
Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions

 , , ,
, , ,  , , ,
, , , 
Abstract
1. Introduction
2. Cancer Vaccine Antigens
2.1. Tumour Associated Antigens
2.1.1. Overexpressed Antigens
2.1.2. Normal Differentiation Antigens
PAP, PSA and the Sipuleucel-T (Provenge) Vaccine
2.1.3. Cancer-Germline/Cancer Testis Antigens
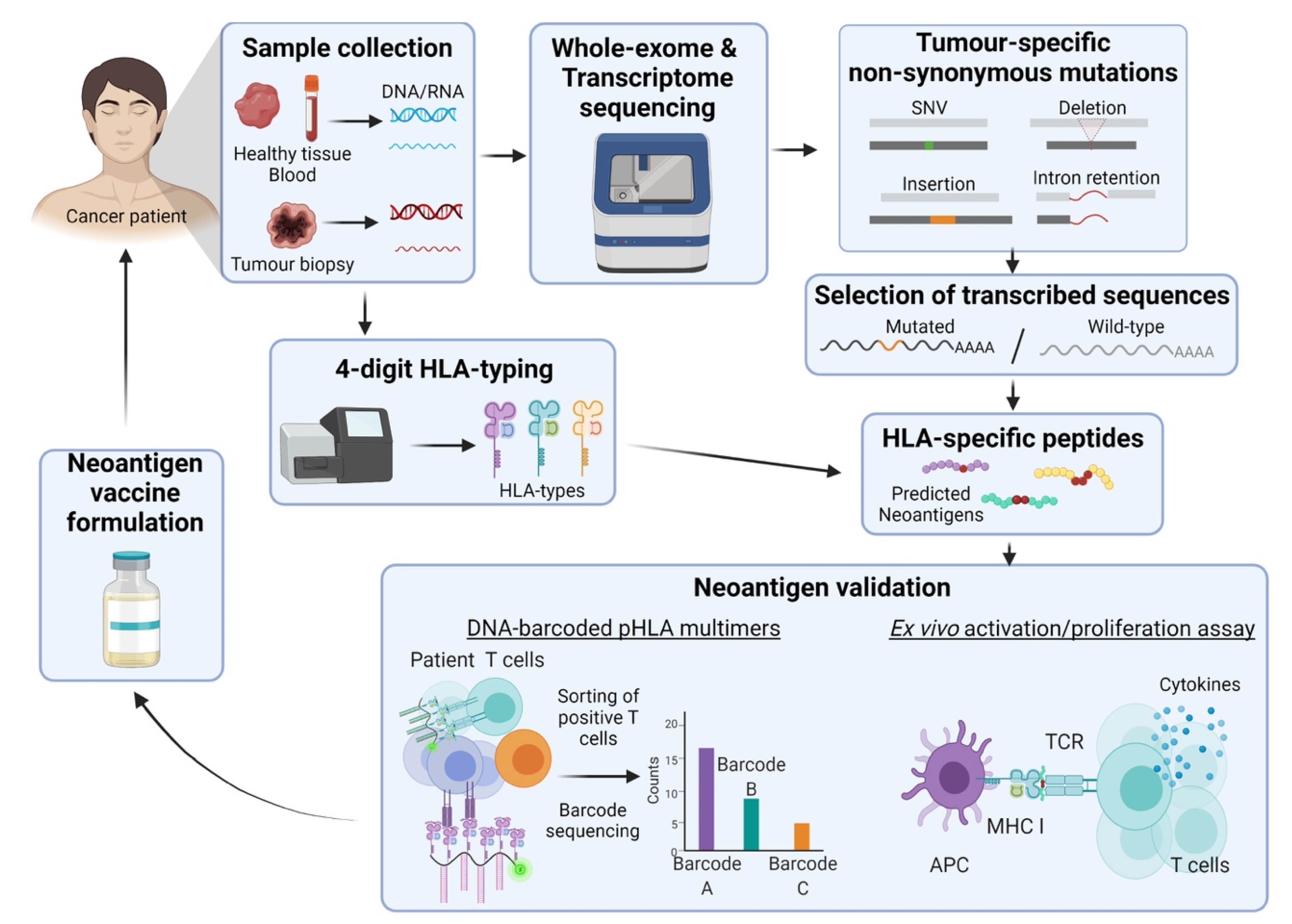
2.2. Neoantigens
2.2.1. Tumour-Specific Antigens
2.2.2. The Framework behind the Development of Neoantigen-Based Cancer Vaccines
2.2.3. Clinical Progress on Neoantigens
2.3. Oncogenic Viral Antigens
2.3.1. HPV
2.3.2. EBV
2.3.3. Hepatitis B and Hepatitis C
3. Vaccine Delivery Systems
3.1. Peptide Vaccines
3.2. Nucleic Acid Vaccines (DNA & mRNA)
3.2.1. Naked DNA Vaccines
3.2.2. Messenger RNA Vaccines, Naked mRNA and DC-Delivery
3.3. Particle Vaccines (LNP and VLP)
3.3.1. Lipid Nanoparticles in Cancer Vaccines Programs
Contemporary Uses of LNPs in Cancer Therapy
3.3.2. Virus Like Particles
3.3.3. Approved Virus Like Particle Therapies for Cancer
Human Papilloma Virus
Hepatocellular Carcinoma
3.4. Cellular Vaccines
3.4.1. Dendritic Cells
New Strategies to Load Antigens in APCs
3.4.2. Whole Cell Vaccines
3.4.3. Bacteria
3.5. Delivery with Viral Vectors
3.6. Adjuvants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Riedel, S. Edward Jenner and the history of smallpox and vaccination. Proc. Bayl Univ Med. Cent. 2005, 18, 21–25. [Google Scholar] [CrossRef]
- FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vaccines-licensed-use-united-states (accessed on 10 February 2021).
