
Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies

 , , ,
, , ,
Abstract
:1. Introduction
2. Binding Modules and Characteristics of Bispecific Antibodies
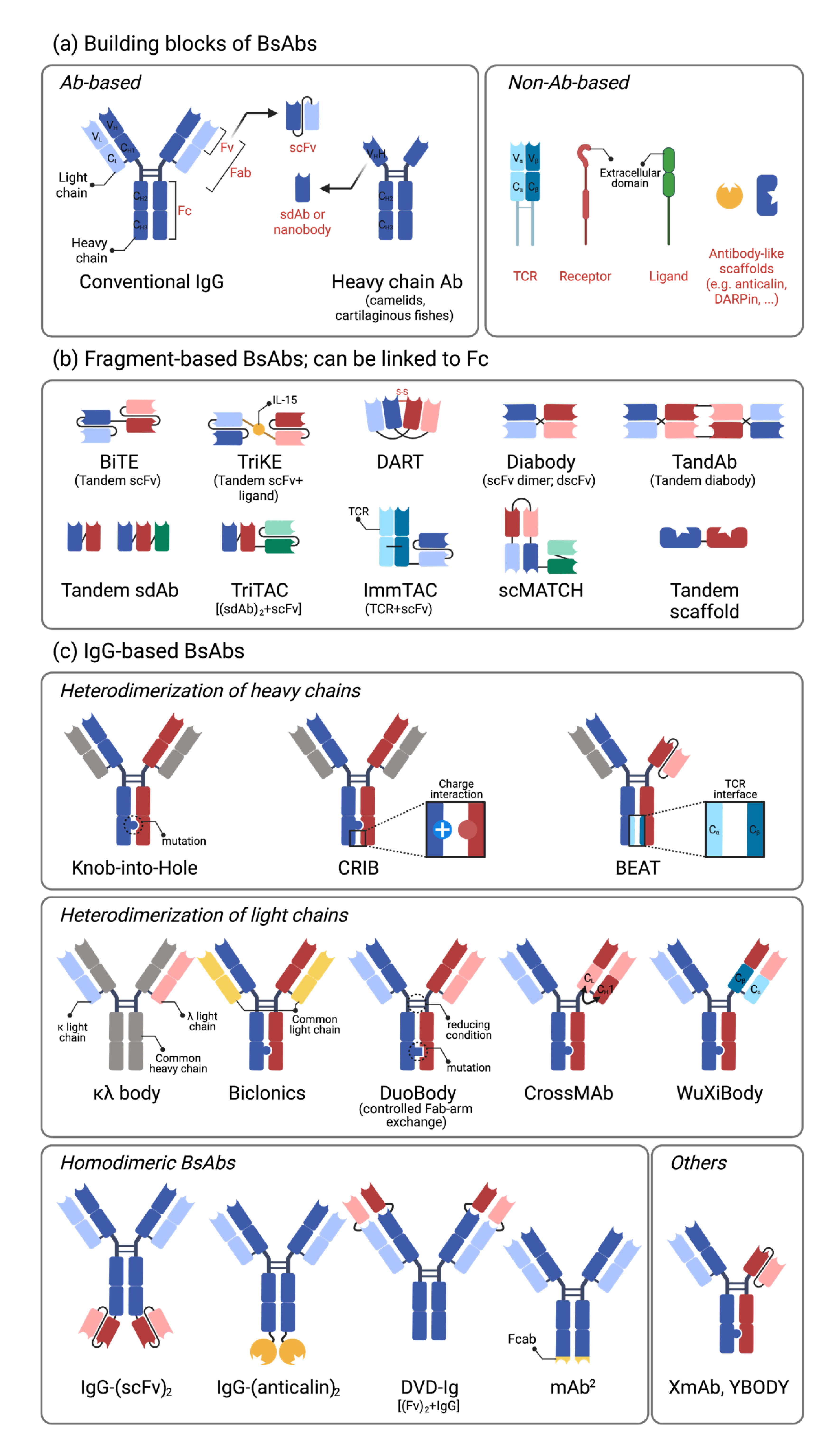
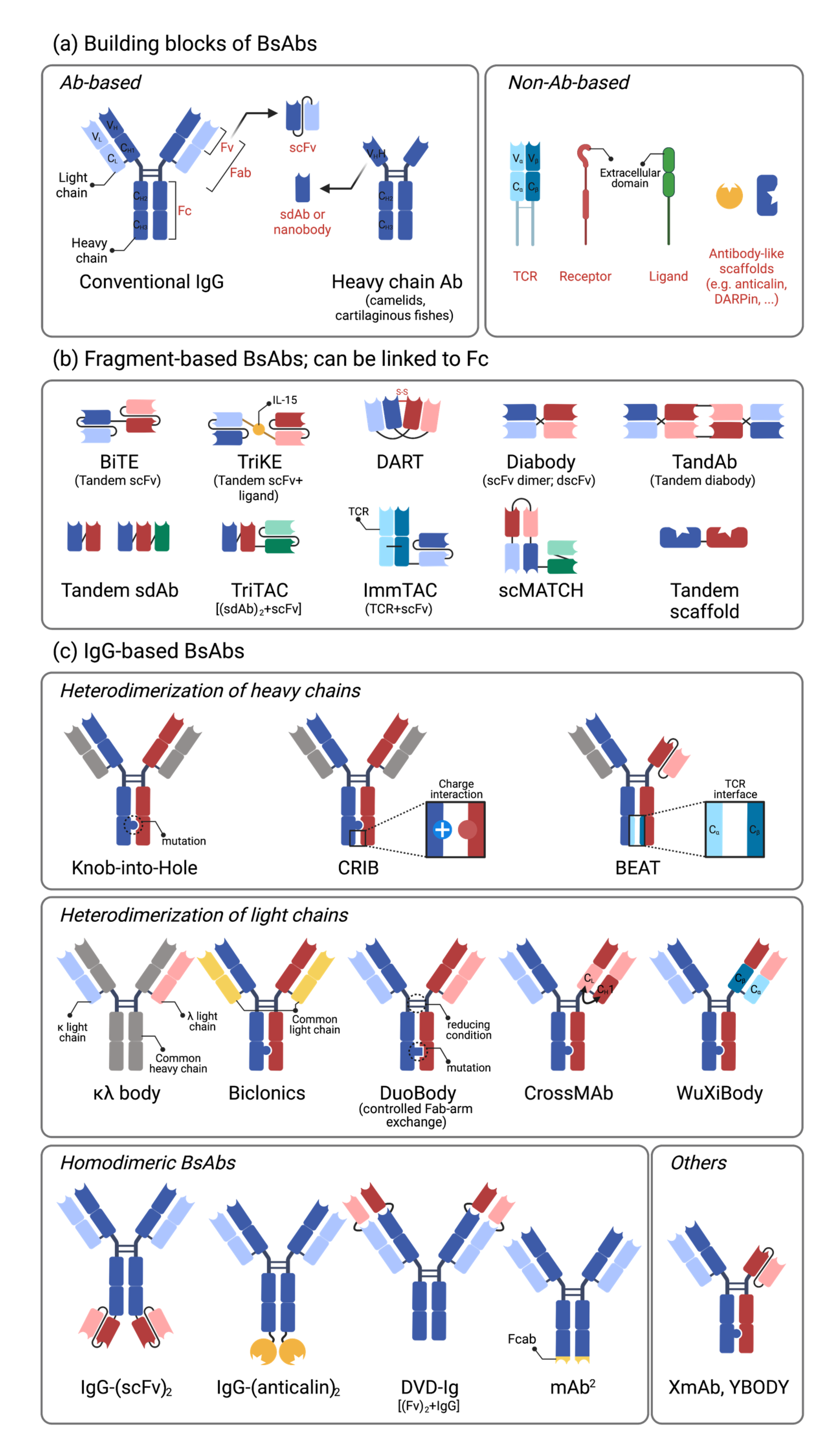
2.1. Building Blocks of Bispecific Antibodies
2.2. Single-Chain Variable Fragment (scFv)-Based Bispecific Antibodies
2.3. IgG-Based Bispecific Antibodies
2.4. Affinity and Valency of Bispecific Antibodies
2.5. Immunogenicity of Bispecific Antibodies
3. Multiple Types of Bispecific Therapeutics in Immuno-Oncology
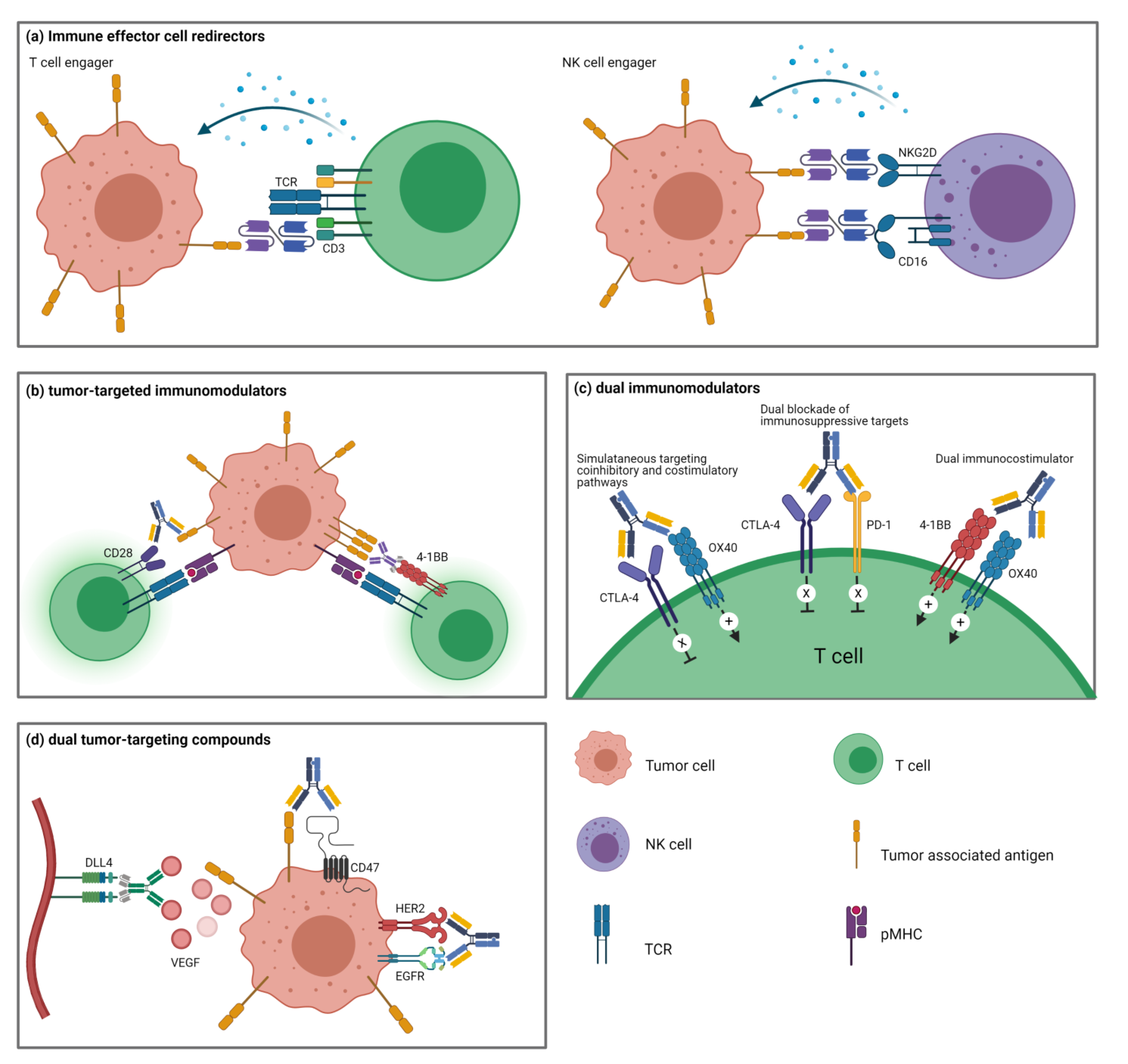
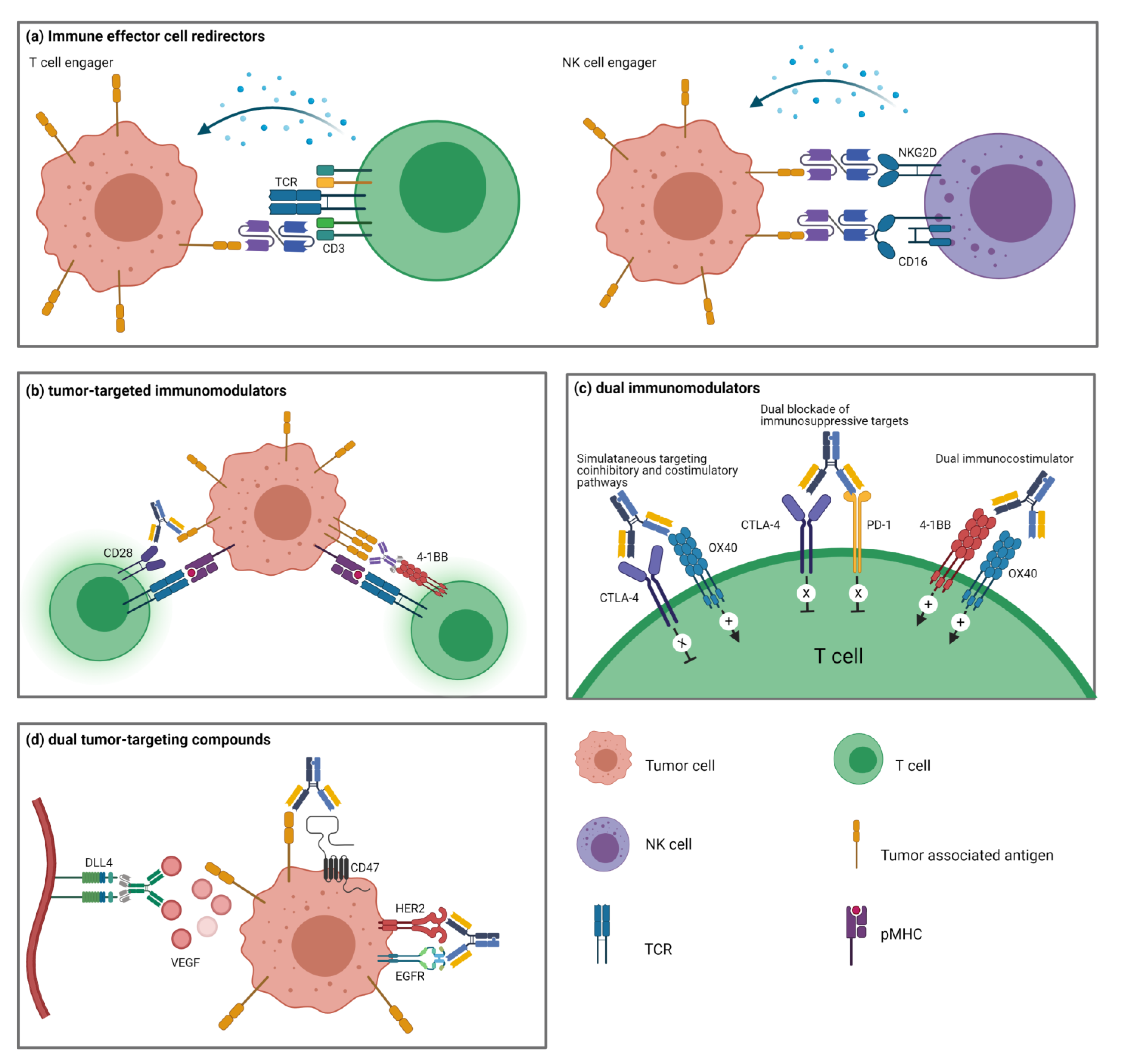
4. Immune Effector Cell Redirectors
4.1. T Cell-Engagers
4.2. NK Cell-Engagers
4.3. Other Immune Cell-Engagers
5. Tumor-Targeted Immunomodulators
5.1. Immunoglobulin Superfamily
5.2. TNFR Superfamily
6. Dual Immunomodulators
6.1. Dual Blockade of Immunosuppressive Targets
6.2. Simultaneous Targeting of Coinhibitory and Costimulatory Pathways
6.3. Dual Stimulation of Costimulatory Pathways
7. Dual Tumor-Targeting BsAbs
7.1. Tumor Receptor Tyrosine Kinase Blockade
7.2. Angiogenesis Inhibition
7.3. Improved Delivery of Payloads
7.4. Tumor-Targeted Tumor Cell Lysis
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Feugier, P. A review of rituximab, the first anti-CD20 monoclonal antibody used in the treatment of B non-Hodgkin’s lymphomas. Future Oncol. 2015, 11, 1327–1342. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Godar, M.; de Haard, H.; Blanchetot, C.; Rasser, J. Therapeutic bispecific antibody formats: A patent applications review (1994-2017). Expert Opin. Ther. Pat. 2018, 28, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Wang, Z.; Moscoso-Castro, M.; D’Souza, P.; Lei, C.; Xu, J.; Gu, J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib. Ther. 2020, 3, 18–62. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.I. Fine-tuning bispecific therapeutics. Pharmacol. Ther. 2020, 212, 107582. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- De Vlieger, D.; Ballegeer, M.; Rossey, I.; Schepens, B.; Saelens, X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies 2019, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Ma, Y.; Zhang, Y.; Hong, S.; Yang, Y.; Fang, W.; Xu, J.; Van, H.; Kong, P.; Yang, F.; et al. The preliminary efficacy and safety data of KN046 in patients failed on prior immune checkpoint inhibitors therapy. J. Clin. Oncol. 2020, 38, 3020. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Patel, M.; Luke, J.; Hamilton, E.; Chmielowski, B.; Blumenschein, G.; Kindler, H.; Bahadur, S.; Santa-Maria, C.; Koucheki, J.; Sun, J.; et al. 313 A phase 1 evaluation of tebotelimab, a bispecific PD-1 x LAG-3 DART® molecule, in combination with margetuximab in patients with advanced HER2+ neoplasms. J. Immunother. Cancer 2020, 8, A193. [Google Scholar] [CrossRef]
- Sharma, M.; Sanborn, R.E.; Cote, G.M.; Bendell, J.C.; Kaul, S.; Chen, F.; Berezhnoy, A.; Moore, P.; Bonvini, E.; Sumrow, B.J.; et al. 1020O A phase I, first-in-human, open-label, dose escalation study of MGD019, an investigational bispecific PD-1 x CTLA-4 DART® molecule in patients with advanced solid tumours. Ann. Oncol. 2020, 31, S704–S705. [Google Scholar] [CrossRef]
- Wu, J.; Fu, J.; Zhang, M.; Liu, D. AFM13: A first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 2015, 8, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayes, P.; Tacken, P.; Wang, S.; Loo, P.-F.v.; Condamine, T.; Maaden, H.v.d.; Rovers, E.; Engels, S.; Fransen, F.; Kulkarni, A.; et al. Abstract 539: A bispecific Fc-silenced IgG1 antibody (MCLA-145) requires PD-L1 binding to activate CD137. Cancer Res. 2019, 79, 539. [Google Scholar] [CrossRef]
- Waite, J.C.; Wang, B.; Haber, L.; Hermann, A.; Ullman, E.; Ye, X.; Dudgeon, D.; Slim, R.; Ajithdoss, D.K.; Godin, S.J.; et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci. Transl. Med. 2020, 12, eaba2325. [Google Scholar] [CrossRef] [PubMed]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12, eaaw7888. [Google Scholar] [CrossRef]
- Hutchings, M.; Lugtenburg, P.; Mous, R.; Clausen, M.R.; Chamuleau, M.; Linton, K.; Rule, S.; Lopez, J.S.; Oliveri, R.S.; DeMarco, D.; et al. Epcoritamab (GEN3013; DuoBody-CD3 × CD20) to induce complete response in patients with relapsed/refractory B-cell non-Hodgkin lymphoma (B-NHL): Complete dose escalation data and efficacy results from a phase I/II trial. J. Clin. Oncol. 2020, 38, 8009. [Google Scholar] [CrossRef]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Killock, D. Engaging results with glofitamab. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Park, E.; Sung, E.; Chung, H.; Lee, Y.; Yoo, J.; Park, M.; Kim, E.; Son, Y.-G.; Choi, H.; Jung, J.; et al. The PD-L1 x 4-1BB bispecific antibody ABL503 shows potent anti-tumor effect through tumor-directed T cell activation. PEGS Boston 2019. [Google Scholar]
