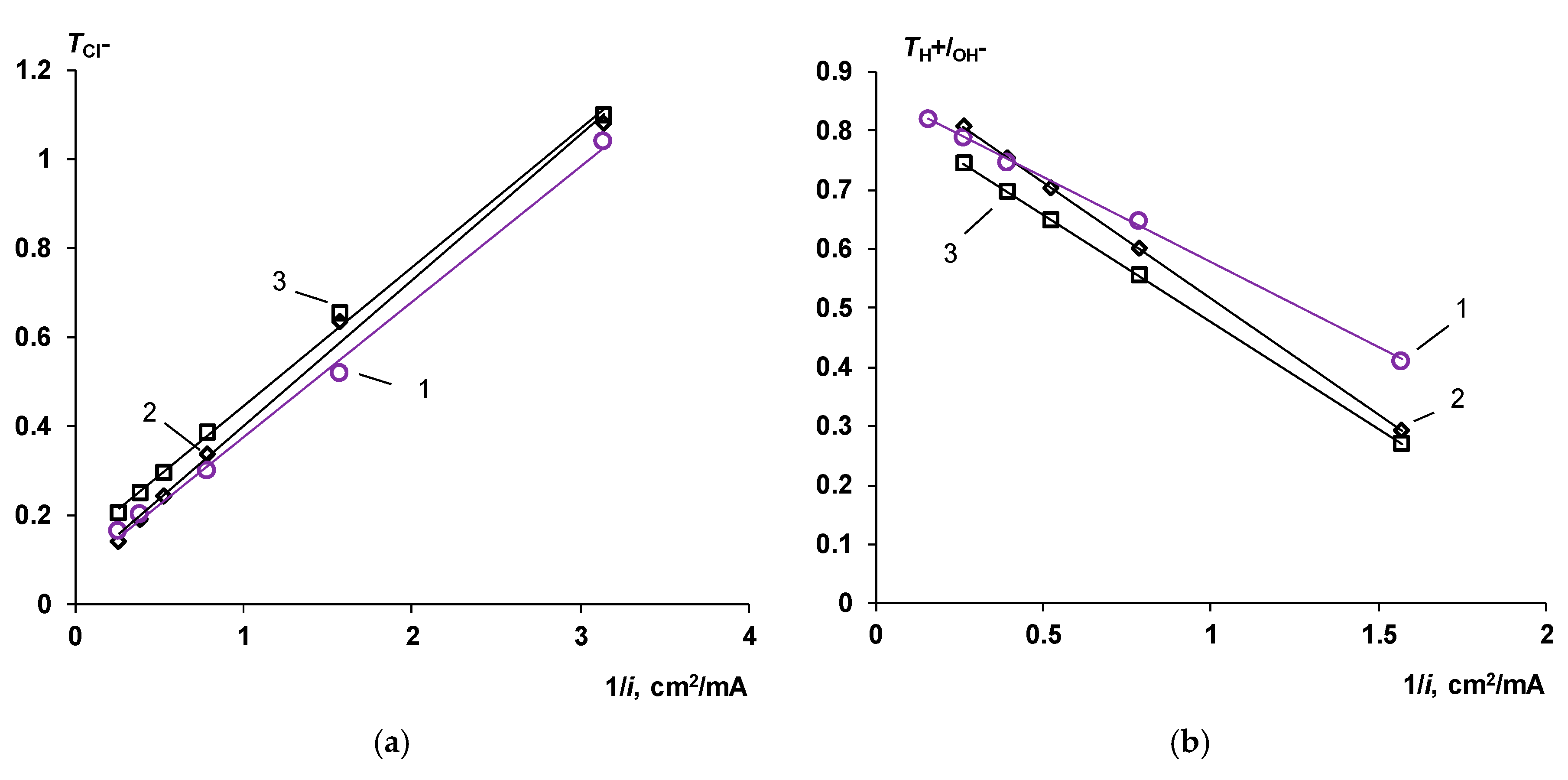
2.2.2. Transport Numbers
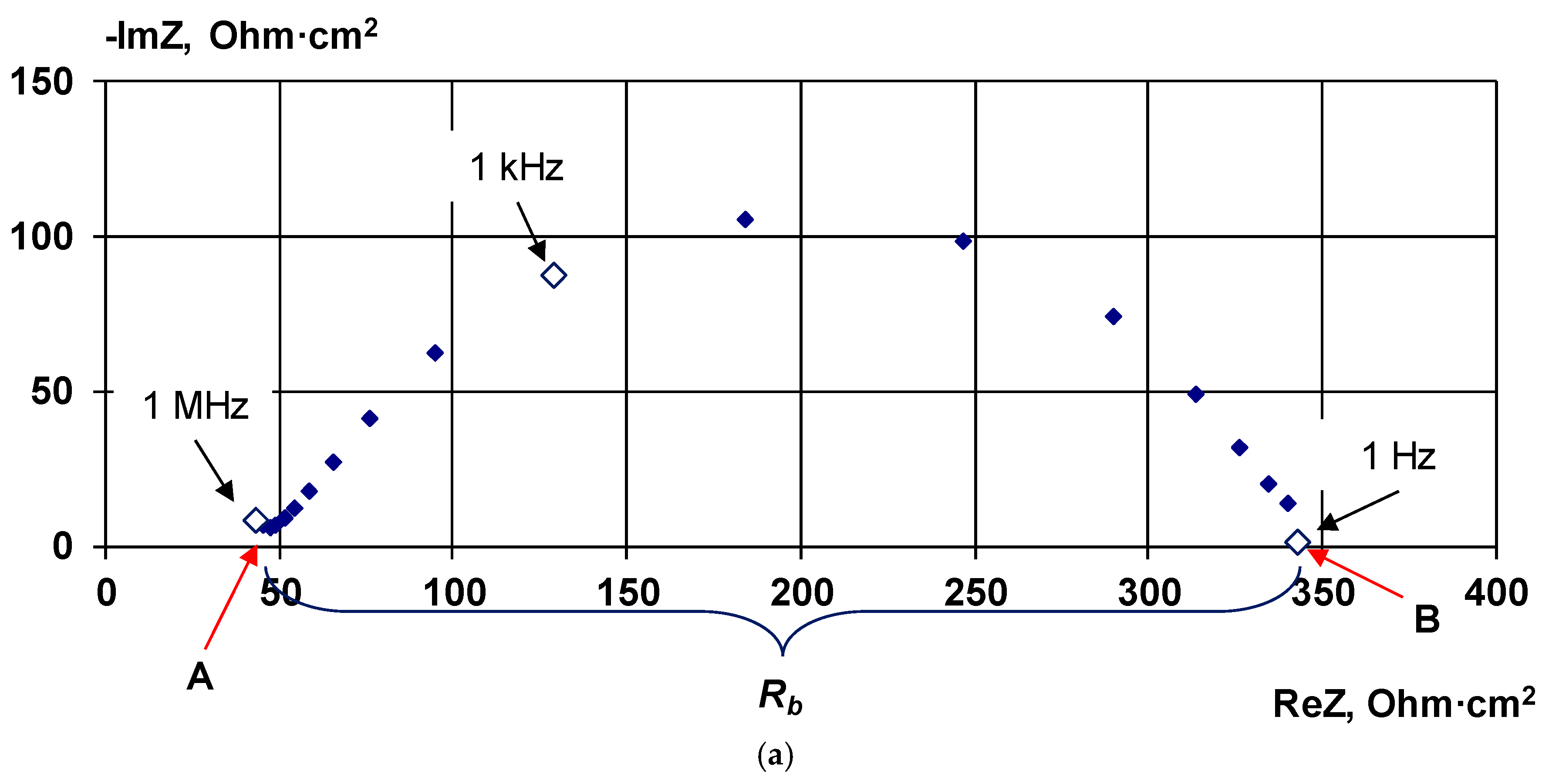
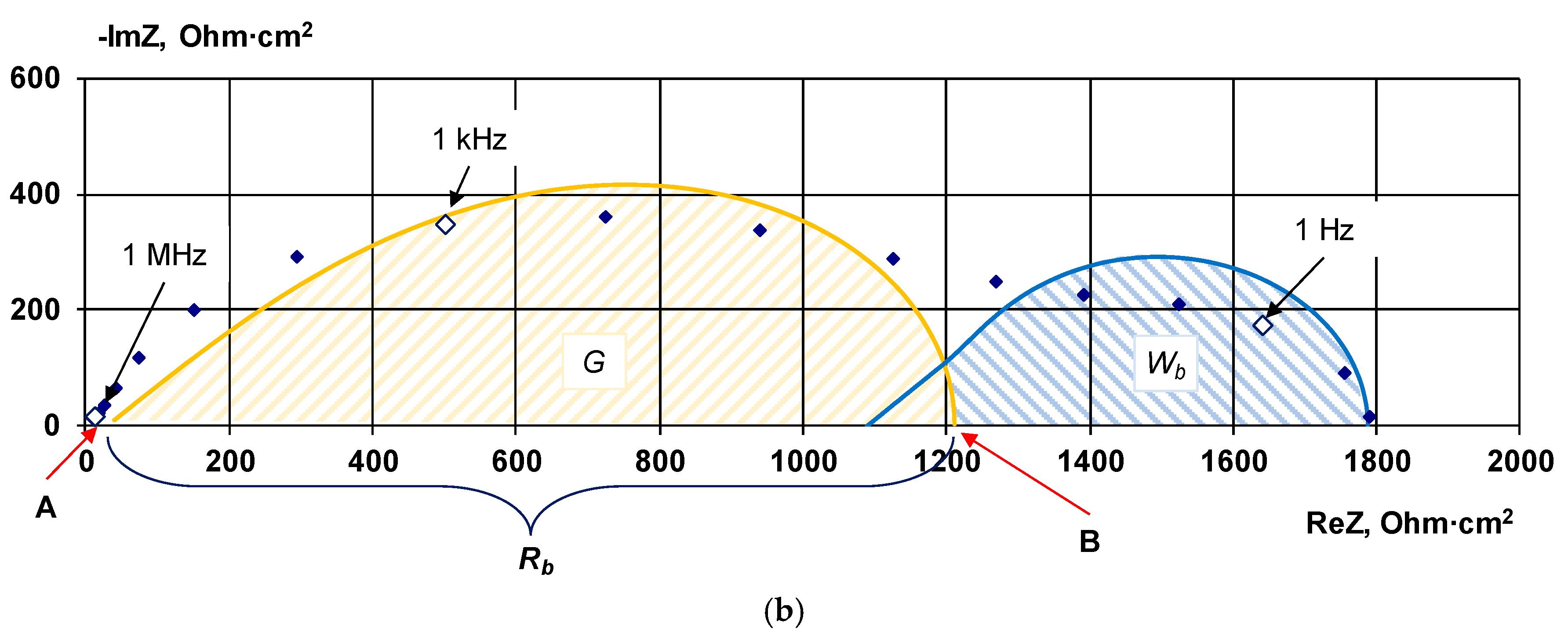
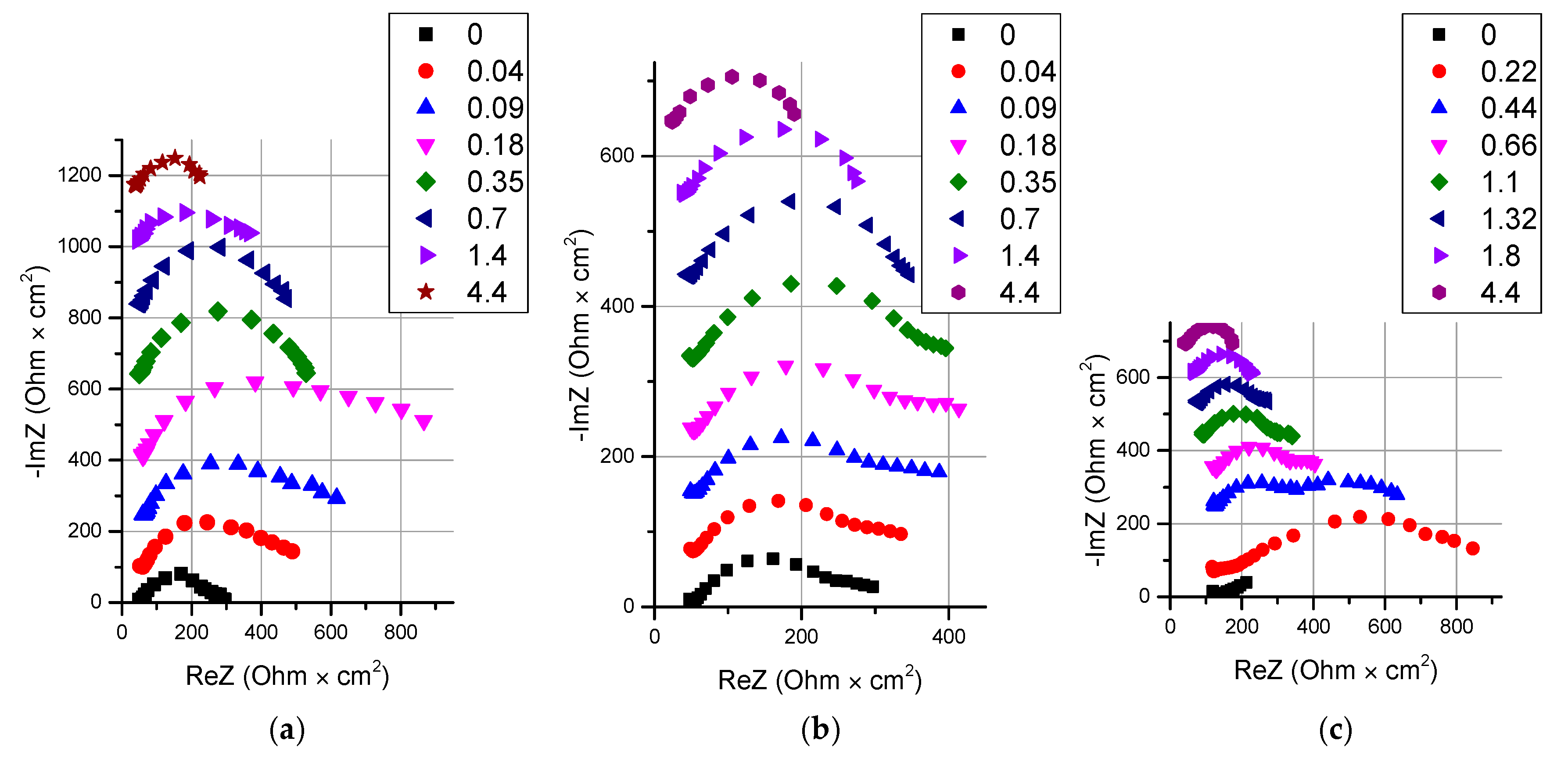
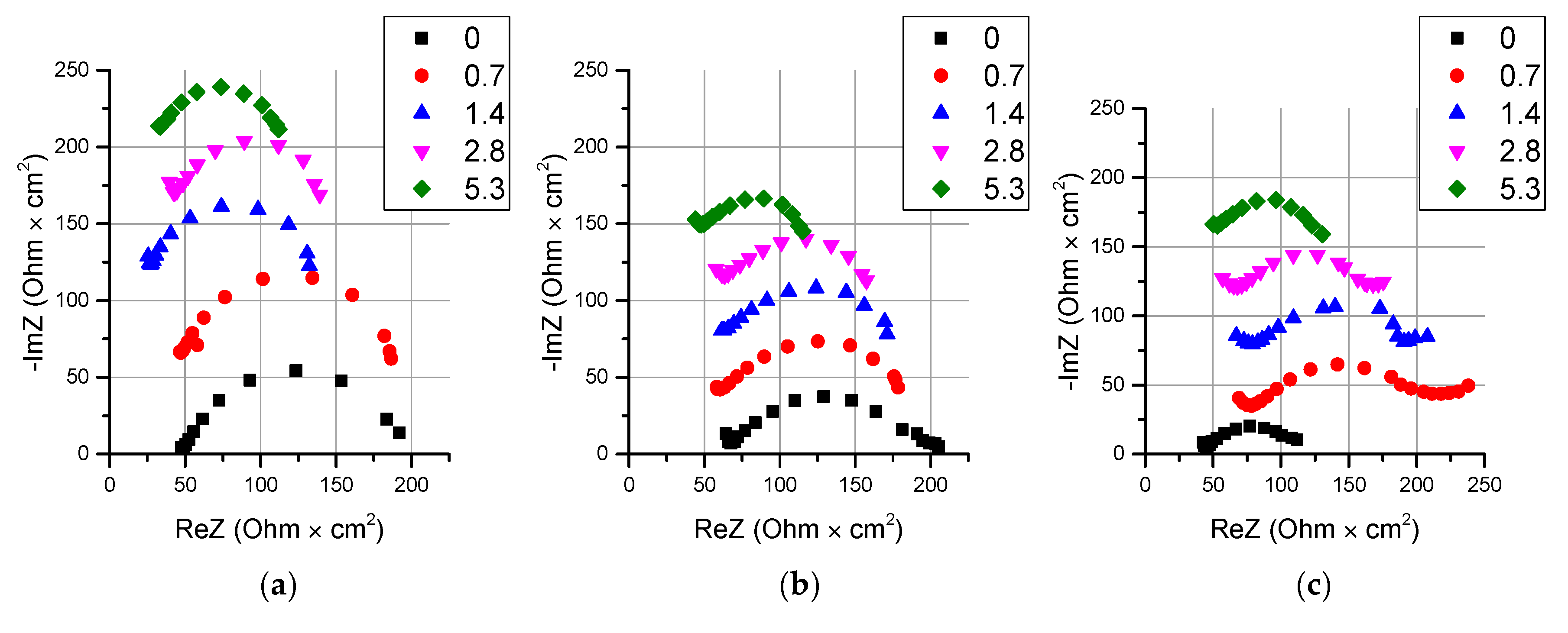
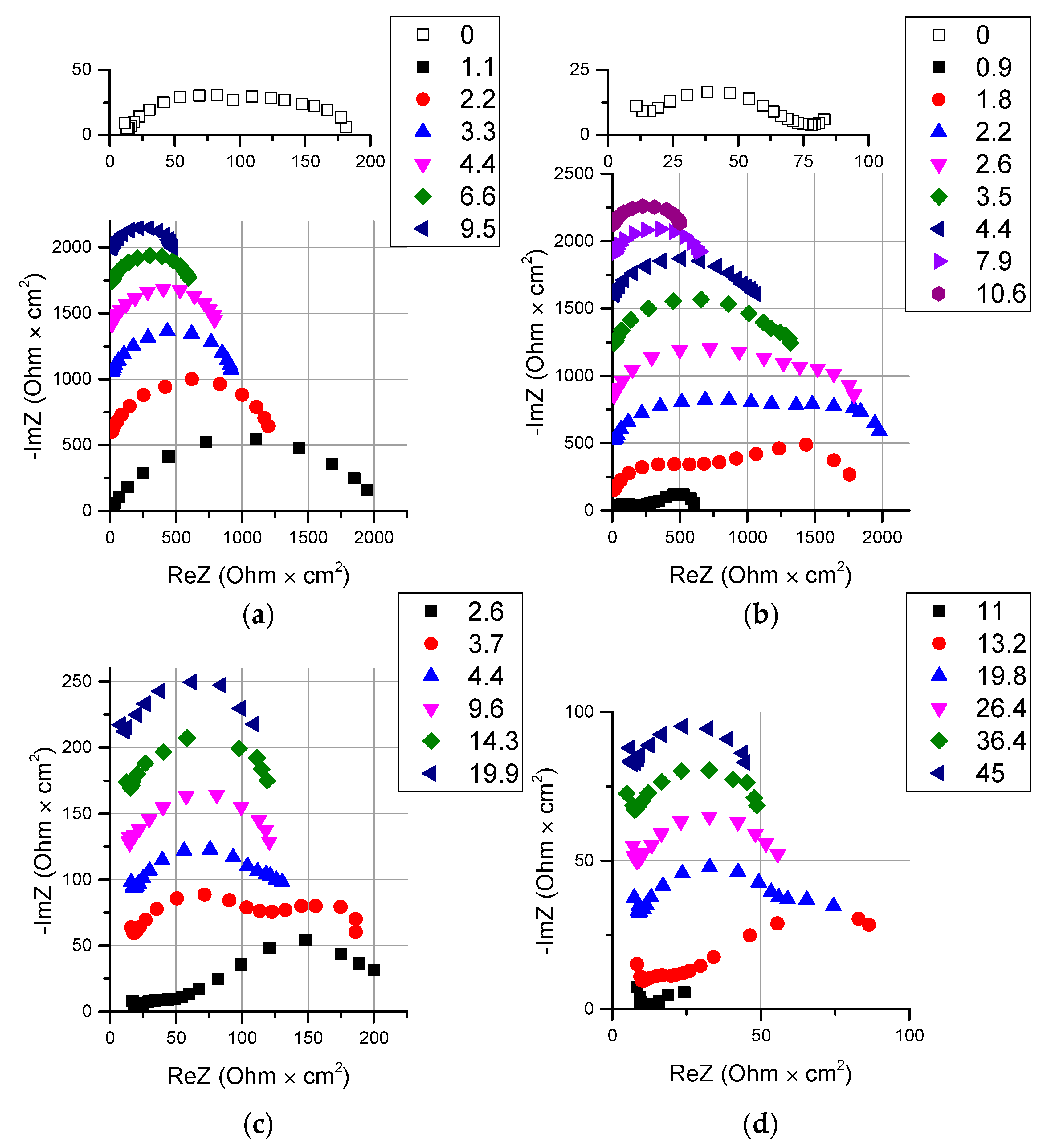
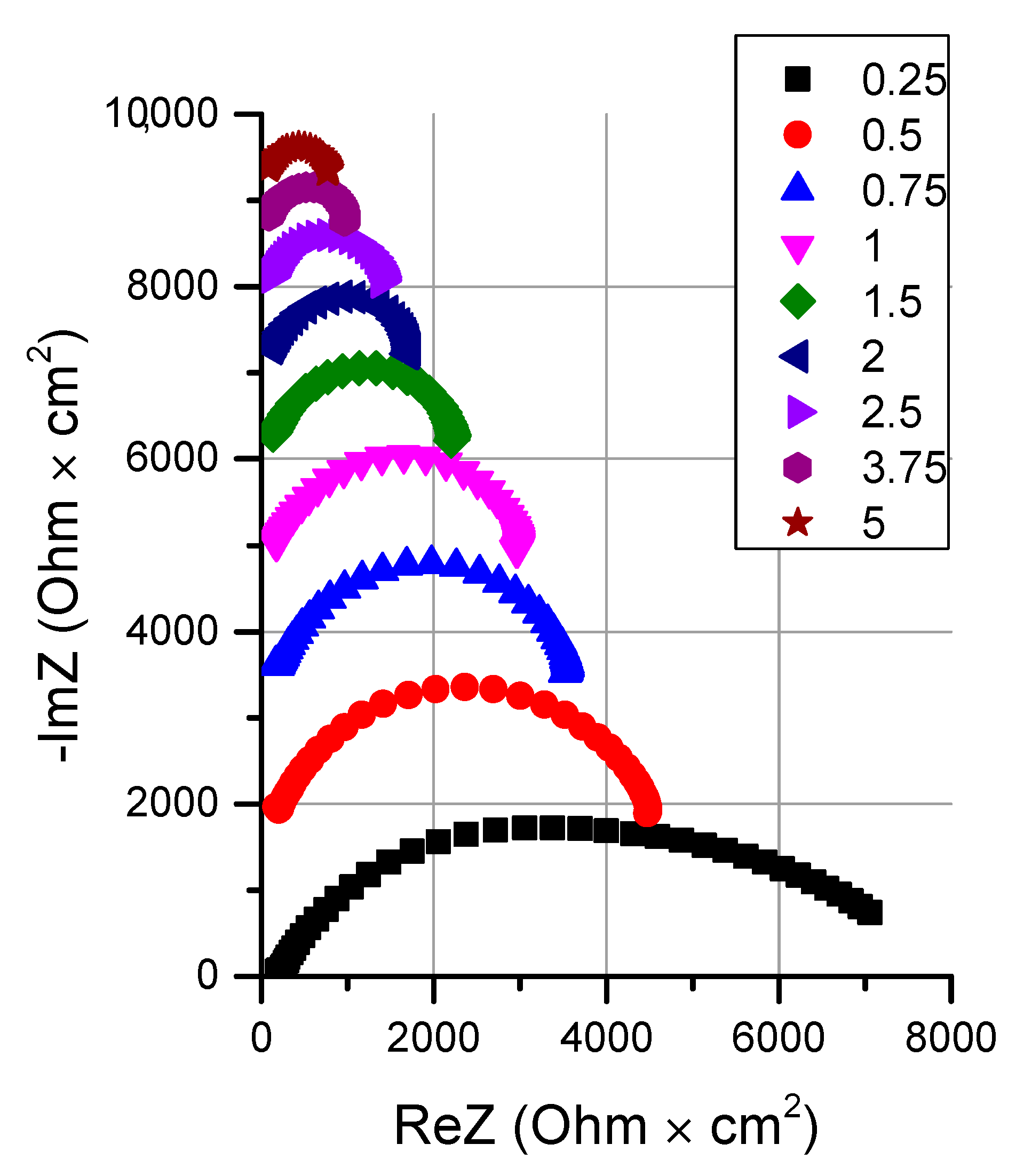
2.2.3. Electrochemical Impedance Spectroscopy
3. Results
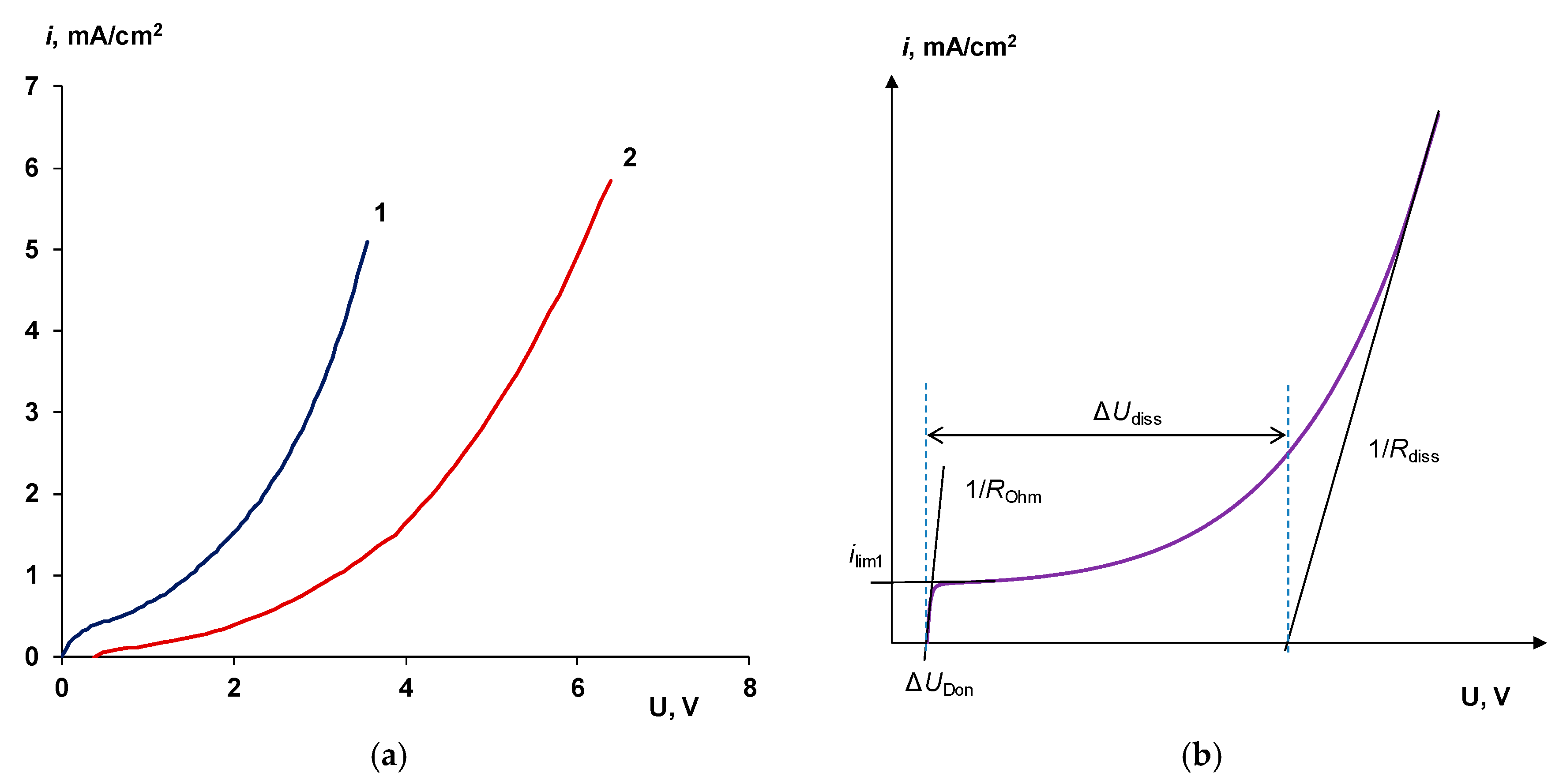
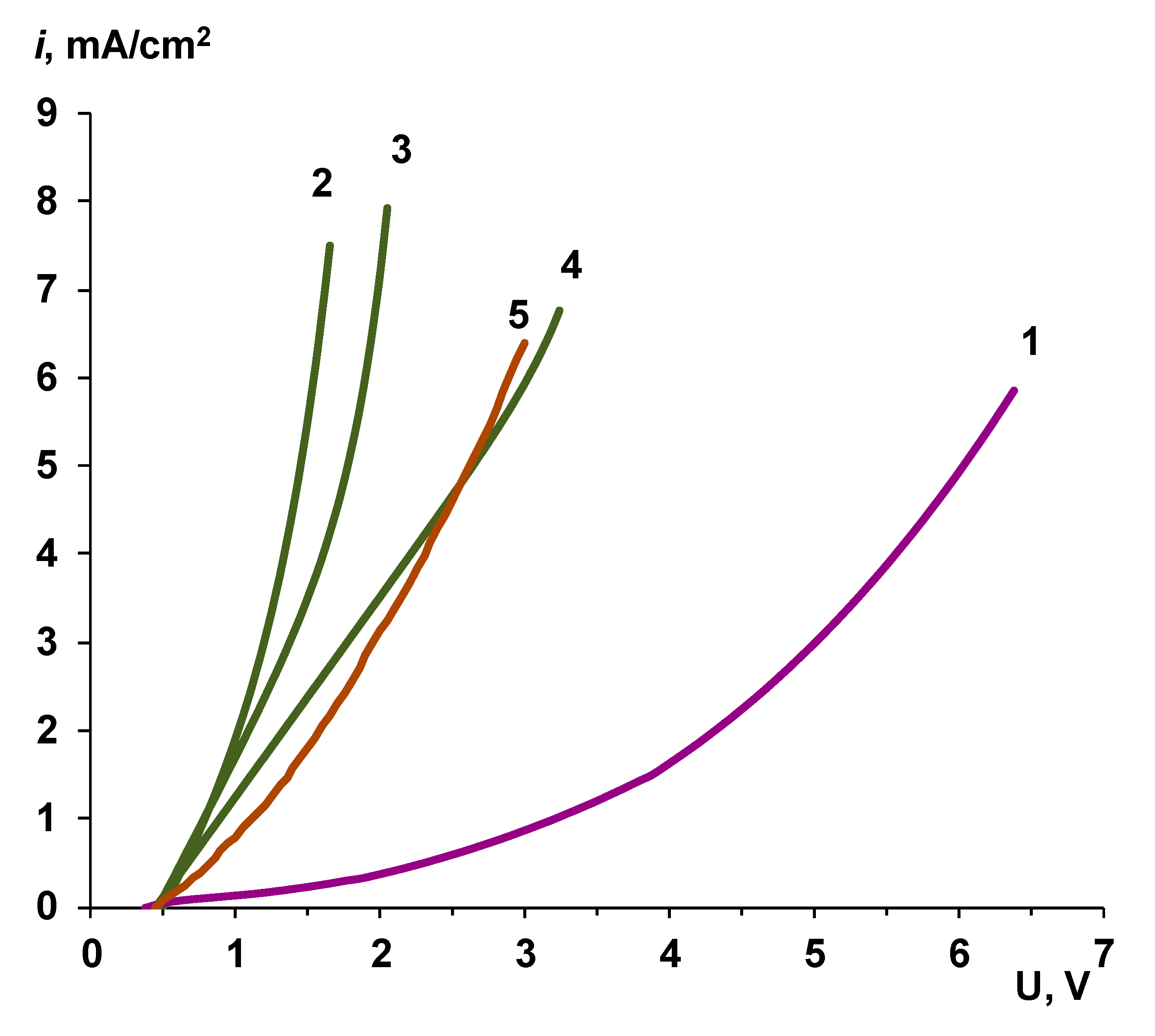
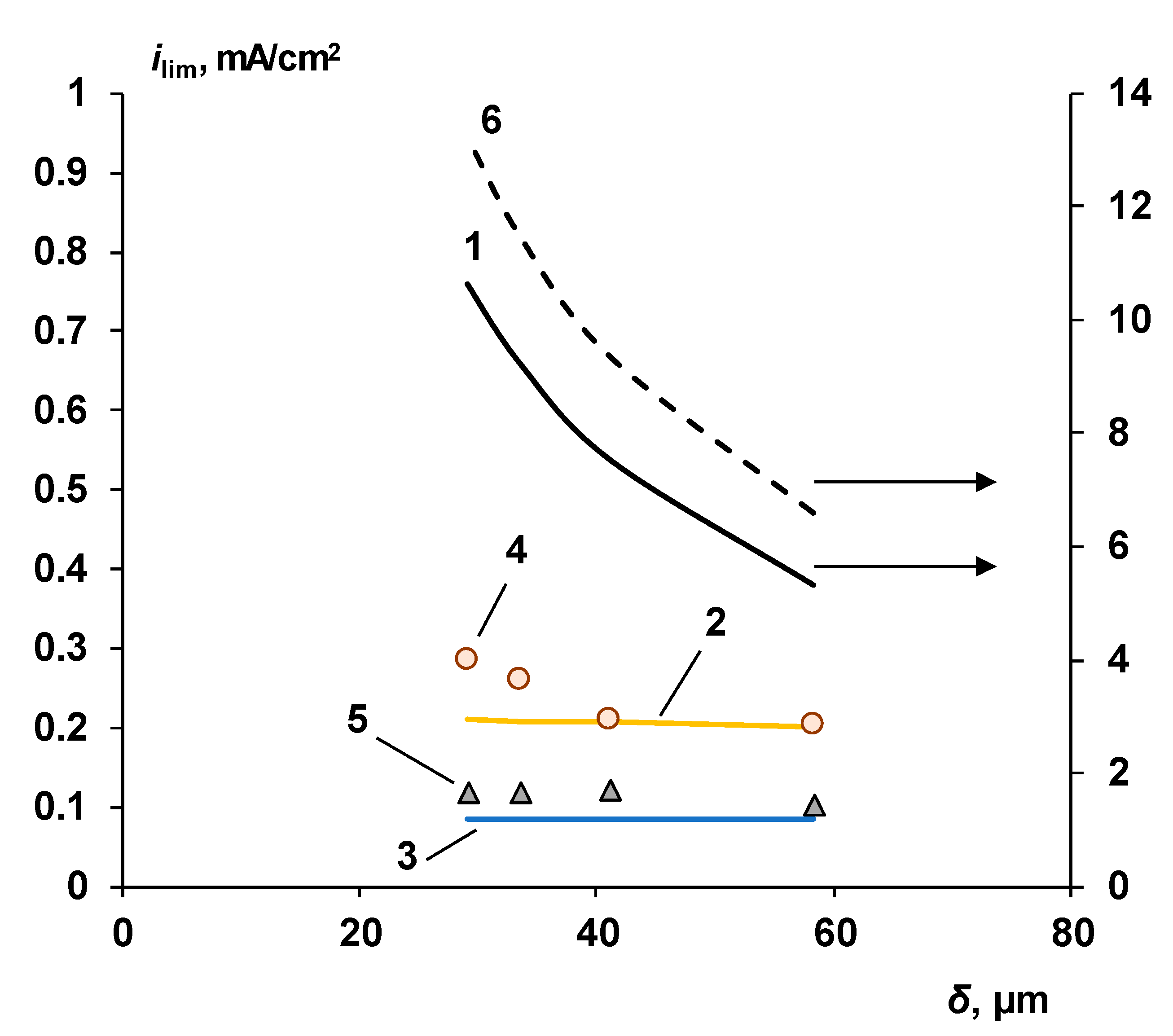
3.1. Cation-Exchange Layer Properties Thickness and Chemical Nature