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, T.; Kierstead, L.S.; Ranieri, E.; Gesualdo, L.; Schena, F.P.; Finke, J.H.; Bukowski, R.M.; Mueller-Berghaus, J.; Kirkwood, J.M.; Kwok, W.W.; et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB10401(+) patients with renal cell carcinoma or melanoma. J. Exp. Med. 2002, 196, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Gussoni, E. Building immune tolerance through DNA vaccination. Proc. Natl Acad. Sci. USA 2018, 115, 9652–9654. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef]
- Samy, K.P.; Brennan, T.V. Dendritic Cell Therapy in Transplantation, Phenotype Governs Destination and Function. Transplantation 2018, 102, 1593–1594. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.-G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm. Ther. 2011, 36, 197–202. [Google Scholar]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.-E.; Bosquée, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef]
- Miles, D.; Roché, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef]
- Petrulio, C.A.; Kaufman, H.L. Development of the PANVAC[trademark]-VF vaccine for pancreatic cancer. Expert Rev. Vaccines 2006, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, T.; Kaufman, H.L.; Marshall, J.L.; Safran, H. Extended survival in second-line pancreatic cancer after therapeutic vaccination. J. Clin. Oncol. 2005, 23, 2576. [Google Scholar] [CrossRef]
- Therion Biologics Corporation Reports Results of Phase 3 PANVAC-VF Trial And Announces Plans For Company Sale. Available online: https://www.biospace.com/article/releases/therion-biologics-corporation-reports-results-of-phase-3-panvac-vf-trial-and-announces-plans-for-company-sale-/ (accessed on 18 March 2021).
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Schneble, E.; van Echo, D.; Ponniah, S.; Peoples, G.E. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann. Oncol. 2014, 25, 1735–1742. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Lu, B.; Melisko, M.; Price Hiller, J.; Bondarenko, I.; Brunt, A.M.; Sergii, G.; Petrakova, K.; Peoples, G.E. Efficacy and Safety Analysis of Nelipepimut-S Vaccine to Prevent Breast Cancer Recurrence: A Randomized, Multicenter, Phase III Clinical Trial. Clin. Cancer Res. 2019, 25, 4248–4254. [Google Scholar] [CrossRef]
- Leffers, N.; Lambeck, A.J.A.; Gooden, M.J.M.; Hoogeboom, B.-N.; Wolf, R.; Hamming, I.E.; Hepkema, B.G.; Willemse, P.H.B.; Molmans, B.H.W.; Hollema, H.; et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int. J. Cancer 2009, 125, 2104–2113. [Google Scholar] [CrossRef]
- Vermeij, R.; Leffers, N.; Hoogeboom, B.-N.; Hamming, I.L.E.; Wolf, R.; Reyners, A.K.L.; Molmans, B.H.W.; Hollema, H.; Bart, J.; Drijfhout, J.W.; et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int. J. Cancer 2012, 131, E670–E680. [Google Scholar] [CrossRef]
- Speetjens, F.M.; Kuppen, P.J.; Welters, M.J.; Essahsah, F.; Voet van den Brink, A.M.; Lantrua, M.G.; Valentijn, A.R.; Oostendorp, J.; Fathers, L.M.; Nijman, H.W.; et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin. Cancer Res. 2009, 15, 1086–1095. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Santegoets, S.J.; Reyners, A.K.; Goedemans, R.; Nijman, H.W.; van Poelgeest, M.I.; van Erkel, A.R.; Smit, V.T.; Daemen, T.A.; van der Hoeven, J.J.; et al. A phase 1/2 study combining gemcitabine, Pegintron and p53 SLP vaccine in patients with platinum-resistant ovarian cancer. Oncotarget 2015, 6, 32228–32243. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, S.H.; Menon, A.G.; Redeker, A.; Bonnet, M.C.; Drijfhout, J.W.; Tollenaar, R.A.; van de Velde, C.J.; Moingeon, P.; Kuppen, P.J.; Offringa, R.; et al. Induction of p53-specific immune responses in colorectal cancer patients receiving a recombinant ALVAC-p53 candidate vaccine. Clin. Cancer Res. 2002, 8, 1019–1027. [Google Scholar] [PubMed]
- Hardwick, N.R.; Frankel, P.; Ruel, C.; Kilpatrick, J.; Tsai, W.; Kos, F.; Kaltcheva, T.; Leong, L.; Morgan, R.; Chung, V.; et al. p53-Reactive T Cells Are Associated with Clinical Benefit in Patients with Platinum-Resistant Epithelial Ovarian Cancer After Treatment with a p53 Vaccine and Gemcitabine Chemotherapy. Clin. Cancer Res. 2018, 24, 1315. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Lilleby, W.; Gaudernack, G.; Brunsvig, P.F.; Vlatkovic, L.; Schulz, M.; Mills, K.; Hole, K.H.; Inderberg, E.M. Phase I/IIa clinical trial of a novel hTERT peptide vaccine in men with metastatic hormone-naive prostate cancer. Cancer Immunol. Immunother. 2017, 66, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Ciuleanu, T.; Domine, M.; Szczesna, A.; Bover, I.; Cobo, M.; Kentepozidis, N.; Zarogoulidis, K.; Kalofonos, C.; Kazarnowisz, A.; et al. Clinical activity of a htert (vx-001) cancer vaccine as post-chemotherapy maintenance immunotherapy in patients with stage IV non-small cell lung cancer: Final results of a randomised phase 2 clinical trial. Br. J. Cancer 2020, 122, 1461–1466. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. CII 2016, 65, 1339–1352. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Belal, A.; Qiu, J.; Mogensen, K.; Schilero, C.; Hutson, A.; et al. SurVaxM with standard therapy in newly diagnosed glioblastoma: Phase II trial update. J. Clin. Oncol. 2019, 37, 2016. [Google Scholar] [CrossRef]