- Jiang, W.; Fang, L.; Wang, Z.; Guo, T.B.; Park, E.; Sung, E.; Jung, J. Abstract 5644: Claudin 18.2 × 4-1BB bispecific antibody induced potent tumor inhibition through tumor-specific 4-1BB activation. Cancer Res. 2020, 80, 5644. [Google Scholar] [CrossRef]
- Rothe, C.; Skerra, A. Anticalin® Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Hinner, M.J.; Aiba, R.S.B.; Jaquin, T.J.; Berger, S.; Dürr, M.C.; Schlosser, C.; Allersdorfer, A.; Wiedenmann, A.; Matschiner, G.; Schüler, J.; et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion PRS-343. Clin. Cancer Res. 2019, 25, 5878–5889. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.; Wong, D.; Hu-Lieskovan, S.; Papadopoulos, K.; Morrow, M.; Grabowska, U.; Gliddon, D.; Holz, J.-B.; LoRusso, P. 395 A first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1, in patients with advanced cancer and resistance to PD-(L)1 therapy. J. Immunother. Cancer 2020, 8, A240. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Hickingbottom, B.; Clynes, R.; Desjarlais, J.; Li, C.; Ding, Y. Preliminary safety and pharmacodynamic (PD) activity of XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in a phase I dose escalation study of patients with selected advanced solid tumors. J. Clin. Oncol. 2020, 38, e15001. [Google Scholar] [CrossRef]
- Zhang, J.; Yi, J.; Zhou, P. Development of bispecific antibodies in China: Overview and prospects. Antib. Ther. 2020, 3, 126–145. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.L.; Cole, D.; Kenefeck, R.; Okelly, I.; Lepore, M.; Jakobsen, B.K. Novel TCR-based biologics: Mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 2019, 77, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Mack, M.; Riethmüller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Lakins, M.A.; Koers, A.; Giambalvo, R.; Hughes, R.; Marshall, S.; Wydro, M.; Gradinaru, C.; Batey, S.; Gliddon, D.; Morrow, M.; et al. Abstract 4547: Clustering CD137 via cell-expressed PD-L1 crosslinking avoided Fc-mediated agonism and resulted in safe and potent conditional lymphocyte activation. Cancer Res. 2020, 80, 4547. [Google Scholar] [CrossRef]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions—Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Staflin, K.; Zuch de Zafra, C.L.; Schutt, L.K.; Clark, V.; Zhong, F.; Hristopoulos, M.; Clark, R.; Li, J.; Mathieu, M.; Chen, X.; et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight 2020, 5, e133757. [Google Scholar] [CrossRef] [Green Version]
- Krishna, M.; Nadler, S.G. Immunogenicity to Biotherapeutics—The Role of Anti-drug Immune Complexes. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Franquiz, M.J.; Short, N.J. Blinatumomab for the Treatment of Adult B-Cell Acute Lymphoblastic Leukemia: Toward a New Era of Targeted Immunotherapy. Biologics 2020, 14, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.; Bendell, J.; Tolcher, A.; Hurvitz, S.; Patnaik, A.; Shroff, R.; Pohlmann, P.; Zettl, M.; Hahn, N.; Krishnamurthy, A.; et al. O82 A phase 1 dose escalation study of PRS-343, a HER2/4–1BB bispecific molecule, in patients with HER2-positive malignancies. J. Immunother. Cancer 2020, 8, A1–A2. [Google Scholar] [CrossRef]
- Jurichson, J. Aptevo Therapeutics and MorphoSys End Joint Development and Commercialization Agreement for MOR209/ES414; Aptevo Therapeutics Inc.: Seattle, WA, USA, 2017. [Google Scholar]
- Jurchison, S. Aptevo Therapeutics Reports Third Quarter 2018 Financial Results; Aptevo Therapeutics Inc.: Seattle, WA, USA, 2018. [Google Scholar]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Dufner, V.; Sayehli, C.M.; Chatterjee, M.; Hummel, H.D.; Gelbrich, G.; Bargou, R.C.; Goebeler, M.-E. Long-term outcome of patients with relapsed/refractory B-cell non-Hodgkin lymphoma treated with blinatumomab. Blood Adv. 2019, 3, 2491–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.G.; Marmé, F.; Moldenhauer, G.; Lindhofer, H.; Hennig, M.; Spannagl, R.; Essing, M.M.; Linke, R.; Seimetz, D. Humoral response to catumaxomab correlates with clinical outcome: Results of the pivotal phase II/III study in patients with malignant ascites. Int. J. Cancer 2012, 130, 2195–2203. [Google Scholar] [CrossRef]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers 2019, 11, 971. [Google Scholar] [CrossRef] [Green Version]