Chemical Nature of the CEL
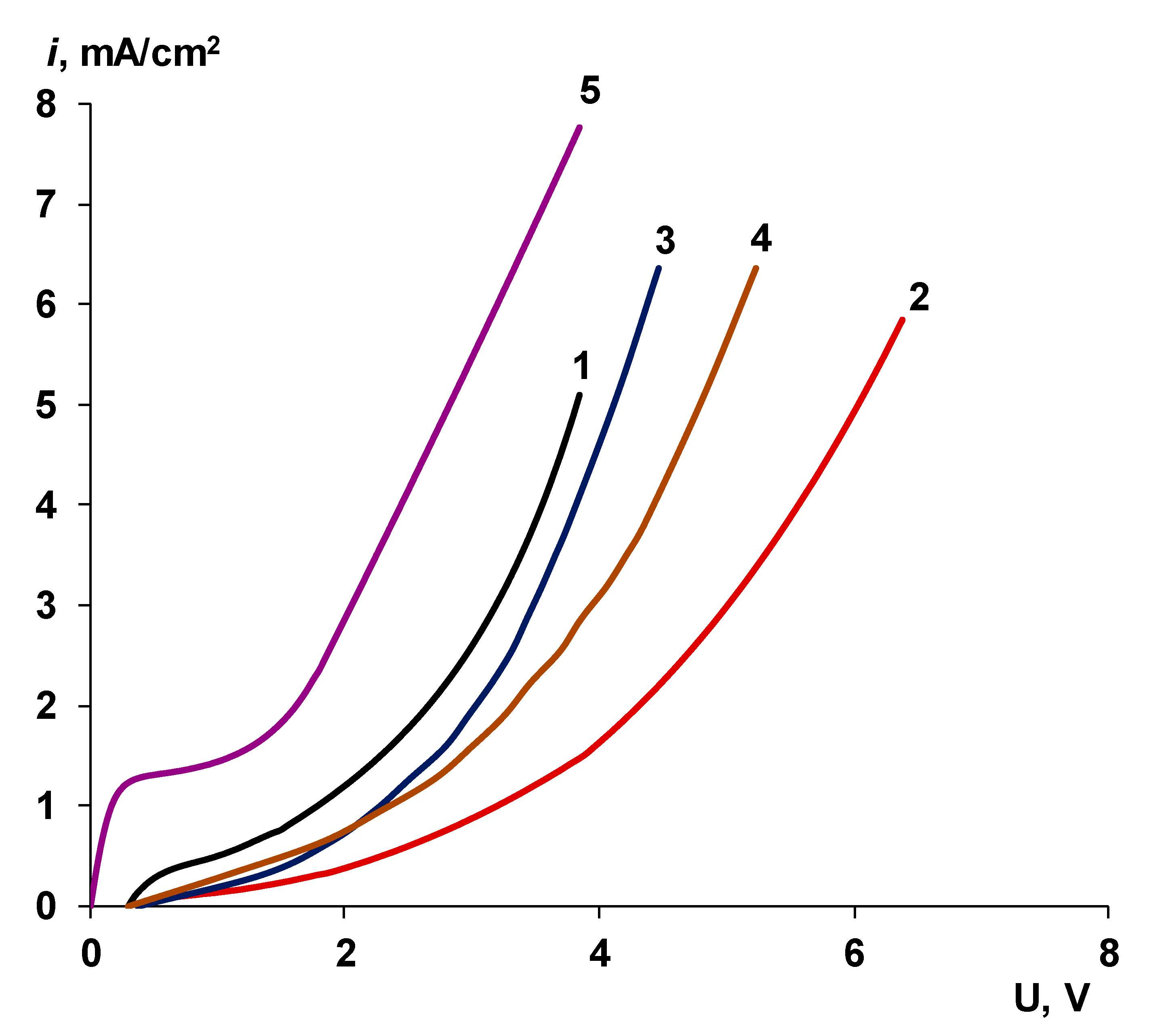
3.2. Catalyst Effect on the Water Splitting Kinetics
3.2.1. Chemical Nature of the Catalyst
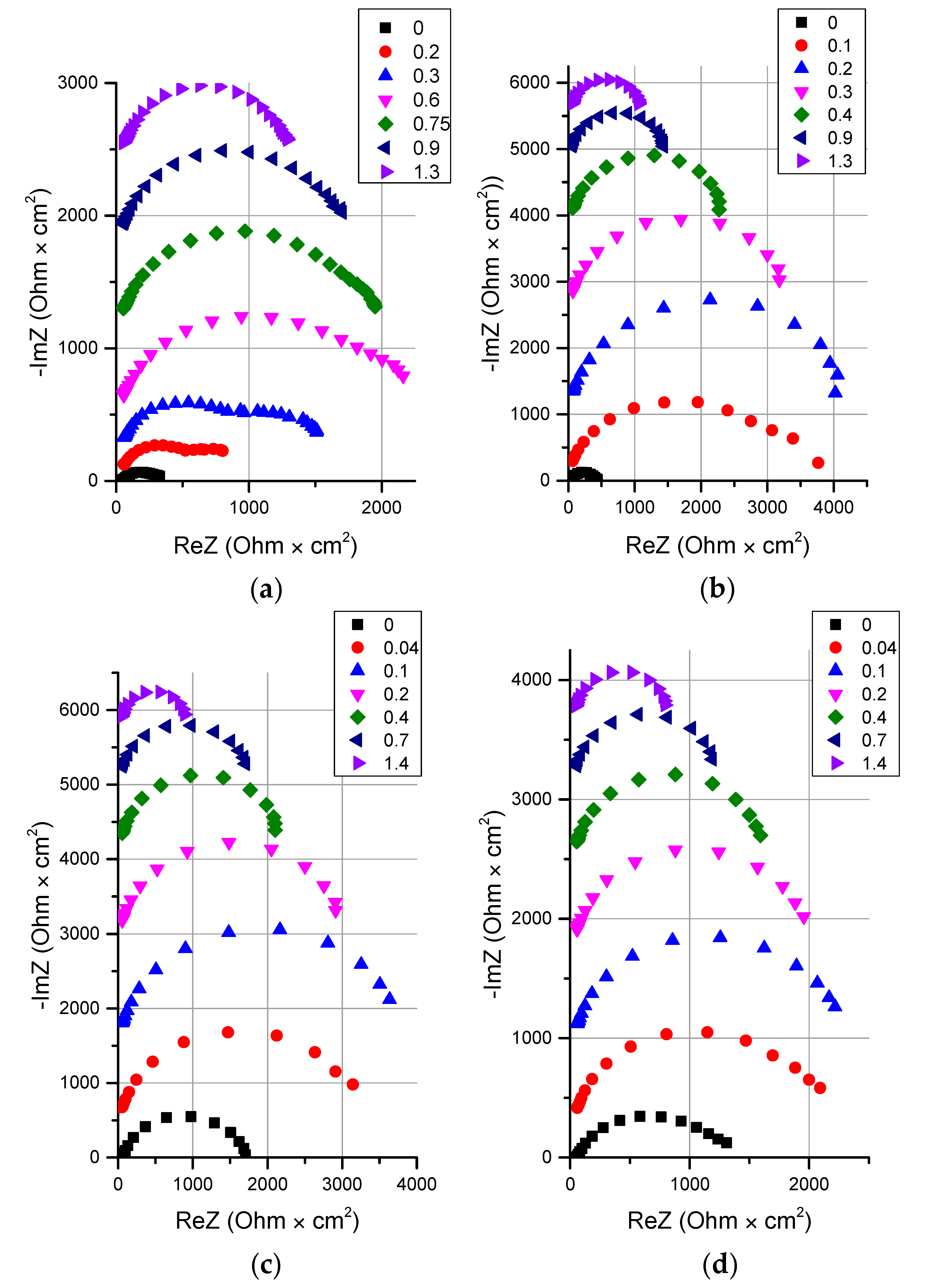
3.2.2. Influence of the Particle Size of Chromium (III) Hydroxide on the Rate of the Water Splitting Reaction
3.3. Electrochemical Properties in Solutions with High Concentrations
3.4. Thin Anion-Exchange Layer
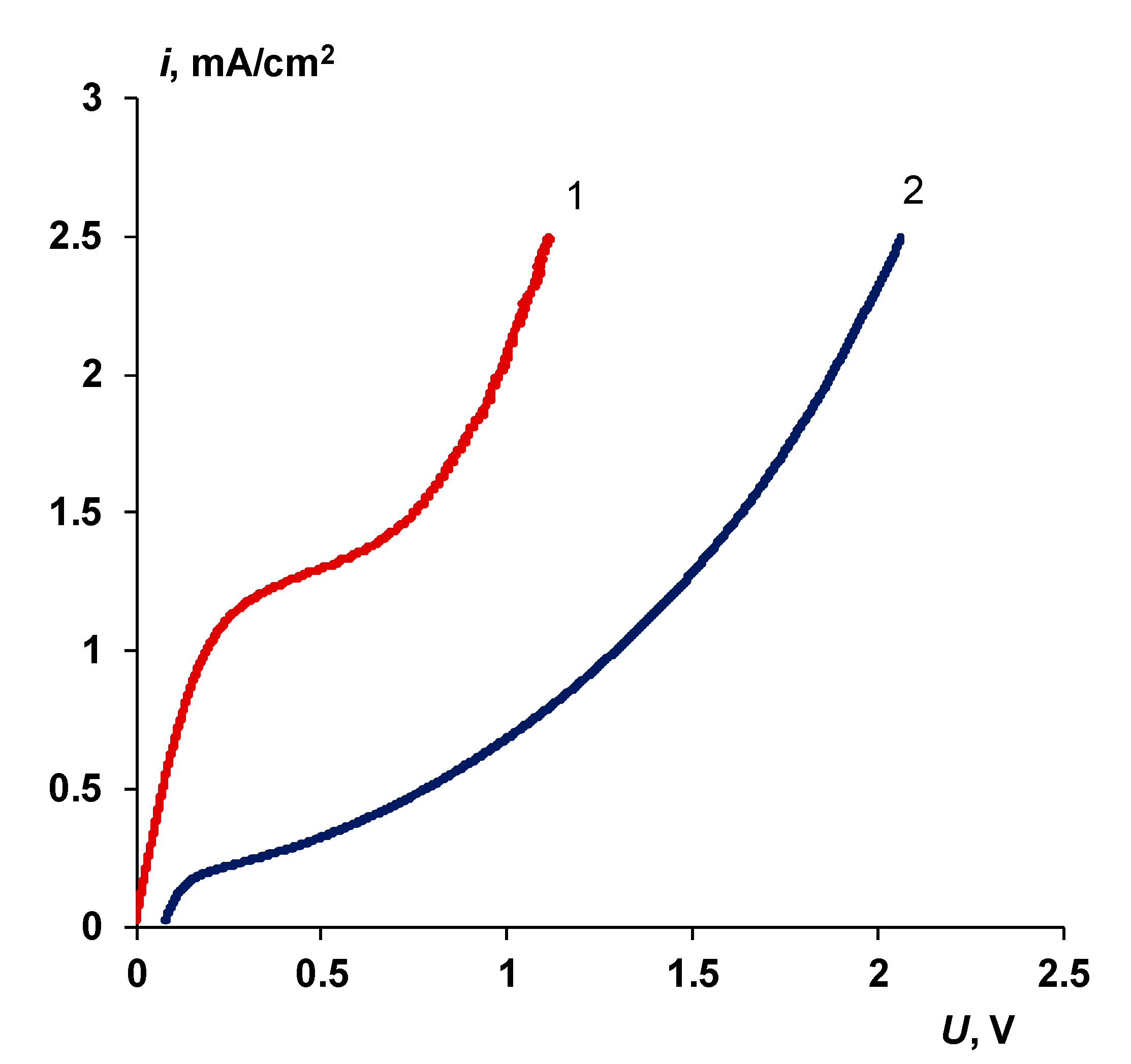
3.5. Comparison with Commercial Bipolar Membranes
4. Discussion