- Lennerz, V.; Gross, S.; Gallerani, E.; Sessa, C.; Mach, N.; Boehm, S.; Hess, D.; von Boehmer, L.; Knuth, A.; Ochsenbein, A.F.; et al. Immunologic response to the survivin-derived multi-epitope vaccine EMD640744 in patients with advanced solid tumors. Cancer Immunol. Immunother. 2014, 63, 381–394. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Dreno, B.; Thompson, J.F.; Smithers, B.M.; Santinami, M.; Jouary, T.; Gutzmer, R.; Levchenko, E.; Rutkowski, P.; Grob, J.-J.; Korovin, S.; et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 916–929. [Google Scholar] [CrossRef]
- Ishihara, M.; Tono, Y.; Miyahara, Y.; Muraoka, D.; Harada, N.; Kageyama, S.; Sasaki, T.; Hori, Y.; Soga, N.; Uchida, K.; et al. First-in-human phase I clinical trial of the NY-ESO-1 protein cancer vaccine with NOD2 and TLR9 stimulants in patients with NY-ESO-1-expressing refractory solid tumors. Cancer Immunol. Immunother. 2020, 69, 663–675. [Google Scholar] [CrossRef]
- Cebon, J.S.; Gore, M.; Thompson, J.F.; Davis, I.D.; McArthur, G.A.; Walpole, E.; Smithers, M.; Cerundolo, V.; Dunbar, P.R.; MacGregor, D.; et al. Results of a randomized, double-blind phase II clinical trial of NY-ESO-1 vaccine with ISCOMATRIX adjuvant versus ISCOMATRIX alone in participants with high-risk resected melanoma. J. Immunother. Cancer 2020, 8, e000410. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Bauman, J.; Burris, H.; Clarke, J.; Patel, M.; Cho, D.; Gutierrez, M.; Julian, R.; Scott, A.; Cohen, P.; Frederick, J.; et al. Safety, Tolerability, and Immunogenicity of Mrna-4157 in Combination with Pembrolizumab in Subjects with Unresectable Solid Tumors (Keynote-603): An Update. J. Immunother. Cancer 2020, 8, A477. [Google Scholar] [CrossRef]
- Bagarazzi, M.L.; Yan, J.; Morrow, M.P.; Shen, X.; Parker, R.L.; Lee, J.C.; Giffear, M.; Pankhong, P.; Khan, A.S.; Broderick, K.E.; et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci. Transl. Med. 2012, 4, 155ra138. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef]
- Rosales, R.; López-Contreras, M.; Rosales, C.; Magallanes-Molina, J.-R.; Gonzalez-Vergara, R.; Arroyo-Cazarez, J.M.; Ricardez-Arenas, A.; Del Follo-Valencia, A.; Padilla-Arriaga, S.; Guerrero, M.V.; et al. Regression of human papillomavirus intraepithelial lesions is induced by MVA E2 therapeutic vaccine. Hum. Gene. 2014, 25, 1035–1049. [Google Scholar] [CrossRef]
- Welters, M.J.P.; Kenter, G.G.; Piersma, S.J.; Vloon, A.P.G.; Löwik, M.J.G.; Berends-van der Meer, D.M.A.; Drijfhout, J.W.; Valentijn, A.R.P.M.; Wafelman, A.R.; Oostendorp, J.; et al. Induction of Tumor-Specific CD4+ and CD8+ T-Cell Immunity in Cervical Cancer Patients by a Human Papillomavirus Type 16 E6 and E7 Long Peptides Vaccine. Clin. Cancer Res. 2008, 14, 178. [Google Scholar] [CrossRef]
- van Poelgeest, M.I.E.; Welters, M.J.P.; van Esch, E.M.G.; Stynenbosch, L.F.M.; Kerpershoek, G.; van Persijn van Meerten, E.L.; van den Hende, M.; Löwik, M.J.G.; Berends-van der Meer, D.M.A.; Fathers, L.M.; et al. HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. J. Transl. Med. 2013, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Cohen, R.B.; Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Knoblock, D.M.; Bauml, J.M.; Weinstein, G.S.; Lin, A.; Boyer, J.; et al. Immunotherapy Targeting HPV16/18 Generates Potent Immune Responses in HPV-Associated Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 110. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Woo, W.P.; Finlayson, N.; Liu, H.Y.; McGrath, M.; Ladwa, R.; Brauer, M.; Xu, Y.; Hanson, S.; Panizza, B.; et al. A phase 1, single centre, open label, escalating dose study to assess the safety, tolerability and immunogenicity of a therapeutic human papillomavirus (HPV) DNA vaccine (AMV002) for HPV-associated head and neck cancer (HNC). Cancer Immunol. Immunother. 2021, 70, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.Y.; Chan, S.L.; Ho, R.; Wong, W.-L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding Epstein-Barr Virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef] [PubMed]