- Bortoletto, N.; Scotet, E.; Myamoto, Y.; D’Oro, U.; Lanzavecchia, A. Optimizing anti-CD3 affinity for effective T cell targeting against tumor cells. Eur. J. Immunol. 2002, 32, 3102–3107. [Google Scholar] [CrossRef]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative Target Affinities of T-Cell–Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, A.; Liu, Q.; Yuan, X.; Xu, H.; Jiao, D.; Pestell, R.G.; Han, X.; Wu, K. Recent advances of bispecific antibodies in solid tumors. J. Hematol. Oncol. 2017, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. ImmunoTherapy Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.Y.; Miranda, Y.; Phung, S.; Chen, H.; Rashid, R.; Endo, N.A.; Chan, E.W.; Pong, E.; Bonzon, C.; Muchhal, U.S.; et al. Immunotherapy with Long-Lived Anti-CD38 × Anti-CD3 Bispecific Antibodies Stimulates Potent T Cell-Mediated Killing of Human Myeloma Cell Lines and CD38+ Cells in Monkeys: A Potential Therapy for Multiple Myeloma. Blood 2014, 124, 4727. [Google Scholar] [CrossRef]
- Moore, G.L.; Lee, S.-H.; Schubbert, S.; Miranda, Y.; Rashid, R.; Pong, E.; Phung, S.; Chan, E.W.; Chen, H.; Endo, N.; et al. Tuning T Cell Affinity Improves Efficacy and Safety of Anti-CD38 × Anti-CD3 Bispecific Antibodies in Monkeys—A Potential Therapy for Multiple Myeloma. Blood 2015, 126, 1798. [Google Scholar] [CrossRef]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Umaña, P.; Herter, S.; Colombetti, S.; Sam, J.; Le Clech, M.; Freimoser-Grundschober, A.; Richard, M.; Nicolini, V.; Gerdes, C.; et al. CD20 Tcb (RG6026), a Novel “2:1” T Cell Bispecific Antibody for the Treatment of B Cell Malignancies. Blood 2016, 128, 1836. [Google Scholar] [CrossRef]
- Hernandez-Hoyos, G.; Sewell, T.; Bader, R.; Bannink, J.; Chenault, R.A.; Daugherty, M.; Dasovich, M.; Fang, H.; Gottschalk, R.; Kumer, J.; et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2155–2165. [Google Scholar] [CrossRef] [Green Version]
- Banaszek, A.; Bumm, T.G.P.; Nowotny, B.; Geis, M.; Jacob, K.; Wölfl, M.; Trebing, J.; Kucka, K.; Kouhestani, D.; Gogishvili, T.; et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun. 2019, 10, 5387. [Google Scholar] [CrossRef] [PubMed]
- Boustany, L.M.; Wong, L.; White, C.W.; Diep, L.; Huang, Y.; Liu, S.; Richardson, J.H.; Kavanaugh, W.M.; Irving, B.A. Abstract A164: EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Mol. Cancer Ther. 2018, 17, A164. [Google Scholar] [CrossRef]
- Geiger, M.; Stubenrauch, K.-G.; Sam, J.; Richter, W.F.; Jordan, G.; Eckmann, J.; Hage, C.; Nicolini, V.; Freimoser-Grundschober, A.; Ritter, M.; et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat. Commun. 2020, 11, 3196. [Google Scholar] [CrossRef]
- Chiang, S.C.C.; Theorell, J.; Entesarian, M.; Meeths, M.; Mastafa, M.; Al-Herz, W.; Frisk, P.; Gilmour, K.C.; Ifversen, M.; Langenskiöld, C.; et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 2013, 121, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, N.C.; Mathew, P.A. NKp44 and Natural Cytotoxicity Receptors as Damage-Associated Molecular Pattern Recognition Receptors. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Chretien, A.-S.; Le Roy, A.; Vey, N.; Prebet, T.; Blaise, D.; Fauriat, C.; Olive, D. Cancer-Induced Alterations of NK-Mediated Target Recognition: Current and Investigational Pharmacological Strategies Aiming at Restoring NK-Mediated Anti-Tumor Activity. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Hogarth, P.M.; Pietersz, G.A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 2012, 11, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Gantke, T.; Weichel, M.; Herbrecht, C.; Reusch, U.; Ellwanger, K.; Fucek, I.; Eser, M.; Müller, T.; Griep, R.; Molkenthin, V.; et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng. Des. Sel. 2017, 30, 673–684. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Oh, F.; Lenvik, A.J.; Lebeau, A.M.; Panyam, J.; Miller, J.S.; Vallera, D.A. Engineering of Anti-CD133 Trispecific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity. Cancer Res Treat 2017, 49, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontos, F.; Kurokawa, T.; Vallera, D.A.; Ferrone, S.; Ferrone, C.R. IL-15/B7-H3 TriKEs-Based Immunotherapy for Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2019, 229, S176. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e1716. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fury, M.G.; Lipton, A.; Smith, K.M.; Winston, C.B.; Pfister, D.G. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol. Immunother. 2008, 57, 155–163. [Google Scholar] [CrossRef]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des Devel 2018, 12, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Corgnac, S.; Perret, R.; Derré, L.; Zhang, L.; Stirnemann, K.; Zauderer, M.; Speiser, D.E.; Mach, J.-P.; Romero, P.; Donda, A. CD1d-antibody fusion proteins target iNKT cells to the tumor and trigger long-term therapeutic responses. Cancer Immunol. Immunother. 2013, 62, 747–760. [Google Scholar] [CrossRef] [Green Version]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Park, S.-H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 2020, 20. [Google Scholar] [CrossRef]
- Tacke, M.; Hanke, G.; Hanke, T.; Hünig, T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: Evidence for functionally distinct forms of CD28. Eur. J. Immunol. 1997, 27, 239–247. [Google Scholar] [CrossRef]