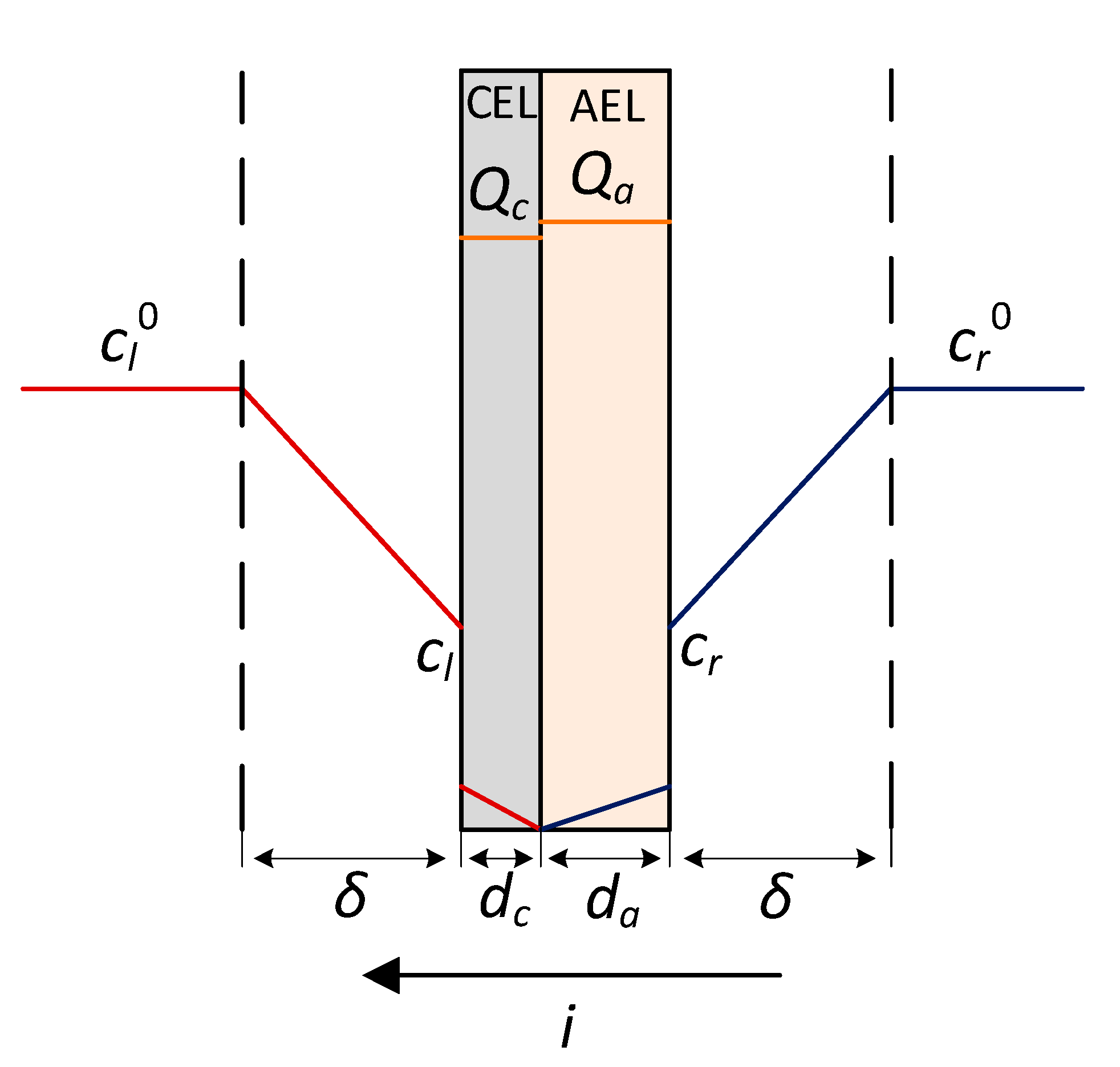
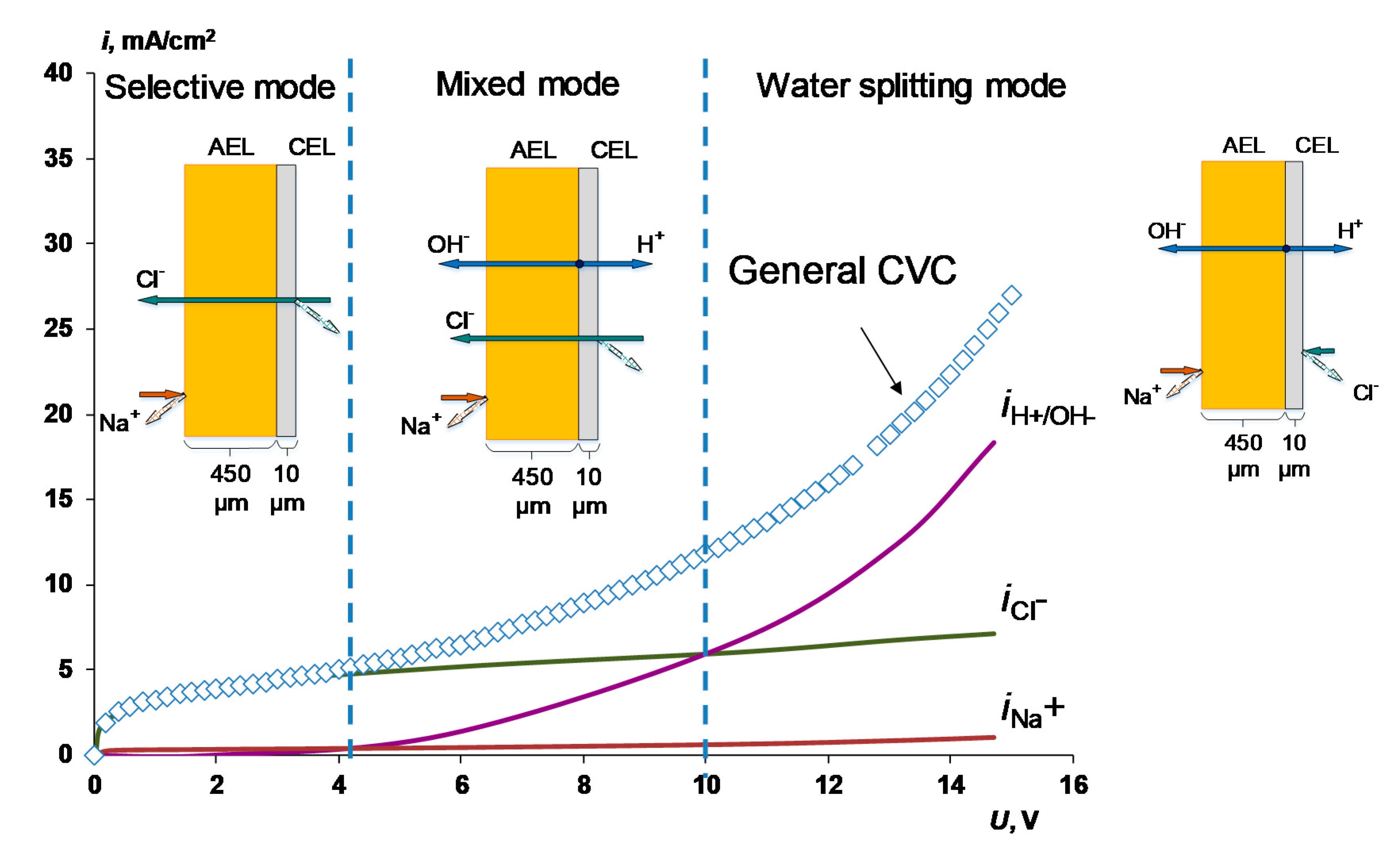
4.1. The Nature of the Limiting Current on Bilayer Membrane
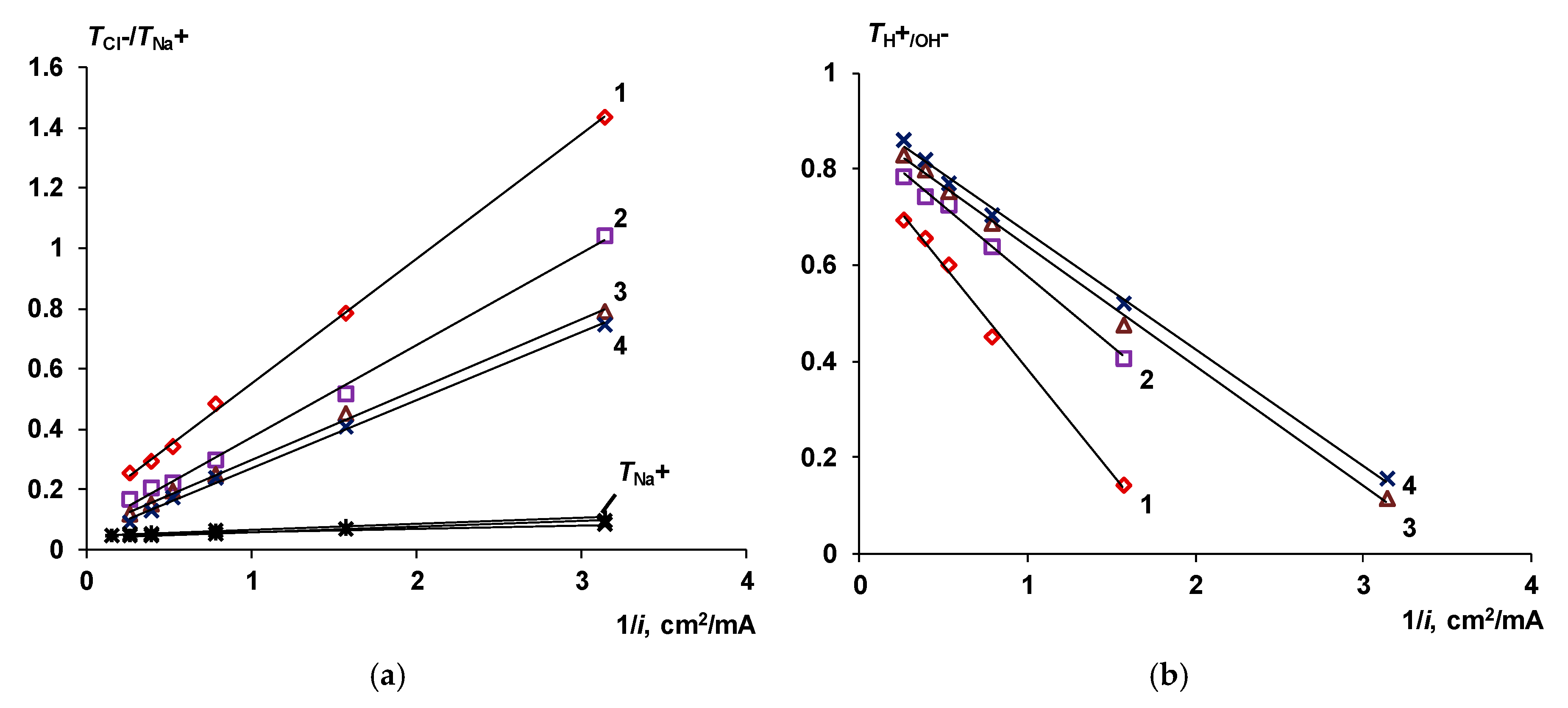
The Co-Ion Leakage through Membranes BM-ac
4.2. The Water Splitting Reaction Kinetics

4.3. Future Perspectives for Bilayer Membranes
5. Conclusions
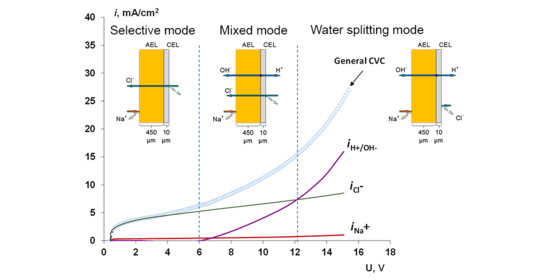
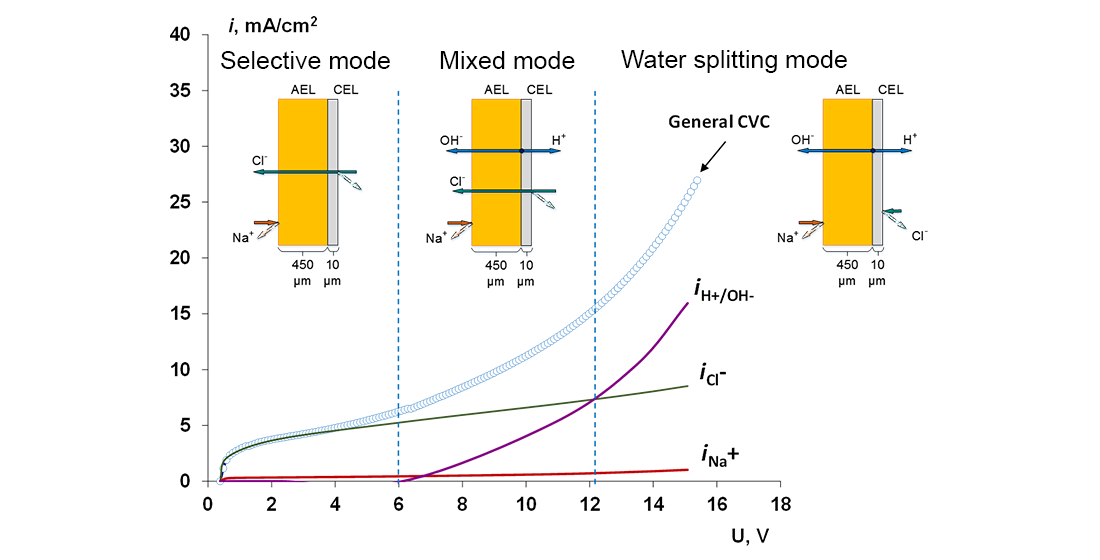
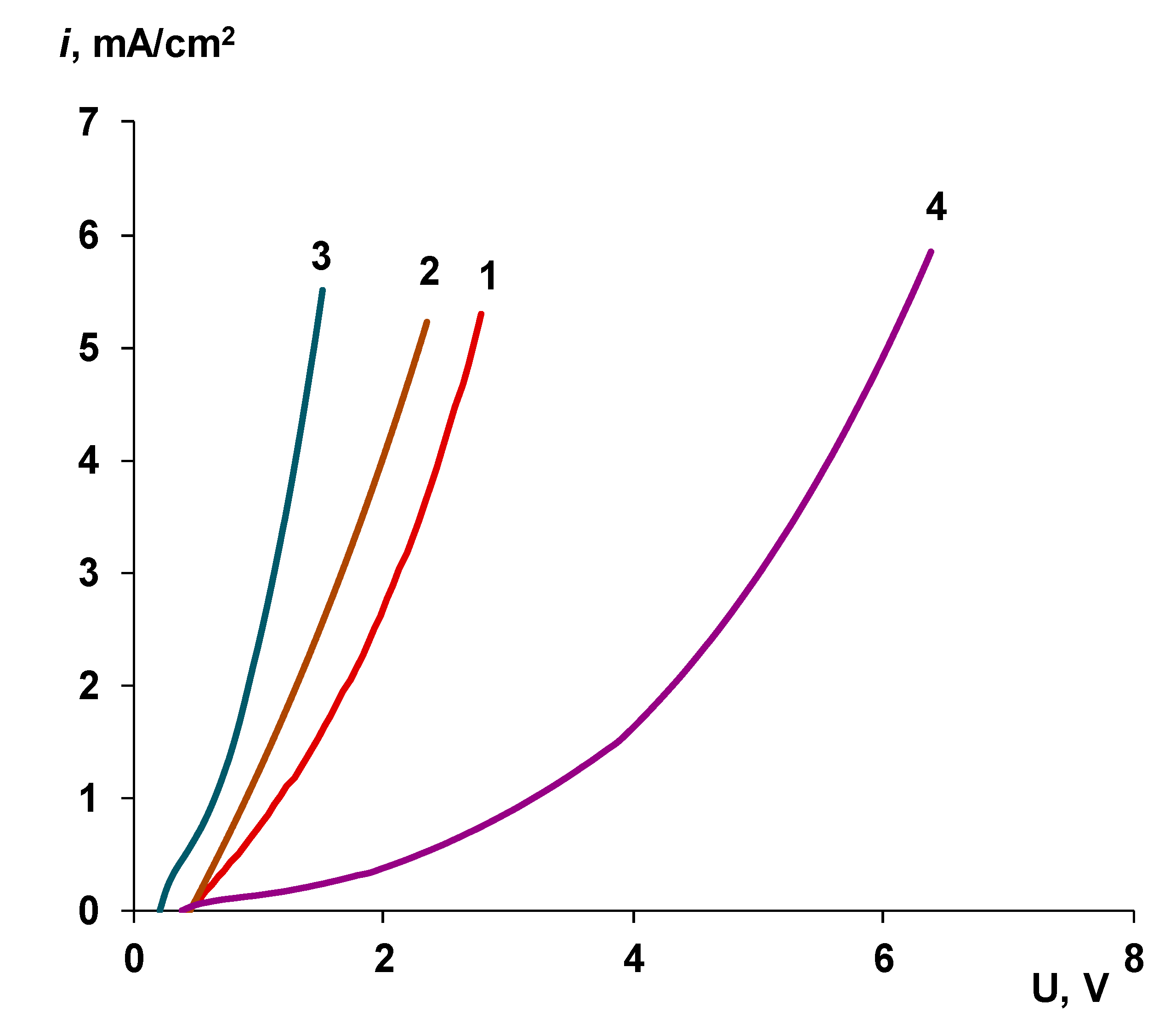
- When a thin layer of an ion-polymer is deposited onto a membrane-substrate with a matrix charge opposite to that of a matrix of the membrane-substrate, an abrupt change in the electrochemical behavior of the resulting bilayer membrane occurs. There is a sharp decrease in the value of the limiting current, which is caused by a change in the nature of the limiting current in electromembrane systems with bilayer membranes. The limiting current in a system with a bilayer membrane is determined not by diffusion restrictions in the solution, but by the electrodiffusion of co-ions (relative to the charge of the thin layer matrix) in the ion-polymer film deposited on the substrate membrane. Previously, we observed such changes only for membranes with a thin cation-exchange layer; however, the same effects are also caused by the deposition of a thin layer of anion-exchanger on the surface of the cation-exchange membrane-substrate. The second important feature is a complete transition to the “generation mode” in an overlimiting state, while for the original Ralex membranes, a mixing of the modes of electroconvection and generation of H+/OH− ions is noted [91].