- Chia, W.K.; Wang, W.W.; Teo, M.; Tai, W.M.; Lim, W.T.; Tan, E.H.; Leong, S.S.; Sun, L.; Chen, J.J.; Gottschalk, S.; et al. A phase II study evaluating the safety and efficacy of an adenovirus-ΔLMP1-LMP2 transduced dendritic cell vaccine in patients with advanced metastatic nasopharyngeal carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Yutani, S.; Ueshima, K.; Abe, K.; Ishiguro, A.; Eguchi, J.; Matsueda, S.; Komatsu, N.; Shichijo, S.; Yamada, A.; Itoh, K.; et al. Phase II Study of Personalized Peptide Vaccination with Both a Hepatitis C Virus-Derived Peptide and Peptides from Tumor-Associated Antigens for the Treatment of HCV-Positive Advanced Hepatocellular Carcinoma Patients. J. Immunol. Res. 2015, 2015, 473909. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Gulley, J.L.; Pico-Navarro, C. Revised Overall Survival Analysis of a Phase II, Randomized, Double-Blind, Controlled Study of PROSTVAC in Men With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 124. [Google Scholar] [CrossRef]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Cockle, P.; Joe, B.; Risini, W.; White, P.; Jooss, K. Abstract LB-093: Vaccine based immunotherapy regimen (VBIR) for the treatment of prostate cancer. Cancer Res. 2016, 76, LB-093. [Google Scholar] [CrossRef]
- Higano, C. A phase III trial of GVAX immunotherapy for prostate cancer versus docetaxel plus prednisone in asymptomatic, castration-resistant prostate cancer (CRPC). In Proceedings of the 2009 Genitourinary Cancer Symposium, American Society of Clinical Oncology (ASCO), Orlando, FL, USA, 26–28 February 2009. [Google Scholar]
- Arlen, P.M.; Mohebtash, M.; Madan, R.A.; Gulley, J.L. Promising novel immunotherapies and combinations for prostate cancer. Future Oncol. 2009, 5, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Small, E. A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel versus docetaxel plus prednisone in symptomatic, castration-resistant prostate cancer (CRPC). In Proceedings of the 2009 Genitourinary Cancer Symposium, American Society of Clinical Oncology (ASCO), Orlando, FL, USA, 26–28 February 2009. [Google Scholar]
- Sosman, J.A.; Unger, J.M.; Liu, P.Y.; Flaherty, L.E.; Park, M.S.; Kempf, R.A.; Thompson, J.A.; Terasaki, P.I.; Sondak, V.K. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: Impact of HLA class I antigen expression on outcome. J. Clin. Oncol. 2002, 20, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.L.; Mozzillo, N.; Thompson, J.F.; Kelley, M.C.; Faries, M.; Wagner, J.; Schneebaum, S.; Schuchter, L.; Gammon, G.; Elashoff, R. An international, randomized, phase III trial of bacillus Calmette-Guerin (BCG) plus allogeneic melanoma vaccine (MCV) or placebo after complete resection of melanoma metastatic to regional or distant sites. J. Clin. Oncol. 2007, 25, 8508. [Google Scholar] [CrossRef]
- Morton, D.L.; Hsueh, E.C.; Essner, R.; Foshag, L.J.; O’Day, S.J.; Bilchik, A.; Gupta, R.K.; Hoon, D.S.B.; Ravindranath, M.; Nizze, J.A.; et al. Prolonged survival of patients receiving active immunotherapy with Canvaxin therapeutic polyvalent vaccine after complete resection of melanoma metastatic to regional lymph nodes. Ann. Surg. 2002, 236, 438–449. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Claessen, A.M.E.; van Tinteren, H.; Gall, H.E.; Ezinga, R.; Meijer, S.; Scheper, R.J.; Meijer, C.J.L.M.; Bloemena, E.; Ransom, J.H.; et al. Active specific immunotherapy for stage II and stage III human colon cancer: A randomised trial. Lancet 1999, 353, 345–350. [Google Scholar] [CrossRef]
- Berd, D.; Maguire, H.C., Jr.; McCue, P.; Mastrangelo, M.J. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: Clinical and immunologic results in 64 patients. J. Clin. Oncol. 1990, 8, 1858–1867. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J., 3rd; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Springer, G.F. T and Tn, general carcinoma autoantigens. Science 1984, 224, 1198. [Google Scholar] [CrossRef]
- Gupta, R.; Leon, F.; Rauth, S.; Batra, S.K.; Ponnusamy, M.P. A Systematic Review on the Implications of O-linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis. Cells 2020, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Papadimitriou, J.; Burchell, J.M.; Graham, R.; Beatson, R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018, 46, 659–668. [Google Scholar] [CrossRef]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for HER2+ breast cancer: A review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef]
- Pallerla, S.; Abdul, A.U.R.M.; Comeau, J.; Jois, S. Cancer Vaccines, Treatment of the Future: With Emphasis on HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 779. [Google Scholar] [CrossRef]