- Attarwala, H. TGN1412: From Discovery to Disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Otz, T.; Große-Hovest, L.; Hofmann, M.; Rammensee, H.G.; Jung, G. A bispecific single-chain antibody that mediates target cell-restricted, supra-agonistic CD28 stimulation and killing of lymphoma cells. Leukemia 2009, 23, 71–77. [Google Scholar] [CrossRef]
- Hedvat, M.; Zeng, V.; Diaz, J.; Bonzon, C.; Avery, K.; Rashid, R.; Leung, I.; Bartlow, N.; Bakhit, C.; Dragovich, M.; et al. 697 Tumor-targeted CD28 costimulatory bispecific antibodies enhance T cell activation in solid tumors. J. Immunother. Cancer 2020, 8, A419. [Google Scholar] [CrossRef]
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; van Ginkel, R.J.; Heskamp, S.; Wierstra, P.J.; Bremer, E.; Helfrich, W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. OncoImmunology 2018, 7, e1466016. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, I.; Hendriks, M.A.J.M.; van Ginkel, R.J.; Samplonius, D.F.; Bremer, E.; Helfrich, W. Bispecific Antibody Approach for Improved Melanoma-Selective PD-L1 Immune Checkpoint Blockade. J. Investig. Dermatol. 2019, 139, 2343–2351.e2343. [Google Scholar] [CrossRef]
- Moran, A.E.; Kovacsovics-Bankowski, M.; Weinberg, A.D. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr. Opin. Immunol. 2013, 25, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-D.; Park, S.; Jeong, S.; Lee, Y.J.; Lee, H.; Kim, C.G.; Kim, K.H.; Hong, S.-M.; Lee, J.-Y.; Kim, S.; et al. 4-1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Hepatology 2020, 71, 955–971. [Google Scholar] [CrossRef] [Green Version]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Williams, J.B.; Horton, B.L.; Zheng, Y.; Duan, Y.; Powell, J.D.; Gajewski, T.F. The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med. 2017, 214, 381–400. [Google Scholar] [CrossRef]
- Jure-Kunkel, M.N.; Calarota, S.; Girit, E.; Abraham, R.; Balimane, P.; Price, K.; Weiner, D.; Hefta, L. Functional characterization of fully human anti-CD137 antibodies. Cancer Res. 2006, 66, 1117. [Google Scholar]
- Fisher, T.S.; Kamperschroer, C.; Oliphant, T.; Love, V.A.; Lira, P.D.; Doyonnas, R.; Bergqvist, S.; Baxi, S.M.; Rohner, A.; Shen, A.C.; et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol. Immunother. 2012, 61, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Ku, G.; Bendell, J.C.; Tolcher, A.W.; Hurvitz, S.A.; Krishnamurthy, A.; El-Khoueiry, A.B.; Patnaik, A.; Shroff, R.T.; Noonan, A.; Hahn, N.M.; et al. 525O A phase I dose escalation study of PRS-343, a HER2/4-1BB bispecific molecule, in patients with HER2-positive malignancies. Ann. Oncol. 2020, 31, S462–S463. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Hüsser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11, eaav5989. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Miller, R.; Nilsson, A.; Ljung, L.; Chunyk, A.; McMahan, C.; Bienvenue, D.; Askmyr, M.; Hernandez-Hoyos, G.; Fritzell, S. 851 Potent tumor-directed T cell activation and in vivo tumor inhibition induced by a 4–1BB x 5T4 ADAPTIR™ bispecific antibody. J. Immunother. Cancer 2020, 8, A507. [Google Scholar] [CrossRef]
- You, G.; Lee, Y.; Kang, Y.-W.; Park, H.W.; Park, K.; Kim, H.; Kim, Y.-M.; Kim, S.; Kim, J.-H.; Moon, D.; et al. B7-H3×4-1BB bispecific antibody augments antitumor immunity by enhancing terminally differentiated CD8+ tumor-infiltrating lymphocytes. Sci. Adv. 2021, 7, eaax3160. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Murphy, J.T.; Wolchok, J.D. Modulation of GITR for cancer immunotherapy. Curr. Opin. Immunol. 2012, 24, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Burckhart, T.; Thiel, M.; Nishikawa, H.; Wüest, T.; Müller, D.; Zippelius, A.; Ritter, G.; Old, L.; Shiku, H.; Renner, C. Tumor-specific Crosslinking of GITR as Costimulation for Immunotherapy. J. Immunother. 2010, 33, 925–934. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Cohen, D.; Belmar, N.A.; Choi, D.; Tan, S.S.; Sho, M.; Akamatsu, Y.; Kim, H.; Iyer, R.; Cabel, J.; et al. A Bispecific Molecule Targeting CD40 and Tumor Antigen Mesothelin Enhances Tumor-Specific Immunity. Cancer Immunol. Res. 2019, 7, 1864–1875. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and efficacy of a monovalent bispecific PD-1/CTLA-4 antibody that enhances CTLA-4 blockade on PD-1+ activated T cells. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Mazor, Y.; Elder, M.; Hasani, S.; Wang, B.; Mosely, S.; Jones, D.; Hansen, A.; Yang, C.; Wu, Y.; et al. Abstract 2776: MEDI5752: A novel bispecific antibody that preferentially targets CTLA-4 on PD-1 expressing T-cells. Cancer Res. 2018, 78, 2776. [Google Scholar] [CrossRef]
- Anderson Ana, C.; Joller, N.; Kuchroo Vijay, K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [Green Version]