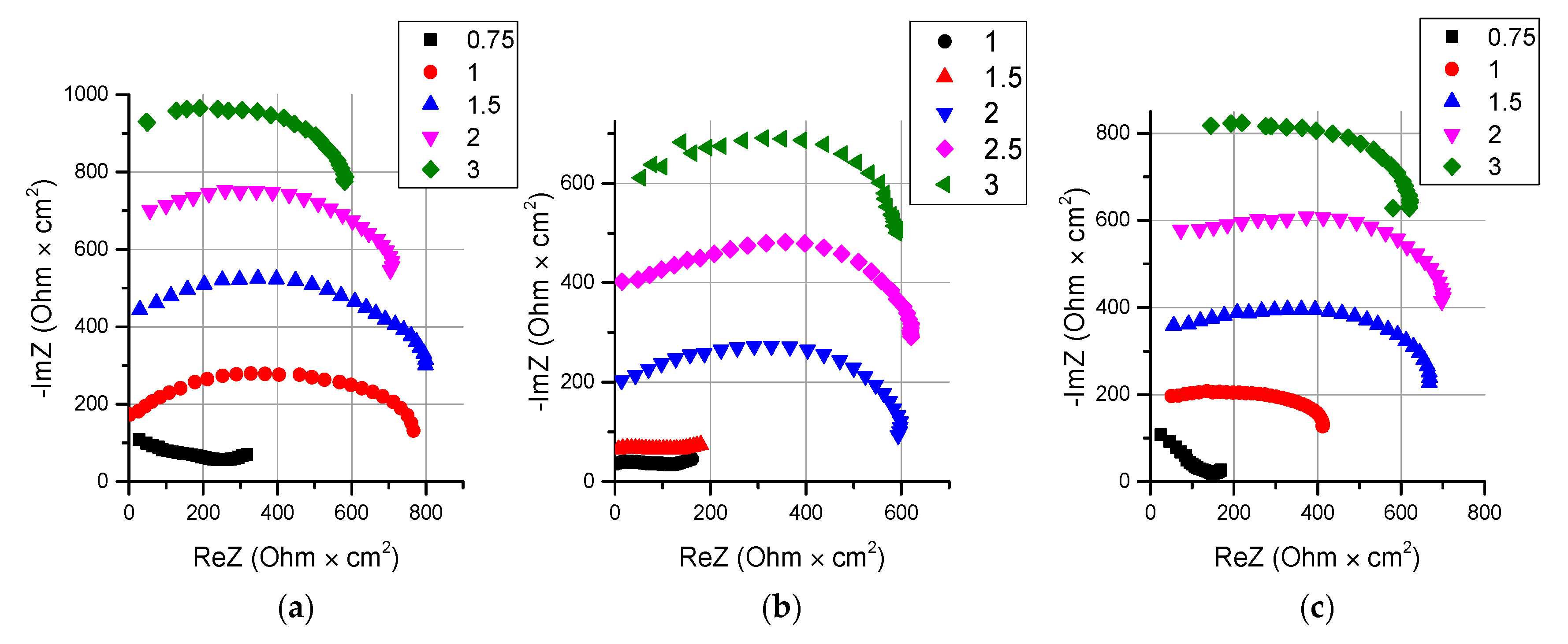
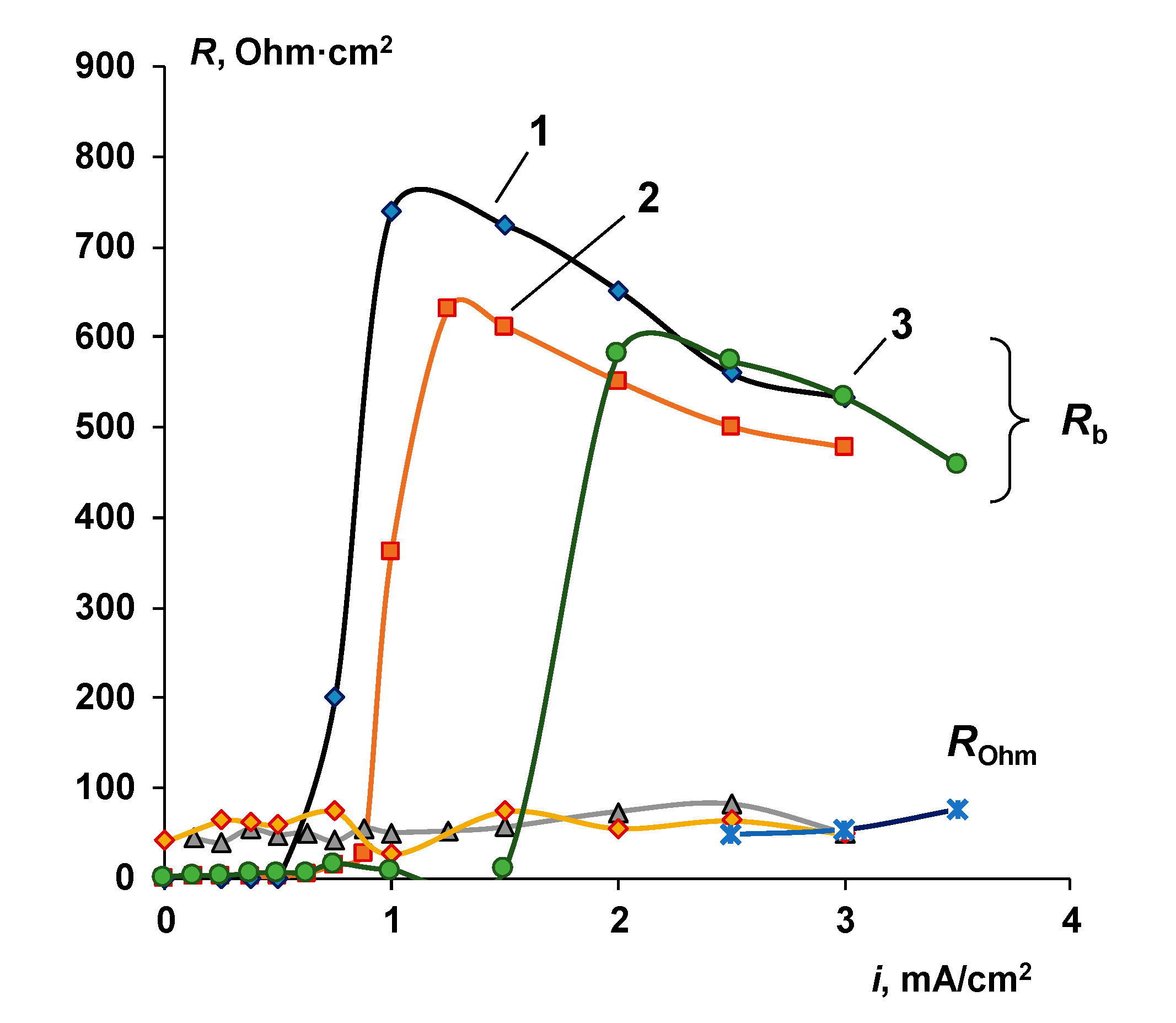
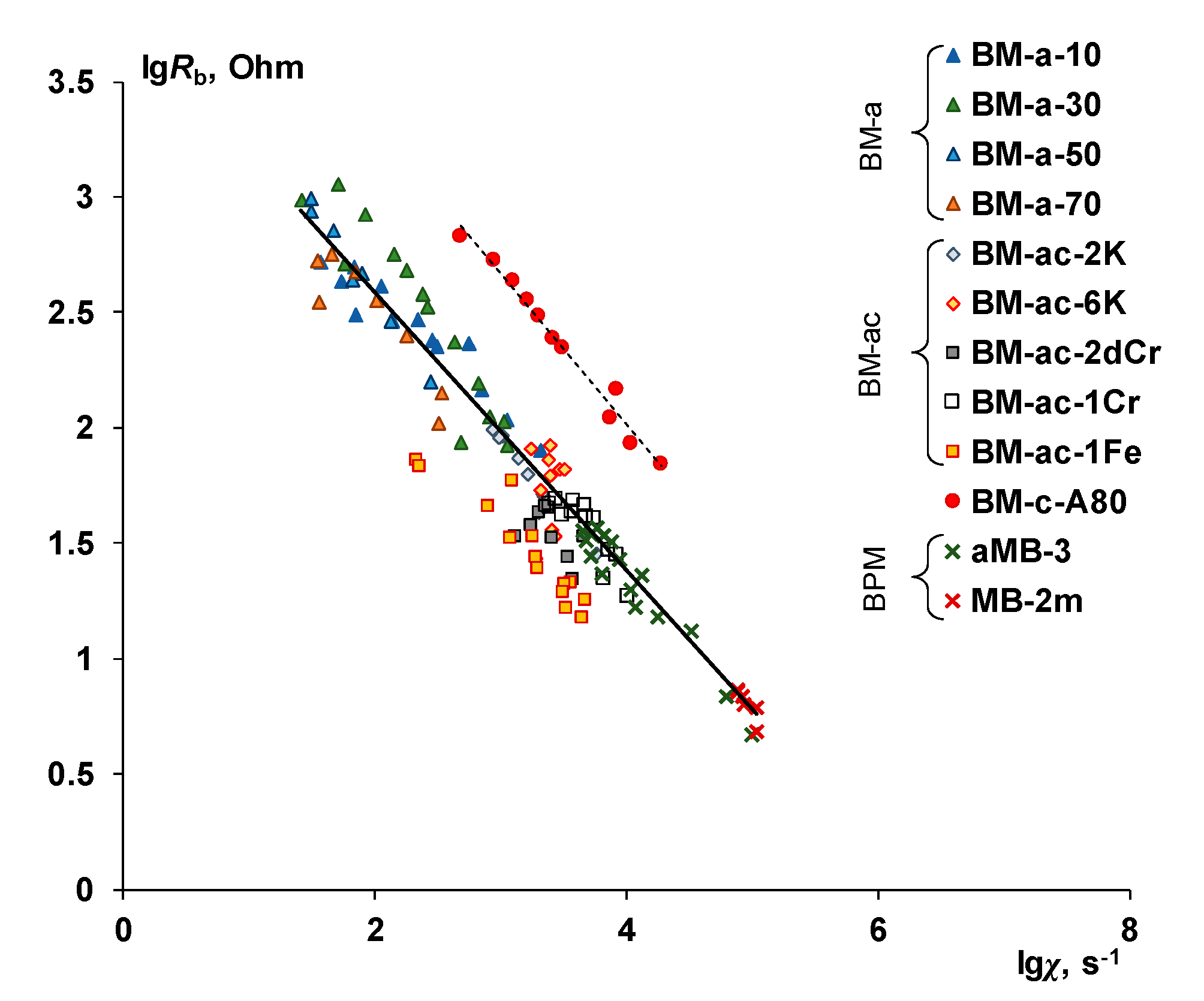
- It is shown that irrespective of the nature of the catalyst or its absence, as well as the thickness of the layers forming the membrane, the process of water splitting in the bipolar region gives a linear relationship between lgRb and lgχ. At the same time, for all studied membranes, a slight deviation of the slope of dependence from the theoretical value of ½ is observed, which may indicate an incomplete correspondence of modern theoretical concepts of the electrochemical spectrum of the impedance of a bipolar (bilayer) membrane as the Gerischer impedance.
- Water splitting catalysts of different chemical nature have different effects on the transport properties of bilayer membranes. The use of non-conductive materials (iron (III) and chromium (III) hydroxides) does not lead to a significant flux of salt ions, compared with the original bilayer membrane without a catalyst, which, together with the high catalytic activity of these substances, allows the use of bilayer membranes instead of bipolar in solutions, with a concentration of up to 0.5 M. At the same time, the use of a phosphoric acid ion-polymer catalyst leads to voltage/selectivity trade-off: on the one hand, the potential drop across membrane significantly decreases, on the other, the limiting current increases.
- Under certain conditions for bilayer membranes, it is possible to achieve a significant transfer of salt ions. These effects are especially pronounced in membrane samples with a phosphoric acid catalyst for the water splitting reaction. The significant contribution of the transport of salt ions, simultaneously with the intensive water splitting, makes it possible to use such membranes in processes that simultaneously require the transfer of salt ions and a change in pH. For example, when processing fruit juices, preparing water for heat power engineering, and carrying out the kinetic separation of inorganic and organic electrolytes. Studying the functioning of bilayer membranes in mixed-mode processes seems to be the most interesting in the future.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AEL | anion-exchange layer | |
| CEL | cation-exchange layer | |
| BM-a | bilayer membranes with thin cation-exchange layer and without a water splitting catalyst | |
| BM-ac | bilayer membranes with thin cation-exchange layer and with a water splitting catalyst | |
| SPEEK | sulfonated poly (ether ether) ketone | |
| SPS | sulfonated polystyrene | |
| SPTFE | sulfonated polytetrafluoroethylene | |
| Parameter | Dimensions | Description |
| Pj | L/s | permeability coefficient of acid or alkali |
| Δci | mol/L | difference in concentration of acid or alkali in the depth of the solution on both sides of the membrane |
| S | cm2 | membrane area |
| mol/L | concentrations of sodium and chloride ions in the solution, at the outlet the cell, and in feed solution | |
| effective transport number of sodium or chloride ion | ||
| Tw | effective transport number of H+ and OH− ions | |
| W | L/s | volume flow rate of a solution |
| i | mA/cm2 | current density |
| ilim | mA/cm2 | limiting current density |
| Ohm·cm2 | Resistances of the bipolar region, ohmic resistance and total resistance of the bilayer membrane | |
| V | overvoltage of the bipolar region | |
| rad/s | angular frequency | |
| χ | 1/s | non-equilibrium effective rate constant of the water splitting reaction |
| cl, cr | mol/L | concentration of electrolyte on the membrane/solution boundary to the left and to the right of the membrane |
| mol/L | bulk concentration of the electrolyte on the left side of the membrane | |
| characteristic thicknesses of the CEL and AEL layers respectively | ||
| dc, da | m | thickness of the CEL and AEL layers respectively |
| Qc, Qa | mol/m3 | ion-exchange capacity of the CEL and AEL layers respectively |
| Dc, Da | m2/s | co-ion diffusion coefficient inside the CEL and AEL layers respectively |
| Dsol | m2/s | salt diffusion coefficient in the solution |
| m | diffusion boundary layer thickness | |
| mA/cm2 | limiting current determined by a thin cation-exchange layer | |
| mA/cm2 | limiting current determined by the electrodiffusion of ions in the solution in the absence of a film on the surface of the substrate membrane | |
| α | ratio of the values of the limiting currents in the membrane and in the diffusion layer | |
| electromigration transport number of hydrogen ion in the CEL | ||
| mol/m3 | concentration of hydrogen ion at the CEL/AEL interface | |
| m2/s | hydrogen ion diffusion coefficient inside the CEL | |
| mA/cm2 | current carried by the hydrogen and hydroxyl ions | |
| mA/cm2 | current of the water recombination reaction in the absence of an external electric field | |
| β | m/V | empirical parameter, which can be related to a characteristic length |
| 1/s | total effective rate constant of the pseudomonomolecular water splitting reaction in the space charge region in the absence of an electric field | |
| Em | V/m | electric field strength at the CEL/AEL interface |
| relative permittivity of the space charge region | ||
| F/m | absolute dielectric constant of vacuum | |
| R | J/(mol·K) | universal gas constant |
| T | K | temperature |
| F | A·s/mol | Faraday constant |
References
- Bazinet, L.; Lamarche, F.; Ippersiel, D. Bipolar-membrane electrodialysis: Applications of electrodialysis in the food industry. Trends Food Sci. Technol. 1998, 9, 107–113. [Google Scholar] [CrossRef]
- Strathmann, H. Electrodialysis, a mature technology with a multitude of new applications. Desalination 2010, 264, 268–288. [Google Scholar] [CrossRef]
- Xu, T. Electrodialysis processes with bipolar membranes (EDBM) in environmental protection—A review. Resour. Conserv. Recycl. 2002, 37, 1–22. [Google Scholar]
- Gutiérrez, L.-F.; Bazinet, L.; Hamoudi, S.; Belkacemi, K. Production of lactobionic acid by means of a process comprising the catalytic oxidation of lactose and bipolar membrane electrodialysis. Sep. Purif. Technol. 2013, 109, 23–32. [Google Scholar] [CrossRef]
- Jaroszek, H.; Dydo, P. Ion-exchange membranes in chemical synthesis—A review. Open Chem. 2016, 14, 1–19. [Google Scholar] [CrossRef]
- Pärnamäe, R.; Mareev, S.; Nikonenko, V.; Melnikov, S.; Sheldeshov, N.; Zabolotskii, V.; Hamelers, H.V.M.; Tedesco, M. Bipolar membranes: A review on principles, latest developments, and applications. J. Memb. Sci. 2020, 617, 118538. [Google Scholar] [CrossRef]
- Readi, O.M.K.; Gironès, M.; Wiratha, W.; Nijmeijer, K. On the isolation of single basic amino acids with electrodialysis for the production of biobased chemicals. Ind. Eng. Chem. Res. 2013, 52, 1069–1078. [Google Scholar] [CrossRef]
- Nir, O.; Sengpiel, R.G.; Wessling, M. Closing the cycle: Phosphorus removal and recovery from diluted effluents using acid resistive membranes. Chem. Eng. J. 2018, 346, 640–648. [Google Scholar] [CrossRef]
- Gurreri, L.; Tamburini, A.; Cipollina, A.; Micale, G. Electrodialysis Applications in Wastewater Treatment for Environmental Protection and Resources Recovery: A Systematic Review on Progress and Perspectives. Membranes 2020, 10, 146. [Google Scholar] [CrossRef]
- Shi, L.; Hu, Y.; Xie, S.; Wu, G.; Hu, Z.; Zhan, X. Recovery of nutrients and volatile fatty acids from pig manure hydrolysate using two-stage bipolar membrane electrodialysis. Chem. Eng. J. 2018, 334, 134–142. [Google Scholar] [CrossRef]
- Patel, A.; Mungray, A.A.K.; Mungray, A.A.K. Technologies for the recovery of nutrients, water and energy from human urine: A review. Chemosphere 2020, 259, 127372. [Google Scholar] [CrossRef]
- Melnikova, E.D.; Pismenskaya, N.D.; Bazinet, L.; Mikhaylin, S.; Nikonenko, V.V. Effect of ampholyte nature on current-voltage characteristic of anion-exchange membrane. Electrochim. Acta 2018, 285, 185–191. [Google Scholar] [CrossRef]
- Rybalkina, O.; Tsygurina, K.; Melnikova, E.; Mareev, S.; Moroz, I.; Nikonenko, V.; Pismenskaya, N. Partial fluxes of phosphoric acid anions through anion-exchange membranes in the course of NaH2PO4 solution electrodialysis. Int. J. Mol. Sci. 2019, 20, 3593. [Google Scholar] [CrossRef]
- Melnikov, S.; Kolot, D.; Nosova, E.; Zabolotskiy, V. Peculiarities of transport-structural parameters of ion-exchange membranes in solutions containing anions of carboxylic acids. J. Memb. Sci. 2018, 557, 1–12. [Google Scholar] [CrossRef]
- Lee, H.J.; Hong, M.K.; Han, S.D.; Cho, S.H.; Moon, S.H. Fouling of an anion exchange membrane in the electrodialysis desalination process in the presence of organic foulants. Desalination 2009, 238, 60–69. [Google Scholar] [CrossRef]
- Ghalloussi, R.; Garcia-Vasquez, W.; Chaabane, L.; Dammak, L.; Larchet, C.; Deabate, S.V.; Nevakshenova, E.; Nikonenko, V.V.; Grande, D. Ageing of ion-exchange membranes in electrodialysis: A structural and physicochemical investigation. J. Memb. Sci. 2013, 436, 68–78. [Google Scholar] [CrossRef]
- Qasim, M.; Badrelzaman, M.; Darwish, N.N.; Darwish, N.A.; Hilal, N. Reverse osmosis desalination: A state-of-the-art review. Desalination 2019, 459, 59–104. [Google Scholar] [CrossRef]
- Pinnau, I.; Freeman, B.D. Formation and Modification of Polymeric Membranes: Overview. In Membrane Formation and Modification; ACS Publications: Washington, DC, USA, 1999; pp. 1–22. [Google Scholar]
- Frilette, V.J. Preparation and Characterization of Bipolar Ion Exchange Membranes. J. Phys. Chem. 1956, 60, 435–439. [Google Scholar] [CrossRef]
- Leitz, F.B. Cationic-Anionic Ion-Exchange Membrane. U.S. Patent 3,562,139, 9 February 1971. [Google Scholar]
- Kollsman, P. Antipolarization Membrane Having Anionic and Cationic Areas. U.S. Patent 3,227,662, 4 January 1966. [Google Scholar]