- Dillon, P.M.; Brenin, C.M.; Slingluff, C.L., Jr. Evaluating Nelipepimut-S in the Treatment of Breast Cancer: A Short Report on the Emerging Data. Breast Cancer 2020, 12, 69–75. [Google Scholar] [CrossRef]
- Yan, W.-L.; Shen, K.-Y.; Tien, C.-Y.; Chen, Y.-A.; Liu, S.-J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Surget, S.; Khoury, M.P.; Bourdon, J.-C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Oncol. Targets Ther. 2013, 7, 57–68. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Vermeij, R.; Leffers, N.; van der Burg, S.H.; Melief, C.J.; Daemen, T.; Nijman, H.W. Immunological and Clinical Effects of Vaccines Targeting p53-Overexpressing Malignancies. J. Biomed. Biotechnol. 2011, 2011, 702146. [Google Scholar] [CrossRef]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, L.; Matmati, S.; Coulon, S. Solving the Telomere Replication Problem. Genes 2017, 8, 55. [Google Scholar] [CrossRef]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef]
- Cong, Y.-S.; Wen, J.; Bacchetti, S. The Human Telomerase Catalytic Subunit hTERT: Organization of the Gene and Characterization of the Promoter. Hum. Mol. Genet. 1999, 8, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int J. Mol. Sci 2019, 20, 1823. [Google Scholar] [CrossRef]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef]
- Stauber, R.H.; Mann, W.; Knauer, S.K. Nuclear and Cytoplasmic Survivin: Molecular Mechanism, Prognostic, and Therapeutic Potential. Cancer Res. 2007, 67, 5999. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef]
- Fernández, J.G.; Rodríguez, D.A.; Valenzuela, M.; Calderon, C.; Urzúa, U.; Munroe, D.; Rosas, C.; Lemus, D.; Díaz, N.; Wright, M.C.; et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced β-catenin/Tcf-Lef dependent transcription. Mol. Cancer 2014, 13, 209. [Google Scholar] [CrossRef]
- Li, F.; Aljahdali, I.; Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study? J. Exp. Clin. Cancer Res. 2019, 38, 368. [Google Scholar] [CrossRef]
- Bright, R.K.; Bright, J.D.; Byrne, J.A. Overexpressed oncogenic tumor-self antigens. Hum. Vaccin. Immunother. 2014, 10, 3297–3305. [Google Scholar] [CrossRef]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment. Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid – from bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef]
- Bakker, A.B.; Schreurs, M.W.; de Boer, A.J.; Kawakami, Y.; Rosenberg, S.A.; Adema, G.J.; Figdor, C.G. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J. Exp. Med. 1994, 179, 1005–1009. [Google Scholar] [CrossRef]
- Muniyan, S.; Chaturvedi, N.K.; Dwyer, J.G.; Lagrange, C.A.; Chaney, W.G.; Lin, M.-F. Human prostatic acid phosphatase: Structure, function and regulation. Int. J. Mol. Sci. 2013, 14, 10438–10464. [Google Scholar] [CrossRef]
- Luchter-Wasyl, E.; Ostrowski, W. Subunit structure of human prostatic acid phosphatase. Biochim. Biophys. Acta (BBA) Protein Struct. 1974, 365, 349–359. [Google Scholar] [CrossRef]
- Cunha, A.C.; Weigle, B.; Kiessling, A.; Bachmann, M.; Rieber, E.P. Tissue-specificity of prostate specific antigens: Comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett. 2006, 236, 229–238. [Google Scholar] [CrossRef]
- Goldfarb, D.A.; Stein, B.S.; Shamszadeh, M.; Petersen, R.O. Age-related changes in tissue levels of prostatic acid phosphatase and prostate specific antigen. J. Urol. 1986, 136, 1266–1269. [Google Scholar] [CrossRef]
- Rönnberg, L.; Vihko, P.; Sajanti, E.; Vihko, R. Clomiphene citrate administration to normogonadotropic subfertile men: Blood hormone changes and activation of acid phosphatase in seminal fluid. Int. J. 1981, 4, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.I.; Aijaz, A.; Ahmad, F. Structural and functional analysis of human prostatic acid phosphatase. Expert Rev. Anticancer Ther. 2010, 10, 1055–1068. [Google Scholar] [CrossRef]
- Abrahamsson, P.A.; Lilja, H.; Falkmer, S.; Wadströ, L.B. Immunohistochemical distribution of the three predominant secretory proteins in the parenchyma of hyperplastic and neoplastic prostate glands. Prostate 1988, 12, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.Y.; Byun, J. Emerging roles of human prostatic Acid phosphatase. Biomol 2013, 21, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Peshwa, M.V.; Shi, J.D.; Ruegg, C.; Laus, R.; van Schooten, W.C. Induction of prostate tumor-specific CD8+ cytotoxic T-lymphocytes in vitro using antigen-presenting cells pulsed with prostatic acid phosphatase peptide. Prostate 1998, 36, 129–138. [Google Scholar] [CrossRef]
- Laus, R.; Yang, D.M.; Ruegg, C.L.; Shapero, M.H.; Slagle, P.H.; Small, E.; Burch, P.; Valone, F.H. Dendritic cell immunotherapy of prostate cancer: Preclinical models and early clinical experience. Cancer Res. Ther. Control. 2001, 11, 1–10. [Google Scholar]