- Zhong, T.; Huang, Z.; Pang, X.; Chen, N.; Jin, X.; Xia, Y.; Wang, Z.M.; Li, B.; Xia, Y. 702 Dual blockade of the PD-1 checkpoint pathway and the adenosinergic negative feedback signaling pathway with a PD-1/CD73 bispecific antibody for cancer immune therapy. J. Immunother. Cancer 2020, 8, A422. [Google Scholar] [CrossRef]
- Kvarnhammar, A.M.; Veitonmäki, N.; Hägerbrand, K.; Dahlman, A.; Smith, K.E.; Fritzell, S.; von Schantz, L.; Thagesson, M.; Werchau, D.; Smedenfors, K.; et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J. Immunother. Cancer 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, M.; Pravin, J.; Rodrigues, L.; Uhlenbroich, S.; Everett, K.L.; Wollerton, F.; Morrow, M.; Tuna, M.; Brewis, N. CD137/OX40 Bispecific Antibody Induces Potent Antitumor Activity that Is Dependent on Target Coengagement. Cancer Immunol. Res. 2020, 8, 781–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2016, 8, 3980–4000. [Google Scholar] [CrossRef] [Green Version]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Cui, J. Anti-angiogenesis: Opening a new window for immunotherapy. Life Sci. 2020, 258, 118163. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Moek, K.L.; de Groot, D.J.A.; de Vries, E.G.E.; Fehrmann, R.S.N. The antibody–drug conjugate target landscape across a broad range of tumour types. Ann. Oncol. 2017, 28, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- de Goeij, B.E.C.G.; Vink, T.; ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblett, K.J.; Hammond, P.W.; Barnscher, S.D.; Fung, V.K.; Davies, R.H.; Wickman, G.R.; Hernandez, A.; Ding, T.; Galey, A.S.; Winters, G.C.; et al. Abstract 3914: ZW49, a HER2-targeted biparatopic antibody-drug conjugate for the treatment of HER2-expressing cancers. Cancer Res. 2018, 78, 3914. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Dheilly, E.; Moine, V.; Broyer, L.; Salgado-Pires, S.; Johnson, Z.; Papaioannou, A.; Cons, L.; Calloud, S.; Majocchi, S.; Nelson, R.; et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol. Ther. 2017, 25, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Walczak, H. Death Receptor–Ligand Systems in Cancer, Cell Death, and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Brünker, P.; Wartha, K.; Friess, T.; Grau-Richards, S.; Waldhauer, I.; Koller, C.F.; Weiser, B.; Majety, M.; Runza, V.; Niu, H.; et al. RG7386, a Novel Tetravalent FAP-DR5 Antibody, Effectively Triggers FAP-Dependent, Avidity-Driven DR5 Hyperclustering and Tumor Cell Apoptosis. Mol. Cancer Ther. 2016, 15, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.; Blay, J.-Y.; Cassier, P.; Bauer, T.; Terret, C.; Mueller, C.; Morel, A.; Chesne, E.; Xu, Z.-x.; Tessier, J.; et al. Abstract A092: Phase 1 trial of RO6874813, a novel bispecific FAP-DR5 antibody, in patients with solid tumors. Mol. Cancer Ther. 2018, 17, A092. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Category | Antibody Format/Platform | Structure | Company | Product | Target |
|---|---|---|---|---|---|
| Single domain antibody (sdAb)-based BsAbs [16,17,18] |  | Alphamab | KN046 | PD-L1 × CTLA-4 | |
| Inhibrix | INBRX-105 | PD-L1 × 4-1BB | |||
| Single-chain variable fragment (scFv)-based BsAbs [19,20,21,22,23,24,25,26,27] | Bispecific T-cell Engager (BiTE) | 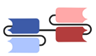 | Amgen | Blinatumomab (Blincyto) | CD19 × CD3 |
| AMG330 | CD33 × CD3 | ||||
| AMG420 | BCMA × CD3 | ||||
| Bayor | BAY2010112 | PSMA × CD3 | |||
| BiTE-Fc/Half-life extended BiTE (HLE BiTE) | 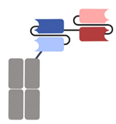 | Amgen | AMG701 (Pavurutamab) | BCMA × CD3 | |
| AMG673 | CD33 × CD3 | ||||
| AMG757 | DLL3 × CD3 | ||||
| Dual affinity retargeting (DART) | 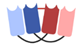 | Macrogenics | MGD006 (Flotetuzumab) | CD123 × CD3 | |
| DART-Fc | 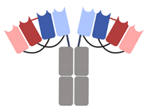 | Macrogenics | MGD013 (Tebotelimab) | PD-1 × LAG-3 | |
| MGD019 | PD-1 × CTLA-4 | ||||
| Tandem Diabody (TandAb) | 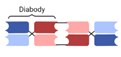 | Affimed | AFM13 | CD30 × CD16A | |
| Amphivena | AMV564 | CD33 × CD3 | |||
| Trispecific Killer Engager (TriKE) | 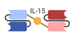 | GT Biopharma | GTB-3550 | CD33 × CD16, IL-15 | |
| IgG-based, heterodimeric bispecifics [28,29,30,31,32,33,34] | Common light chain (Biclonics, Veloci-Bi) |  | Merus | MCLA-145 | PD-L1 × 4-1BB |
| Regeneron | REGN1979 | CD20 × CD3 | |||
| REGN5678 | PSMA × CD28 | ||||
| DuoBody; Controlled Fam-arm exchange |  | Janssen | Teclistamab (JNJ-64007957) | BCMA × CD3 | |
| Genmab-Abbvie | Epcoritamab (GEN3013) | CD20 × CD3 | |||
| Genmab-Biontech | BNT312 (GEN1042) | CD40 × 4-1BB | |||
| BNT311 (GEN1046) | PD-L1 × 4-1BB | ||||
| 1 + 1 CrossMAb | 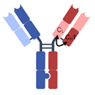 | Roche | RO7247669 | PD-1 × LAG-3 | |
| Mosunetuzumab | CD20 × CD3 | ||||
| Cevostamab | FcRH5 × CD3 | ||||
| 2 + 1 CrossMAb | 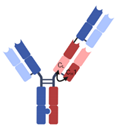 | Roche | Glofitamab | CD20 × CD3 | |