- Antonov, Y.A.; Ponomarev, M.I.; Volkov, S.A.; Grebenyuk, V.D. Production of alkali with simultaneous water desaltination in electrodialyzer with semi- bipolar membranes. Sov. J. Water Chem. Technol. (Engl. Transl. Khimiya Tekhnologiya Vody) 1983, 5, 454–456. [Google Scholar]
- Shendrik, O.R.; Ponomarev, M.I.; Grebenyuk, V.D. Modification of monopolar ion exchange membranes for hydrogen and hydroxyl ions generation. J. Appl. Chem. USSR 1986, 59, 1486–1488. [Google Scholar]
- Ramireza, P.; Manzanaresb, J.A.; Mafe, S. Water dissociation effects in ion transport through anion exchange membranes with thin cationic exchange surface films. Berl. Bunsenges Physycal Chem. 1991, 95, 499–503. [Google Scholar] [CrossRef]
- Bukhovets, A.E.; Eliseeva, T.V.; Oren, Y. Fouling of anion-exchange membranes in electrodialysis of aromatic amino acid solution. J. Memb. Sci. 2010, 364, 339–343. [Google Scholar] [CrossRef]
- Slouka, Z.; Senapati, S.; Yan, Y.; Chang, H.C. Charge inversion, water splitting, and vortex suppression due to DNA sorption on ion-selective membranes and their ion-current signatures. Langmuir 2013. [Google Scholar] [CrossRef]
- Senapati, S.; Slouka, Z.; Shah, S.S.; Behura, S.K.; Shi, Z.; Stack, M.S.; Severson, D.W.; Chang, H.C. An ion-exchange nanomembrane sensor for detection of nucleic acids using a surface charge inversion phenomenon. Biosens. Bioelectron. 2014, 60, 92–100. [Google Scholar] [CrossRef]
- Balster, J.H.; Sumbharaju, R.; Srikantharajah, S.; Pünt, I.; Stamatialis, D.F.; Jordan, V.; Wessling, M. Asymmetric bipolar membrane: A tool to improve product purity. J. Memb. Sci. 2007, 287, 246–256. [Google Scholar] [CrossRef]
- Zabolotskii, V.I.; Sheldeshov, N.V.; Melnikov, S.S. Effect of cation-exchange layer thickness on electrochemical and transport characteristics of bipolar membranes. J. Appl. Electrochem. 2013, 43, 1117–1129. [Google Scholar] [CrossRef]
- Zabolotsky, V.; Utin, S.; Bespalov, A.; Strelkov, V. Modification of asymmetric bipolar membranes by functionalized hyperbranched polymers and their investigation during pH correction of diluted electrolytes solutions by electrodialysis. J. Memb. Sci. 2015, 494, 188–195. [Google Scholar] [CrossRef]
- Abdu, S.; Sricharoen, K.; Wong, J.E.; Muljadi, E.S.; Melin, T.; Wessling, M. Catalytic polyelectrolyte multilayers at the bipolar membrane interface. Appl. Mater. Interfaces 2013, 5, 10445–10455. [Google Scholar] [CrossRef]
- Abdu, S.; Wessling, M. Layer-by-Layer Modification of Cation Exchange Membranes Controls Ion Selectivity and Water Splitting. Appl. Mater. Interfaces 2014, 3, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Rijnaarts, T.; Reurink, D.M.; Radmanesh, F.; de Vos, W.M.; Nijmeijer, K. Layer-by-layer coatings on ion exchange membranes: Effect of multilayer charge and hydration on monovalent ion selectivities. J. Memb. Sci. 2019, 570–571, 513–521. [Google Scholar] [CrossRef]
- Ahmad, M.; Tang, C.; Yang, L.; Yaroshchuk, A.; Bruening, M.L. Layer-by-layer modification of aliphatic polyamide anion-exchange membranes to increase Cl−/SO42− selectivity. J. Memb. Sci. 2019, 578, 209–219. [Google Scholar] [CrossRef]
- White, N.; Misovich, M.; Yaroshchuk, A.; Bruening, M.L. Coating of Nafion membranes with polyelectrolyte multilayers to achieve high monovalent/divalent cation electrodialysis selectivities. ACS Appl. Mater. Interfaces 2015, 7, 6620–6628. [Google Scholar] [CrossRef] [PubMed]
- Decher, G.; Hong, J.-D. Buildup of ultrathin multilayer films by a self-assembly process, 1 consecutive adsorption of anionic and cationic bipolar amphiphiles on charged surfaces. Makromol. Chemie. Macromol. Symp. 1991, 46, 321–327. [Google Scholar] [CrossRef]
- Decher, G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Decher, G.; Eckle, M.; Schmitt, J.; Struth, B. Layer-by-layer assembled multicomposite films. Curr. Opin. Colloid Interface Sci. 1998, 3, 32–39. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Tanioka, A.; Miyasaka, K. Enzyme immobilization in an asymmetric charged membrane. J. Memb. Sci. 1988, 38, 223–236. [Google Scholar] [CrossRef]
- Fu, R.; Xu, T.; Yang, W.; Pan, Z. Preparation of a mono-sheet bipolar membrane by simultaneous irradiation grafting polymerization of acrylic acid and chloromethylstyrene. J. Appl. Polym. Sci. 2003, 90, 572–576. [Google Scholar] [CrossRef]
- Golubenko, D.; Yaroslavtsev, A. Development of surface-sulfonated graft anion-exchange membranes with monovalent ion selectivity and antifouling properties for electromembrane processes. J. Memb. Sci. 2020, 612, 118408. [Google Scholar] [CrossRef]
- Wang, M.; Jia, Y.X.; Yao, T.T.; Wang, K.K. The endowment of monovalent selectivity to cation exchange membrane by photo-induced covalent immobilization and self-crosslinking of chitosan. J. Memb. Sci. 2013, 442, 39–47. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y. Synthesis and applications of one-dimensional nano-structured polyaniline: An overview. Mater. Sci. Eng. B 2006, 134, 9–19. [Google Scholar] [CrossRef]
- Xiao, X.; Shehzad, M.A.; Yasmin, A.; Ge, Z.; Liang, X.; Sheng, F.; Ji, W.; Ge, X.; Wu, L.; Xu, T. Anion permselective membranes with chemically-bound carboxylic polymer layer for fast anion separation. J. Memb. Sci. 2020, 614, 118553. [Google Scholar] [CrossRef]
- Afsar, N.U.; Shehzad, M.A.; Irfan, M.; Emmanuel, K.; Sheng, F.; Xu, T.; Ren, X.; Ge, L.; Xu, T. Cation exchange membrane integrated with cationic and anionic layers for selective ion separation via electrodialysis. Desalination 2019, 458, 25–33. [Google Scholar] [CrossRef]
- Yan, Z.; Zhu, L.; Li, Y.C.C.; Wycisk, R.J.J.; Pintauro, P.N.N.; Hickner, M.A.A.; Mallouk, T.E.E. The balance of electric field and interfacial catalysis in promoting water dissociation in bipolar membranes. Energy Environ. Sci. 2018, 11, 2235–2245. [Google Scholar] [CrossRef]
- Ariono, D.; Gede Wenten, I. Surface modification of ion-exchange membranes: Methods, characteristics, and performance. J. Appl. Polym. Sci. 2017, 134, 45540. [Google Scholar] [CrossRef]
- Luo, T.; Abdu, S.; Wessling, M. Selectivity of ion exchange membranes: A review. J. Memb. Sci. 2018, 555, 429–454. [Google Scholar] [CrossRef]
- Afsar, N.U.; Ji, W.; Wu, B.; Shehzad, M.A.; Ge, L.; Xu, T. SPPO-based cation exchange membranes with a positively charged layer for cation fractionation. Desalination 2019, 472, 114145. [Google Scholar] [CrossRef]
- Ahmad, M.; Yaroshchuk, A.; Bruening, M.L. Moderate pH changes alter the fluxes, selectivities and limiting currents in ion transport through polyelectrolyte multilayers deposited on membranes. J. Memb. Sci. 2020, 616, 118570. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Jia, Y.X.; Wang, X.L. The improvement of comprehensive transport properties to heterogeneous cation exchange membrane by the covalent immobilization of polyethyleneimine. Sep. Purif. Technol. 2015, 140, 69–76. [Google Scholar] [CrossRef]
- Pourcelly, G. Electrodialysis with bipolar membranes: Principles, optimization, and applications. Russ. J. Electrochem. 2002, 38, 919–926. [Google Scholar] [CrossRef]
- Huang, C.; Xu, T.; Zhang, Y.; Xue, Y.; Chen, G. Application of electrodialysis to the production of organic acids: State-of-the-art and recent developments. J. Memb. Sci. 2007, 288, 1–12. [Google Scholar] [CrossRef]
- Wilhelm, F.G.; Pünt, I.; Van Der Vegt, N.F.A.; Wessling, M.; Strathmann, H. Optimisation strategies for the preparation of bipolar membranes with reduced salt ion leakage in acid—Base electrodialysis. J. Memb. Sci. 2001, 182, 13–28. [Google Scholar] [CrossRef]
- Hao, J.; Yu, L.; Chen, C.; Li, L.; Jiang, W. Preparation of bipolar membranes. J. Appl. Polym. Sci. 2001, 80, 1658–1663. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, H. Preparation of a bipolar membrane by photografting polymerization. Front. Chem. China 2008, 3, 10–13. [Google Scholar] [CrossRef]
- Xue, Y.; Wang, N.; Huang, C.; Cheng, Y.; Xu, T. Catalytic water dissociation at the intermediate layer of a bipolar membrane: The role of carboxylated Boltorn® H30. J. Memb. Sci. 2009, 344, 129–135. [Google Scholar] [CrossRef]
- Balster, J.H.; Srinkantharajah, S.; Sumbharaju, R.; Pünt, I.; Lammertink, R.G.H.G.H.; Stamatialis, D.F.F.; Wessling, M. Tailoring the interface layer of the bipolar membrane. J. Memb. Sci. 2010, 365, 389–398. [Google Scholar] [CrossRef]
- Rajesh, A.M.; Chakrabarty, T.; Prakash, S.; Shahi, V.K. Effects of metal alkoxides on electro-assisted water dissociation across bipolar membranes. Electrochim. Acta 2012, 66, 325–331. [Google Scholar] [CrossRef]
- Manohar, M.; Shukla, G.; Pandey, R.P.; Shahi, V.K. Efficient bipolar membrane with protein interfacial layer for optimal water splitting. J. Ind. Eng. Chem. 2017, 47, 141–149. [Google Scholar] [CrossRef]
- Sheldeshov, N.V.; Zabolotskii, V.I.; Bespalov, A.V.; Kovalev, N.V.; Alpatova, N.V.; Akimova, A.V.; Mochalova, T.V.; Kovaleva, V.I.; Boyarishcheva, A.Y. The influence of catalytic additives on electrochemical properties of bipolar membranes. Pet. Chem. 2017, 57, 518–522. [Google Scholar] [CrossRef]
- Simons, R. Preparation of a high performance bipolar membrane. J. Memb. Sci. 1993, 78, 13–23. [Google Scholar] [CrossRef]
- Mischi, E.; Mantione, D.; Pastacaldi, A.; Botte, L. Method for Making a Bipolar Membrane and Use of Resulting Bipolar Membrane. U.S. Patent 6,924,318, 2 August 2005. [Google Scholar]
- Peng, F.; Peng, S.; Huang, C.; Xu, T. Modifying bipolar membranes with palygorskite and FeCl3. J. Memb. Sci. 2008, 322, 122–127. [Google Scholar] [CrossRef]
- Li, S.-D.; Wang, C.-C.; Chen, C.-Y. Preparation and characterization of a novel bipolar membrane by plasma-induced polymerization. J. Memb. Sci. 2008, 318, 429–434. [Google Scholar] [CrossRef]