- Abd Hamid, M.; Peng, Y.; Dong, T. Human cancer germline antigen-specific cytotoxic T cell—what can we learn from patient. Cell. Mol. Immunol. 2020, 17, 684–692. [Google Scholar] [CrossRef]
- Fijak, M.; Meinhardt, A. The testis in immune privilege. Immunol. Rev. 2006, 213, 66–81. [Google Scholar] [CrossRef]
- Doyle, J.M.; Gao, J.; Wang, J.; Yang, M.; Potts, P.R. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 2010, 39, 963–974. [Google Scholar] [CrossRef]
- Simpson, A.J.G.; Caballero, O.L.; Jungbluth, A.; Chen, Y.-T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Weon, J.L.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef]
- Hao, Y.-H.; Doyle, J.M.; Ramanathan, S.; Gomez, T.S.; Jia, D.; Xu, M.; Chen, Z.J.; Billadeau, D.D.; Rosen, M.K.; Potts, P.R. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell 2013, 152, 1051–1064. [Google Scholar] [CrossRef]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.-H.; White, M.A.; Potts, P.R. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Bastin, J.; Gajewski, T.; Coulie, P.G.; Boël, P.; De Smet, C.; Traversari, C.; Townsend, A.; Boon, T. A peptide encoded by human gene MAGE-3 and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur. J. Immunol. 1994, 24, 3038–3043. [Google Scholar] [CrossRef]
- Gaugler, B.; Van den Eynde, B.; van der Bruggen, P.; Romero, P.; Gaforio, J.J.; De Plaen, E.; Lethé, B.; Brasseur, F.; Boon, T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 1994, 179, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Hemminger, J.A.; Ewart Toland, A.; Scharschmidt, T.J.; Mayerson, J.L.; Kraybill, W.G.; Guttridge, D.C.; Iwenofu, O.H. The cancer-testis antigen NY-ESO-1 is highly expressed in myxoid and round cell subset of liposarcomas. Mod. Pathol. 2013, 26, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Kock, K.; Nielsen, O.; Ditzel, H.J. MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development. Hum. Reprod. 2007, 22, 953–960. [Google Scholar] [CrossRef]
- Nicholaou, T.; Ebert, L.; Davis, I.D.; Robson, N.; Klein, O.; Maraskovsky, E.; Chen, W.; Cebon, J. Directions in the immune targeting of cancer: Lessons learned from the cancer-testis Ag NY-ESO-1. Immunol. Cell Biol. 2006, 84, 303–317. [Google Scholar] [CrossRef]
- Kisseleva-Romanova, E.; Lopreiato, R.; Baudin-Baillieu, A.; Rousselle, J.-C.; Ilan, L.; Hofmann, K.; Namane, A.; Mann, C.; Libri, D. Yeast homolog of a cancer-testis antigen defines a new transcription complex. Embo J. 2006, 25, 3576–3585. [Google Scholar] [CrossRef]
- Cho, H.J.; Caballero, O.L.; Gnjatic, S.; Andrade, V.C.; Colleoni, G.W.; Vettore, A.L.; Outtz, H.H.; Fortunato, S.; Altorki, N.; Ferrera, C.A.; et al. Physical interaction of two cancer-testis antigens, MAGE-C1 (CT7) and NY-ESO-1 (CT6). Cancer Immun. Arch. 2006, 6, 12. [Google Scholar]
- Jäger, E.; Chen, Y.-T.; Drijfhout, J.W.; Karbach, J.; Ringhoffer, M.; Jäger, D.; Arand, M.; Wada, H.; Noguchi, Y.; Stockert, E.; et al. Simultaneous Humoral and Cellular Immune Response against Cancer–Testis Antigen NY-ESO-1: Definition of Human Histocompatibility Leukocyte Antigen (HLA)-A2–binding Peptide Epitopes. J. Exp. Med. 1998, 187, 265–270. [Google Scholar] [CrossRef]
- Zarour, H.M.; Storkus, W.J.; Brusic, V.; Williams, E.; Kirkwood, J.M. NY-ESO-1 Encodes DRB1*0401-restricted Epitopes Recognized by Melanoma-reactive CD4+ T Cells. Cancer Res. 2000, 60, 4946. [Google Scholar]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Prehn, R.T.; Main, J.M. Immunity to Methylcholanthrene-Induced Sarcomas. JNCI J. Natl. Cancer Inst. 1957, 18, 769–778. [Google Scholar] [CrossRef] [PubMed]
- De Plaen, E.; Lurquin, C.; Van Pel, A.; Mariamé, B.; Szikora, J.P.; Wölfel, T.; Sibille, C.; Chomez, P.; Boon, T. Immunogenic (tum-) variants of mouse tumor P815: Cloning of the gene of tum- antigen P91A and identification of the tum- mutation. Proc. Natl. Acad. Sci. USA 1988, 85, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Karpanen, T.; Olweus, J. The Potential of Donor T-Cell Repertoires in Neoantigen-Targeted Cancer Immunotherapy. Front. Immunol. 2017, 8, 1718. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. Bmc. Bioinform. 2019, 20, 264. [Google Scholar] [CrossRef] [PubMed]
- Scheetz, L.; Park, K.S.; Li, Q.; Lowenstein, P.R.; Castro, M.G.; Schwendeman, A.; Moon, J.J. Engineering patient-specific cancer immunotherapies. Nat. Biomed. Eng. 2019, 3, 768–782. [Google Scholar] [CrossRef] [PubMed]
- The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [CrossRef]