| RG7802 (RO6958688) | CEA × CD3 | ||||
| Celgene | CC-93269 | BCMA × CD3 | |||
| IgG-based, homodimeric bispecifics [35,36,37,38,39] | Grabody, IgG-scFv | 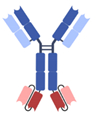 | ABL Bio | ABL503 | PD-L1 × 4-1BB |
| ABL111 | Claudin 18.2 × 4-1BB | ||||
| IgG-anticalin fusion protein | 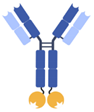 | Pieris | PRS-343 | HER2 × 4-1BB | |
| Fc region with antigen binding (Fcab), mAb2 | 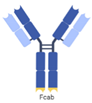 | F-Star | FS118 | PD-L1 × LAG-3 | |
| Others [40,41,42,43] | XmAb, Fab + scFv + Fc | 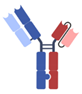 | Xencor | Vibecotamab (XmAb 14045) | CD123 × CD3 |
| XmAb20717 | PD-1 × CTLA-4 | ||||
| Bispecific Engagement by Antibodies based on TCR (BEAT) |  | Glenmark Pharmaceuticals | GBR1342 | CD38 × CD3 |
| Name | Targets | Developer | Format | Highest Phase | Clinical Trials |
|---|---|---|---|---|---|
| AFM13 | CD30 × CD16A | Affimed GmbH | Fv + (Fv)2 + Fv, 2 + 2 TandAb | II | NCT04074746 and 5 studies |
| AFM24 | EGFR × CD16A | Affimed GmbH | Fv + (Fv)2 + Fv, 2 + 2 TandAb | I/II | NCT04259450 |
| AFM26, RO7297089 | BCMA × CD16A | Affimed GmbH, Genentech | Fv + (Fv)2 + Fv, 2 + 2 TandAb | I | NCT04434469 |
| Solitomab, AMG110, MT110 | EpCAM × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE | I | NCT00635596 |
| AMG160 | PSMA × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE | I | NCT03792841 NCT04631601 |
| AMG199 | MUC17 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE | I | NCT04117958 |
| AMG330 | CD33 × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE | I | NCT02520427 NCT04478695 |
| AMG427 | FLT3 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE | I | NCT03541369 |
| AMG562 | CD19 × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE | I | NCT03571828 |
| AMG596 | EGFRvIII × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE | I | NCT03296696 |
| AMG673 | CD33 × CD3 | Amgen | scFv + scFv + Fc, 1+ 1 HLE BiTE | I | NCT03224819 |
| Pavurutamab, AMG701 | BCMA × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE | I | NCT03287908 |
| AMG757 | DLL3 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE | I | NCT03541369 |
| AMG910 | CLDN18.2 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE | I | NCT04260191 |
| Pasotuxizumab, AMG212, BAY2010112 | PSMA × CD3 | Amgen, Bayer AG | scFv + scFv, 1 + 1 BiTE | I | NCT01723475 |
| AMG420, BI836909 | BCMA × CD3 | Amgen, Boehringer Ingelheim | scFv + scFv, 1 + 1 BiTE | I | NCT03836053 |
| AMG424 | CD38 × CD3 | Amgen, Xencor | Fab + scFv + Fc, 1 + 1 XmAb | I | NCT03445663 |
| AMG509 | STEAP1 × CD3 | Amgen, Xencor | (Fab)2 + scFv + Fc, 2 + 1 XmAb | I | NCT04221542 |
| AMV564 | CD33 × CD3 | Amphivena Therapeutics | Fv + (Fv)2 + Fv, 2 + 2 TandAb | I | NCT04128423 and 2 studies |
| APVO436 | CD123 × CD3 | Aptevo Therapeutics | (scFv)2 + (scFv)2 + Fc, 2 + 2 ADAPTIR | I | NCT03647800 |
| CC-93269, EM801 | BCMA × CD3 | Celgene | Fab + (Fab)2 + Fc, 2 + 1 CrossMAb | I | NCT03486067 |
| ERY974 | Glypican-3 × CD3 | Chugai Pharmaceutical | Fab + Fab + Fc, 1 + 1 ART-Ig | I | NCT02748837 |
| A-319 | CD19 × CD3 | Evive Biotech | scFv + Fab, 1 + 1 | I | NCT04056975 |
| GEM333 | CD33 × CD3 | GEMoaB Monoclonals | scFv + scFv, 1 + 1 | I | NCT03516760 |
| GEM3PSCA | PSCA × CD3 | GEMoaB Monoclonals | scFv + scFv, 1 + 1 | I | NCT03927573 |
| RG6160, RO7187797, BFCR4350A | FcRH5 × CD3 | Genentech | Fab + Fab + Fc, 1 + 1 | I | NCT03275103 |
| RG6194, BTRC4017A | HER2 × CD3 | Genentech | undisclosed | I | NCT03448042 |
| RG6296, RO7297089 | BCMA × CD16A | Genentech | Fab + Fab + Fc, 1 + 1 | I | NCT04434469 |
| Mosunetuzumab, RG7828, RO7030816, BTCT4465A | CD20 × CD3 | Genentech, Roche, Chugai | Fab + Fab + Fc, 1 + 1 | II | NCT03677154 and 8 studies |
| GEN1044 | 5T4 × CD3 | Genmab, Abbvie | Fab + Fab + Fc, 1 + 1 DuoBody | I | NCT04424641 |
| Epcoritamab, GEN3013 | CD20 × CD3 | Genmab, Abbvie | Fab + Fab + Fc, 1 + 1 DuoBody | III | NCT03625037 and 5 studies |
| GTB-3550, OXS-3550 | CD33 × CD16, IL-15 | GT Biopharma | scFv + ligand + scFv, 1 + 1 + 1 TriKE | I/II | NCT03214666 |
| HPN424 | PSMA × HSA × CD3 | Harpoon Therapeutics | sdAb + sdAb + scFv, 1 + 1 + 1 TriTAC | I/II | NCT03577028 |
| ISB1302, GBR1302 | HER2 × CD3 | Ichnos Sciences, Glenmark Pharmaceuticals | Fab + scFv + Fc, 1 + 1 BEAT | I/II | NCT02829372 NCT03983395 |
| ISB1342, GBR1342 | CD38 × CD3 | Ichnos Sciences, Glenmark Pharmaceuticals | Fab + scFv + Fc, 1 + 1 BEAT | I/II | NCT03309111 |
| IGM-2323 | CD20 × CD3 | IGM Biosciences | IgM + scFv, 10 + 1 | I | NCT04082936 |
| Tebentafusp, IMCgp100 | gp100/HLA-A * 02:01 × CD3 | Immunocore | TCR + scFv, 1 + 1 ImmTAC | III | NCT03070392 and 5 studies |
| IMC-F106C | PRAME/HLA-A * 02:01 × CD3 | Immunocore | TCR + scFv, 1 + 1 ImmTAC | I/II | NCT04262466 |