- Wang, H.; Ding, F.; Jin, G.; Li, C.; Meng, H. Ultra-thin graphene oxide intermediate layer for bipolar membranes using atomizing spray assembly. Colloids Surfaces A Physicochem. Eng. Asp. 2017, 520, 114–120. [Google Scholar] [CrossRef]
- Kishino, M.; Yuzuki, K.; Fukuta, K. Bipolar Membrane. U.S. Patent Application 16/091,918, 25 April 2019. [Google Scholar]
- Cheng, G.; Zhao, Y.; Li, W.; Zhang, J.; Wang, X.; Dong, C. Performance enhancement of bipolar membranes modified by Fe complex catalyst. J. Memb. Sci. 2019, 589, 117243. [Google Scholar] [CrossRef]
- Melnikov, S.S.; Shapovalova, O.V.; Sheldeshov, N.V.; Zabolotskii, V.I. Effect of d-metal hydroxides on water dissociation in bipolar membranes. Pet. Chem. 2011, 51, 577–584. [Google Scholar] [CrossRef]
- Melnikov, S.S.; Zabolotskii, V.I.; Sheldeshov, N.V.; Achoh, A.; Bondarev, D. Catalysis of water splitting reaction in asymmetric bipolar membranes with different chemical composition of cation-exchange layer. Desalin. Water Treat. 2018, 124, 30–36. [Google Scholar] [CrossRef]
- Melnikov, S.S.; Sheldeshov, N.V.; Zabolotskii, V.I. Theoretical and experimental study of current-voltage characteristics of asymmetric bipolar membranes. Desalin. Water Treat. 2018, 123, 1–13. [Google Scholar] [CrossRef]
- Kang, M.-S.; Tanioka, A.; Moon, S.-H. Effects of Interface Hydrophilicity and Metallic Compounds on Water-Splitting Efficiency in Bipolar Membranes. Korean J. Chem. Eng. 2002, 19, 99–106. [Google Scholar] [CrossRef]
- Kang, M.S.; Choi, Y.J.; Lee, H.J.; Moon, S.H. Effects of inorganic substances on water splitting in ion-exchange membranes: I. Electrochemical characteristics of ion-exchange membranes coated with iron hydroxide/oxide and silica sol. J. Colloid Interface Sci. 2004, 273, 523–532. [Google Scholar] [CrossRef]
- El Moussaoui, R.; Pourcelly, G.; Maeck, M.; Hurwitz, H.D.; Gavach, C. Co-ion leakage through bipolar membranes Influence on I–V responses and water-splitting efficiency. J. Memb. Sci. 1994, 90, 283–292. [Google Scholar] [CrossRef]
- Wilhelm, F.G.; van der Vegt, N.F.A.; Wessling, M.; Strathmann, H. Chronopotentiometry for the advanced current–voltage characterisation of bipolar membranes. J. Electroanal. Chem. 2001, 502, 152–166. [Google Scholar] [CrossRef]
- Hurwitz, H.D.; Dibiani, R. Experimental and theoretical investigations of steady and transient states in systems of ion exchange bipolar membranes. J. Memb. Sci. 2004, 228, 17–43. [Google Scholar] [CrossRef]
- Sharafan, M.V.; Zabolotsky, V.I. Study of electric mass transfer peculiarities in electromembrane systems by the rotating membrane disk method. Desalination 2014, 343, 194–197. [Google Scholar] [CrossRef]
- Zabolotsky, V.I.; Achoh, A.R.; Lebedev, K.A.; Melnikov, S.S. Permselectivity of bilayered ion-exchange membranes in ternary electrolyte. J. Memb. Sci. 2020, 608, 118152. [Google Scholar] [CrossRef]
- Kniaginicheva, E.; Pismenskaya, N.; Melnikov, S.; Belashova, E.; Sistat, P.; Cretin, M.; Nikonenko, V. Water splitting at an anion-exchange membrane as studied by impedance spectroscopy. J. Memb. Sci. 2015, 496, 78–83. [Google Scholar] [CrossRef]
- Pismenskaya, N.D.; Rybalkina, O.A.; Kozmai, A.E.; Tsygurina, K.A.; Melnikova, E.D.; Nikonenko, V.V. Generation of H+ and OH− ions in anion-exchange membrane/ampholyte-containing solution systems: A study using electrochemical impedance spectroscopy. J. Memb. Sci. 2020, 601, 117920. [Google Scholar] [CrossRef]
- Suendo, V.; Minagawa, M.; Tanioka, A. Bipolar Interface Formation of Cationic Surfactant on the Surface of a Cation-Exchange Membrane: Current−Voltage Characteristics in Aqueous Electrolyte Solution. Langmuir 2002, 18, 6266–6273. [Google Scholar] [CrossRef]
- Alcaraz, A.; Holdik, H.; Ruffing, T.; Ramírez, P.; Mafé, S. AC impedance spectra of bipolar membranes: An experimental study. J. Memb. Sci. 1998, 150, 43–56. [Google Scholar] [CrossRef]
- Ashrafi, A.M.; Gupta, N.; Neděla, D. An investigation through the validation of the electrochemical methods used for bipolar membranes characterization. J. Memb. Sci. 2017, 544, 195–207. [Google Scholar] [CrossRef]
- Kovalchuk, V.I.; Zholkovskij, E.K.; Aksenenko, E.V.; Gonzalez-Caballero, F.; Dukhin, S.S. Ionic transport across bipolar membrane and adjacent Nernst layers. J. Memb. Sci. 2006, 284, 255–266. [Google Scholar] [CrossRef]
- Sonin, A.A.; Grossman, G. Ion transport through layered ion exchange membranes. J. Phys. Chem. 1972, 76, 3996–4006. [Google Scholar] [CrossRef]
- Zabolotskii, V.I.; Sharafan, M.V.; Sheldeshov, N.V.; Lovtsov, E.G. Electric mass transport of sodium chloride through cation-exchange membrane MK-40 in dilute sodium chloride solutions: A rotating membrane disk study. Russ. J. Electrochem. 2008, 44, 141–146. [Google Scholar] [CrossRef]
- Melnikov, S.; Shkirskaya, S. Transport properties of bilayer and multilayer surface-modified ion-exchange membranes. J. Memb. Sci. 2019, 590, 117272. [Google Scholar] [CrossRef]
- Zabolotskii, V.I.; Sheldeshov, N.V.; Melnikov, S.S. Heterogeneous bipolar membranes and their application in electrodialysis. Desalination 2014, 342, 183–203. [Google Scholar] [CrossRef]
- Umnov, V.V.; Shel’deshov, N.V.; Zabolotskii, V.I. Current-voltage curve for the space charge region of a bipolar membrane. Russ. J. Electrochem. 1999, 35, 871–878. [Google Scholar]
- Mareev, S.A.; Evdochenko, E.; Wessling, M.; Kozaderova, O.A.; Niftaliev, S.I.; Pismenskaya, N.D.; Nikonenko, V.V. A comprehensive mathematical model of water splitting in bipolar membranes: Impact of the spatial distribution of fixed charges and catalyst at bipolar junction. J. Memb. Sci. 2020, 603, 118010. [Google Scholar] [CrossRef]
- Belloň, T.; Polezhaev, P.; Vobecká, L.; Svoboda, M.; Slouka, Z. Experimental observation of phenomena developing on ion-exchange systems during current-voltage curve measurement. J. Memb. Sci. 2019, 572, 607–618. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
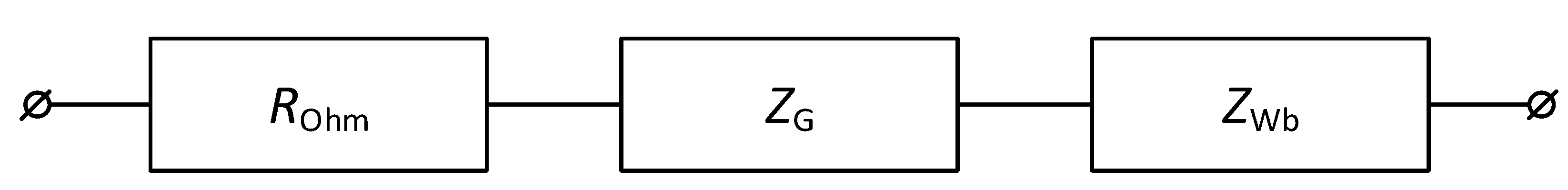{kind=link}
| Membrane Name | Series | AEL | CEL | Thickness of CEL/AEL, Microns | Catalyst | Mass of the Catalyst, mg/cm2 Deposition Time, min |
|---|---|---|---|---|---|---|
| BM-a-10 | BM-a | Ralex AMH-PES | sulfonated polytetrafluoroethylene | 10 | - | - |
| BM-a-30 | 30 | |||||
| BM-a-50 | 50 | |||||
| BM-a-70 | 70 | |||||
| BM-a-SPEEK | sulfonated poly(ether ether)ketone | 30 | ||||
| BM-a-SPS | sulfonated polystyrene | 30 | ||||
| BM-c-A80 | BM-c | Ralex CM-PES | Copolymer* | 80 | ||
| BM-ac-0.2K | BM-ac | Ralex AMH-PES | sulfonated polytetrafluoroethylene | 30 | KF-1 (microdispersion) | 0.2 |
| BM-ac-2K | 2 | |||||
| BM-ac-6K | 6 | |||||
| BM-ac-2dCr | Cr(OH)3 (microdispersion) | 2 | ||||
| BM-ac-2dFe | Fe(OH)3 (microdispersion) | 2 | ||||
| BM-ac-1Cr | Cr(OH)3 (electrodeposition) | 1 ** | ||||
| BM-ac-2Cr | 2 ** | |||||
| BM-ac-10Cr | 10 ** | |||||
| BM-ac-1Fe | Fe(OH)3 (electrodeposition) | 1 ** | ||||
| BM-ac-solFe | Fe(OH)3 (sol) | - |
| Membrane | BM-a-10 | BM-a-30 | BM-a-50 | BM-a-70 |
|---|---|---|---|---|
| CEL thickness, μm | 10 | 30 | 50 | 70 |
| 0.05 ± 0.03 | 0.05 ± 0.03 | 0.04 ± 0.02 | 0.02 ± 0.02 | |
| 0.14 ± 0.03 | 0.09 ± 0.02 | 0.06 ± 0.01 | 0.05 ± 0.01 | |
| 0.81 ± 0.05 | 0.86 ± 0.04 | 0.89 ± 0.03 | 0.91 ± 0.03 | |
| ΔUdiss, V | 2.5 | 4.1 | 3.3 | 2.9 |
| ilim, mA/cm2 | 0.38 | 0.05 | - | - |
| Cation-Exchange Layer | Current Density i, mA/cm2 | ||
|---|---|---|---|
| 2.0 | 2.5 | 3.0 | |
| SPTFE | 3.9 | 5.0 | 5.4 |
| SPEEK | 3.4 | 4.2 | 5.1 |
| SPS | 2.1 | 1.7 | 1.9 |
| Membrane | BM-ac-02K | BM-ac-2K | BM-ac-6K |
|---|---|---|---|
| Amount of catalyst, mg/cm2 | 0.2 | 2 | 6 |
| 0.05 ± 0.01 | 0.05 ± 0.01 | 0.05 ± 0.01 | |
| 0.09 ± 0.02 | 0.13 ± 0.02 | 0.21 ± 0.03 | |
| 0.86 ± 0.02 | 0.81 ± 0.02 | 0.74 ± 0.05 | |
| ΔUdiss, V | 1.6 | 0.9 | 0.75 |
| ilim, mA/cm2 | - | - | 0.25 |
| Membrane | BM-a-30 | BM-ac-2K | ||||
|---|---|---|---|---|---|---|
| Concentration, mol/L | 0.01 | 0.1 | 0.5 | 0.01 | 0.1 | 0.5 |
| 0.05 ± 0.03 | 0.02 ± 0.02 | 0.03 ± 0.02 | 0.05 ± 0.03 | 0.01 ± 0.02 | 0.01 ± 0.03 | |
| 0.09 ± 0.02 | 0.17 ± 0.04 | 0.15 ± 0.05 | 0.13 ± 0.04 | 0.08 ± 0.04 | 0.39 ± 0.04 | |
| 0.86 ± 0.04 | 0.81 ± 0.04 | 0.82 ± 0.07 | 0.82 ± 0.04 | 0.91 ± 0.04 | 0.60 ± 0.04 | |
| ΔUdiss, V | 3.7 | 5.0–5.5 | 9–10 | 1.2 | 1.8 | 2.3 |
| ilim, mA/cm2 | 0.05 | 0.6 | 3.1 | - | 1.8 | 15 |
| Membrane | BM-a-10 | BM-a-30 | BM-a-50 | BM-a-70 | BM-ac-0.2K | BM-ac-2K | BM-ac-6K | BM-ac-2dCr | BM-ac-1Fe | BM-ac-1Cr | BM-ac-2Cr | BM-ac-10Cr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kΣ, s−1 | 3.29 | 2.68 | 4.47 | 6.14 | 16.9 | 18.3 | 19.0 | 38.3 | 9.72 | 92.2 | 38.3 | 4.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melnikov, S.; Bondarev, D.; Nosova, E.; Melnikova, E.; Zabolotskiy, V. Water Splitting and Transport of Ions in Electromembrane System with Bilayer Ion-Exchange Membrane. Membranes 2020, 10, 346. https://doi.org/10.3390/membranes10110346
Melnikov S, Bondarev D, Nosova E, Melnikova E, Zabolotskiy V. Water Splitting and Transport of Ions in Electromembrane System with Bilayer Ion-Exchange Membrane. Membranes. 2020; 10(11):346. https://doi.org/10.3390/membranes10110346
Chicago/Turabian StyleMelnikov, Stanislav, Denis Bondarev, Elena Nosova, Ekaterina Melnikova, and Victor Zabolotskiy. 2020. "Water Splitting and Transport of Ions in Electromembrane System with Bilayer Ion-Exchange Membrane" Membranes 10, no. 11: 346. https://doi.org/10.3390/membranes10110346
APA StyleMelnikov, S., Bondarev, D., Nosova, E., Melnikova, E., & Zabolotskiy, V. (2020). Water Splitting and Transport of Ions in Electromembrane System with Bilayer Ion-Exchange Membrane. Membranes, 10(11), 346. https://doi.org/10.3390/membranes10110346