- Bentzen, A.K.; Marquard, A.M.; Lyngaa, R.; Saini, S.K.; Ramskov, S.; Donia, M.; Such, L.; Furness, A.J.S.; McGranahan, N.; Rosenthal, R.; et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Tamhane, T.; Anjanappa, R.; Saikia, A.; Ramskov, S.; Donia, M.; Svane, I.M.; Jakobsen, S.N.; Garcia-Alai, M.; Zacharias, M.; et al. Empty peptide-receptive MHC class I molecules for efficient detection of antigen-specific T cells. Sci. Immunol. 2019, 4, eaau9039. [Google Scholar] [CrossRef] [PubMed]
- Heldenbrand, J.R.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Hudson, M.E.; Iyer, R.K.; Kalmbach, M.T.; Kendig, K.I.; Klee, E.W.; et al. Recommendations for performance optimizations when using GATK3.8 and GATK4. BMC Bioinform. 2019, 20, 557. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; Stehr, H.; Lee, L.J.; Liu, C.L.; Diehn, M.; Alizadeh, A.A. FACTERA: A practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics 2014, 30, 3390–3393. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Luksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803. [Google Scholar] [CrossRef]
- zur Hausen, H. Viruses in human cancers. Science 1991, 254, 1167. [Google Scholar] [CrossRef]
- Tashiro, H.; Brenner, M.K. Immunotherapy against cancer-related viruses. Cell Res. 2017, 27, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Burd, E.M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef]
- Serrano, B.; Brotons, M.; Bosch, F.X.; Bruni, L. Epidemiology and burden of HPV-related disease. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Weinberg, Robert A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Thompson, M.P.; Kurzrock, R. Epstein-Barr Virus and Cancer. Clin. Cancer Res. 2004, 10, 803. [Google Scholar] [CrossRef] [PubMed]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H., Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin. Microbiol. Rev. 2011, 24, 193–209. [Google Scholar] [CrossRef]
- Amon, W.; Farrell, P.J. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 2005, 15, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Zimber-Strobl, U.; Kempkes, B.; Marschall, G.; Zeidler, R.; Van Kooten, C.; Banchereau, J.; Bornkamm, G.W.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein (LMP1) is not sufficient to maintain proliferation of B cells but both it and activated CD40 can prolong their survival. Embo. J. 1996, 15, 7070–7078. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein–Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.B.; Manet, E.; Gruffat, H.; Busson, P.; Blondel, M.; Fahraeus, R. EBNA1: Oncogenic Activity, Immune Evasion and Biochemical Functions Provide Targets for Novel Therapeutic Strategies against Epstein-Barr Virus- Associated Cancers. Cancers 2018, 109. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Banerjee, A.; Ray, R.B.; Ray, R. Oncogenic potential of hepatitis C virus proteins. Viruses 2010, 2, 2108–2133. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of Viral Hepatitis and Hepatocellular Carcinoma. Gastroenterology 2012, 142, 1264–1273.e1261. [Google Scholar] [CrossRef]
- Pardoll, D.M. Cancer vaccines. Nat. Med. 1998, 4, 525–531. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Mittrucker, H.W.; Visekruna, A.; Huber, M. Heterogeneity in the differentiation and function of CD8+ T cells. Arch. Immunol. Exp. (Warsz) 2014, 62, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Kumai, T.; Kobayashi, H.; Harabuchi, Y.; Celis, E. Peptide vaccines in cancer-old concept revisited. Curr. Opin. Immunol. 2017, 45, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct Mol. Biol 2015, 22, 109–115. [Google Scholar] [CrossRef]
- Schwartz, D.A.; Quinn, T.J.; Thorne, P.S.; Sayeed, S.; Yi, A.K.; Krieg, A.M. CpG motifs in bacterial DNA cause inflammation in the lower respiratory tract. J. Clin. Investig. 1997, 100, 68–73. [Google Scholar] [CrossRef]
- Cai, Y.; Rodriguez, S.; Hebel, H. DNA vaccine manufacture: Scale and quality. Expert Rev. Vaccines 2009, 8, 1277–1291. [Google Scholar] [CrossRef]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.F. DNA vaccine for cancer immunotherapy. Hum. Vaccin Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef]
- Suschak, J.J.; Williams, J.A.; Schmaljohn, C.S. Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum. Vaccin Immunother. 2017, 13, 2837–2848. [Google Scholar] [CrossRef]
- Wolff, J.A.; Robert, W.M.; Phillip, W.; Wang, C.; Gyula, A.; Agnes, J.; Philip, L.F. Direct Gene Transfer into Mouse Muscle in Vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Bonehill, A.; Van Nuffel, A.M.; Corthals, J.; Tuyaerts, S.; Heirman, C.; Francois, V.; Colau, D.; van der Bruggen, P.; Neyns, B.; Thielemans, K. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef]