| IMC-C103C | MAGE-A4/HLA-A * 02:01 × CD3 | Immunocore, Genentech | TCR + scFv, 1 + 1 ImmTAC | I/II | NCT03973333 |
| IMCnyeso, GSK01 | NY-ESO-1/HLA-A * 02:01 × CD3 | Immunocore, GlaxoSmithKline | TCR + scFv, 1 + 1 ImmTAC | I/II | NCT03515551 |
| JNJ-63709178 | CD123 × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody | I | NCT02715011 |
| JNJ-63898081 | PSMA × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody | I | NCT03926013 |
| Teclistamab, JNJ-64007957 | BCMA × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody | II | NCT04557098 and 5 studies |
| Talquetamab, JNJ-64407564 | GPRC5D × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody | II | NCT04634552 and 3 studies |
| JNJ-67571244 | CD33 × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody | I | NCT03915379 |
| MGD007 | gpA33 × CD3 | MacroGenics | Fv + Fv + Fc, 1 + 1 DART-Fc | I | NCT02248805 NCT03531632 |
| Orlotamab, MGD009 | B7-H3 × CD3 | MacroGenics | Fv + Fv + Fc, 1 + 1 DART-Fc | I | NCT02628535 NCT03406949 |
| Duvortuxizumab, MGD011, JNJ-64052781 | CD19 × CD3 | MacroGenics, Janssen Research & Development | Fv + Fv, 1 + 1 DART | I | NCT02743546 NCT02454270 |
| Flotetuzumab, MGD006, S80880 | CD123 × CD3 | MacroGenics, Servier | Fv + Fv, 1 + 1 DART | I | NCT04582864 and 5 studies |
| Tepoditamab, MCLA-117 | CLEC12A × CD3 | Merus | Fab + Fab + Fc, 1 + 1 Biclonics | I | NCT03038230 |
| PF-06671008 | P-cadherin × CD3 | Pfizer | Fv + Fv + Fc, 1 + 1 DART-Fc | I | NCT02659631 |
| PF-06863135 | BCMA × CD3 | Pfizer | Fab + Fab + Fc, 1 + 1 | II | NCT04649359 and 2 studies |
| Odronextamab, REGN1979 | CD20 × CD3 | Regeneron | Fab + Fab + Fc, 1 + 1 | II | NCT03888105 and 2 studies |
| REGN5458 | BCMA × CD3 | Regeneron | Fab + Fab + Fc, 1 + 1 | I/II | NCT03761108 |
| REGN5459 | BCMA × CD3 | Regeneron | Fab + Fab + Fc, 1 + 1 | I | NCT04083534 |
| REGN4018 | MUC16 × CD3 | Regeneron, Sanofi | Fab + Fab + Fc, 1 + 1 | I/II | NCT03564340 NCT04590326 |
| Glofitamab, RO7082859, RG6026 | CD20 × CD3 | Roche | Fab + (Fab)2 + Fc, 2 + 1 CrossMAb | III | NCT03075696 and 8 studies |
| Cibisatamab, RO6958688, RG7802 | CEA × CD3 | Roche, Genentech | Fab + (Fab)2 + Fc, 2 + 1 CrossMAb | I | NCT02650713 and 3 studies |
| SAR440234 | CD123 × CD3 | Sanofi | Fab + Fv + Fc, 1 + 1 | I/II | NCT03594955 |
| TNB-383B | BCMA × CD3 | TeneoBio, AbbVie | sdAb + Fab + Fc, 1 + 1 | I | NCT03933735 |
| M802 | HER2 × CD3 | Wuhan YZY Biopharma | Fab + scFv + Fc, 1 + 1 YBODY | I | NCT04501770 |
| Plamotamab, Xmab13676 | CD20 × CD3 | Xencor | Fab + scFv + Fc, 1 + 1 XmAb | I | NCT02924402 |
| Tidutamab, Xmab18087 | SSTR2 × CD3 | Xencor | Fab + scFv + Fc, 1 + 1 XmAb | I/II | NCT03411915 NCT04590781 |
| Vibecotamab, Xmab14045 | CD123 × CD3 | Xencor, Novartis | Fab + scFv + Fc, 1 + 1 XmAb | I | NCT02730312 |
| Nivatrotamab | GD2 × CD3 | Y-mAbs | IgG + (scFv)2, 2 + 2 BiClone | I/II | NCT04750239 |
| Name | Targets | Developer | Format | Highest Phase | Clinical Trials |
|---|---|---|---|---|---|
| ABBV-428 | MSLN × CD40 | AbbVie | (scFv)2 + (scFv)2 + Fc, 2 + 2 | I | NCT02955251 |
| ABL503 | PD-L1 × 4-1BB | ABL Bio, I-MAB | IgG + (scFv)2, 2 + 2 | I | NCT04762641 |
| AK104 | PD-1 × CTLA-4 | Akeso Biopharma | IgG + (scFv)2, 2 + 2 | II | NCT04172454 and 13 studies |
| ATOR-1015, ADC-1015 | OX40 × CTLA-4 | Alligator Bioscience | IgG + (ligand)2, 2 + 2 | I | NCT03782467 |
| AMG506, MP0310 | FAP × 4-1BB × HSA | Molecular Partners AG, Amgen | (DARPin)3, 1 + 1 + 1 | I | NCT04049903 |
| CDX-527 | PD-1 × CD40 | Celldex | IgG + (scFv)2, 2 + 2 | I | NCT04440943 |
| LY3415244 | PD-L1 × Tim-3 | Eli Lilly | Fab + Fab + Fc, 1 + 1 | I | NCT03752177 |
| LY3434172 | PD-1 × PD-L1 | Eli Lilly | Fab + Fab + Fc, 1 + 1 | I | NCT03936959 |
| FS118 | PD-L1 × LAG-3 | F-star | IgG with Fcab, 2 + 2 mAb2 | I/II | NCT03440437 |
| FS120 | OX40 × 4-1BB | F-star | IgG with Fcab, 2 + 2 mAb2 | I | NCT04648202 |
| FS222 | PD-L1 × 4-1BB | F-star | IgG with Fcab, 2 + 2 mAb2 | I | NCT04740424 |
| GEN1042 | CD40 × 4-1BB | Genmab, BioNTech | Fab + Fab + Fc, 1 + 1 DuoBody | I | NCT04083599 |
| GEN1046, BNT311 | PD-L1 × 4-1BB | Genmab, BioNTech | Fab + Fab + Fc, 1 + 1 DuoBody | I/II | NCT03917381 |
| GS-1423, AGEN1423 | CD73 × TGF-b | Gilead Sciences, Agenus | IgG + TGFβ receptor, 2 + 2 | I | NCT03954704 |
| INBRX-105, ES101 | PD-L1 × 4-1BB | Inhibrx, Elpiscience Biopharma | (sdAb)2 + (sdAb)2 + Fc, 2 + 2 | I | NCT03809624 NCT04009460 |
| IBI318 | PD-1 × PD-L1 | Innovent Biologics | Fab + Fab + Fc, 1 + 1 | II | NCT04777084 and 5 studies |
| KN046 | PD-L1 × CTLA-4 | Jiangsu Alphamab Biopharmaceuticals | (sdAb)2 + (sdAb)2 + Fc, 2 + 2 | III | NCT04040699 and 15 studies |
| SHR-1701 | PD-L1 × TGF-b | Jiangsu HengRui Medicine | IgG + TGFβ receptor, 2 + 2 | II | NCT04650633 and 11 studies |
| MGD019 | PD-1 × CTLA-4 | Macrogenics | (Fv)2 + (Fv)2 + Fc, 2 + 2 DART-Fc | I | NCT03761017 |
| Tebotelimab, MGD013 | PD-1 × LAG-3 | Macrogenics, Zai Lab | (Fv)2 + (Fv)2 + Fc, 2 + 2 DART-Fc | II/III | NCT04212221 and 6 studies |
| MEDI5752 | PD-1 × CTLA-4 | MedImmune | Fab + Fab + Fc, 1 + 1 | I | NCT04522323 and 2 studies |
| Bintrafusp Alfa, M7824 | PD-L1 × TGF-b | Merck KGaA | IgG + TGFβ receptor, 2 + 2 | III | NCT03631706 and 38 studies |
| MCLA-145 | PD-L1 × 4-1BB | Merus, Incyte | Fab + Fab + Fc, 1 + 1 Biclonics | I | NCT03922204 |
| NM21-1480 | PD-L1 × 4-1BB × HSA | Numab | Fv + Fv + Fv, 1 + 1 + 1 scMATCH3 | I/II | NCT04442126 |
| PRS-343 | HER2 × 4-1BB | Pieris Pharmaceuticals | IgG + (anticalin)2, 2 + 2 | I | NCT03330561 NCT03650348 |
| REGN5678 | PSMA × CD28 | Regeneron | Fab + Fab + Fc, 1 + 1 | I/II | NCT03972657 |
| RG6139, RO7247669 | PD-1 × LAG-3 | Roche | Fab + Fab + Fc, 1 + 1 CrossMAb | II | NCT04785820 NCT04140500 |
| RG7769, RO7121661 | PD-1 × Tim-3 | Roche | Fab + Fab + Fc, 1 + 1 CrossMAb | II | NCT04785820 and 2 studies |
| Xmab20717 | PD-1 × CTLA-4 | Xencor | scFv + Fab + Fc, 1 + 1 Xtend XmAb | I | NCT03517488 |
| Xmab22841 | CTLA-4 × LAG-3 | Xencor | scFv + Fab + Fc, 1 + 1 Xtend XmAb | I | NCT03849469 |
| Xmab23104 | PD-1 × ICOS | Xencor | scFv + Fab + Fc, 1 + 1 Xtend XmAb | I | NCT03752398 |
| Name | Targets | Developer | Format | Highest Phase | Clinical Trials |
|---|---|---|---|---|---|
| Dilpacimab, ABT-165 | DLL4 × VEGF | AbbVie | (Fv)2 + IgG, 2 + 2 DVD-Ig | II | NCT01946074 NCT03368859 |
| ABL001, NOV1501, TR009 | DLL4 × VEGF | ABL Bio | IgG + (scFv)2, 2 + 2 | I/II | NCT03292783 NCT04492033 |
| BI836880 | VEGF × Ang-2 | Boehringer Ingelheim | sdAb + sdAb + albumin, 1 + 1 | II | NCT03861234 and 5 studies |
| BI905677 | LRP5/6 | Boehringer Ingelheim | sdAb + sdAb + albumin, 1 + 1 | I | NCT03604445 |
| AK112 | VEGF × PD-1 | Akesobio | IgG + (scFv)2, 2 + 2 | I/II | NCT04736823 and 2 studies |
| KN026 | HER2 × HER2 (Biparatopic) | Jiangsu Alphamab Pharmaceuticals | Fab + Fab + Fc, 1 + 1 | II | NCT04521179 and 6 studies |
| MBS301 | HER2 × HER2 (Biparatopic) | Beijing Mabworks Biotech | Fab + Fab + Fc, 1 + 1 Fab-arm exchange | I | NCT03842085 |
| LY3164530 | MET × EGFR | Eli Lilly and Co | Fab + Fab + Fc, 1 + 1 orthoFab-IgG | I | NCT02221882 |
| EMB-01 | cMET × EGFR | Epimab Biotherapeutics | (Fab)2 + IgG, 2 + 2 FIT-Ig | I/II | NCT03797391 |
| Duligotuzumab, MEHD7945A, RO5541078, RG7597 | EGFR × HER3 | Genentech | Fab + Fab + Fc, 1 + 1 | II | NCT01986166 and 4 studies |
| Amivantamab, JNJ-61186372 | cMET × EGFR | Genmab, Janssen | Fab + Fab + Fc, 1 + 1 DuoBody | III | NCT04077463 and 5 studies |
| HX009 | PD-1 × CD47 | HanxBio | IgG + (ligand)2, 2 + 2 | I | NCT04097769 |
| IMM0306 | CD20 × CD47 | ImmuneOnco | IgG + (ligand)2, 2 + 2 | I | CTR20192612 |
| IBI322 | PD-L1 × CD47 | Innovent | sdAb + Fab + Fc, 1 + 1 | I | NCT04795128 and 2 studies |
| MM-111 | HER2 × HER3 | Merrimack Pharmaceuticals | (scFv)2 + albumin, 1 + 1 | II | NCT01097460 and 3 studies |
| MM-141 | IGF-1R × HER3 | Merrimack Pharmaceuticals | IgG + (scFv)2, 2 + 2 | II | NCT02538627 and 2 studies |
| Zenocutuzumab, MCLA-128,PB4188 | HER2 × HER3 | Merus | Fab + Fab + Fc, 1 + 1 Biclonics | II | NCT02912949 and 2 studies |
| MP0250 | HGF × VEGF × HSA | Molecular Partners AG | (DARPin)4, 1 + 1 + 2 | I/II | NCT03136653 and 2 studies |
| MP0274 | HER2 × HER2 (Biparatopic) | Molecular Partners AG | (DARPin)4, 1 + 1 + 2 | I | NCT03084926 |
| TG-1801, NI-1701 | CD19 × CD47 | NovImmune, TG Therapeutics | Fab + Fab + Fc, 1 + 1 κλ body | I | NCT03804996 NCT04806035 |
| Navicixizumab, OMP-305B83 | DLL4 × VEGF | OncoMed | Fab + Fab + Fc, 1 + 1 common LC | I | NCT03035253 and 2 studies |
| REGN5093 | MET × MET (Biparatopic) | Regeneron | Fab + Fab + Fc, 1 + 1 | I/II | NCT04077099 |
| Vanucizumab, RG7221, RO5520985 | Ang-2 × VEGF-A | Roche | Fab + Fab + Fc, 1 + 1 CrossMAb | II | NCT02665416 and 4 studies |
| Zanidatamab, ZW25 | HER2 × HER2 (Biparatopic) | Zymeworks | Fab + scFv + Fc, 1 + 1 | II | NCT04224272 and 6 studies |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, G.; Won, J.; Lee, Y.; Moon, D.; Park, Y.; Lee, S.H.; Lee, S.-W. Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies. Vaccines 2021, 9, 724. https://doi.org/10.3390/vaccines9070724
You G, Won J, Lee Y, Moon D, Park Y, Lee SH, Lee S-W. Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies. Vaccines. 2021; 9(7):724. https://doi.org/10.3390/vaccines9070724
Chicago/Turabian StyleYou, Gihoon, Jonghwa Won, Yangsoon Lee, Dain Moon, Yunji Park, Sang Hoon Lee, and Seung-Woo Lee. 2021. "Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies" Vaccines 9, no. 7: 724. https://doi.org/10.3390/vaccines9070724
APA StyleYou, G., Won, J., Lee, Y., Moon, D., Park, Y., Lee, S. H., & Lee, S.-W. (2021). Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies. Vaccines, 9(7), 724. https://doi.org/10.3390/vaccines9070724