- Tacken, P.J.; de Vries, I.J.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, F.; Persano, S. Advances in Lipid Nanoparticles for mRNA-Based Cancer Immunotherapy. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Buschmann, M.D.; Carrasco, M.J.; Alishetty, S.; Paige, M.; Alameh, M.G.; Weissman, D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9, 65. [Google Scholar] [CrossRef]
- Scioli Montoto, S.; Muraca, G.; Ruiz, M.E. Solid Lipid Nanoparticles for Drug Delivery: Pharmacological and Biopharmaceutical Aspects. Front. Mol. Biosci. 2020, 7, 587997. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulieres, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.J.; Soria, A.; Machiels, J.P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Speiser, D.E.; Knuth, A.; Bachmann, M.F. Virus-like particles for vaccination against cancer. Wiley Interdiscip. Rev. Nanomed Nanobiotechnol. 2020, 12, e1579. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, J.C.; Perrine, M.; Pericle, F.; Cavallo, F. Virus-Like Particles as an Immunogenic Platform for Cancer Vaccines. Viruses 2020, 488. [Google Scholar] [CrossRef]
- Heffernan, M.E.; Garland, S.M.; Kane, M.A. Global reduction of cervical cancer with human papillomavirus vaccines: Insights from the hepatitis B virus vaccine experience. Sex. Health 2010, 7, 383–390. [Google Scholar] [CrossRef]
- Koutsky, L.A.; Ault, K.A.; Wheeler, C.M.; Brown, D.R.; Barr, E.; Alvarez, F.B.; Chiacchierini, L.M.; Jansen, K.U.; Proof of Principle Study, I. A controlled trial of a human papillomavirus type 16 vaccine. N. Engl. J. Med. 2002, 347, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Szmuness, W.; Stevens, C.E.; Harley, E.J.; Zang, E.A.; Oleszko, W.R.; William, D.C.; Sadovsky, R.; Morrison, J.M.; Kellner, A. Hepatitis B vaccine: Demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N. Engl. J. Med. 1980, 303, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; DeMars, L.R. HPV vaccines—A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; Du, J. Human Papillomavirus Vaccines: An Updated Review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef]
- Zhou, J.; Stenzel, D.J.; Sun, X.Y.; Frazer, I.H. Synthesis and assembly of infectious bovine papillomavirus particles in vitro. J. Gen. Virol. 1993, 74, 763–768. [Google Scholar] [CrossRef]
- Lee, G.H.; Lim, S.G. CpG-Adjuvanted Hepatitis B Vaccine (HEPLISAV-B(R)) Update. Expert. Rev. Vaccines 2021, 1–9. [Google Scholar] [CrossRef]
- Atsmon, J.; Machluf, N.; Yayon-Gur, V.; Sabbah, C.; Spaans, J.N.; Yassin-Rajkumar, B.; Anderson, D.E.; Popovic, V.; Diaz-Mitoma, F. Rapid and high seroprotection rates achieved with a tri-antigenic Hepatitis B vaccine in healthy young adults: Results from a Phase IV study. Vaccine 2021, 39, 1328–1332. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Sharei, A.; Cho, N.; Mao, S.; Jackson, E.; Poceviciute, R.; Adamo, A.; Zoldan, J.; Langer, R.; Jensen, K.F. Cell Squeezing as a Robust, Microfluidic Intracellular Delivery Platform. JOVE J. Vis. Exp. 2013. [Google Scholar] [CrossRef]
- Maloney, M.; Loughhead, S.; Ramakrishnan, A.; Smith, C.; Venkitaraman, A.; Yee, C.; Jacques, M.; Yarar, D.; Sharei, A.; Bernstein, H.; et al. Microfluidics Cell Squeezing Enables Human Pbmcs as Drivers of Antigen-Specific Cd8 T Responses across Broad Range of Antigens for Diverse Clinical Applications. J. Immunother. Cancer 2020, 8, A102. [Google Scholar] [CrossRef]
- Blagovic, K.; Ramakrishnan, A.; Sharei, A.; Bernstein, H.; Seidl, K.; Yarar, D. Activating Antigen Carriers Generated with Microfluidics Cell Squeezing Drive Effective Anti-Tumor Responses. J. Immunother. Cancer 2020, 8, A98–A99. [Google Scholar] [CrossRef]
- Booty, M.; Stockmann, A.; Pryor, O.; Myint, M.; Trumpfheller, C.; Nicolini, V.; Klein, C.; Codarri, L.; Umana, P.; Sharei, A.; et al. Pbmc-Based Cancer Vaccines Generated with Microfluidics Squeezing Demonstrate Synergistic and Durable Tumor Reduction in Combination with Pd1 Checkpoint and Fap Targeted Il-2 Variants. J. Immunother. Cancer 2020, 8, A86. [Google Scholar] [CrossRef]
- Keenan, B.P.; Jaffee, E.M. Whole cell vaccines--past progress and future strategies. Semin. Oncol. 2012, 39, 276–286. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef]
- Harris, J.E.; Ryan, L.; Hoover, H.C., Jr.; Stuart, R.K.; Oken, M.M.; Benson, A.B., 3rd; Mansour, E.; Haller, D.G.; Manola, J.; Hanna, M.G., Jr. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group Study E5283. J. Clin. Oncol. 2000, 18, 148–157. [Google Scholar] [CrossRef]
- Hanna, M.G.; Hoover, H.C.; Vermorken, J.B.; Harris, J.E.; Pinedo, H.M. Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: First randomized phase III trials show promise. Vaccine 2001, 19, 2576–2582. [Google Scholar] [CrossRef]
- Old, L.J.; Clarke, D.A.; Benacerraf, B. Effect of Bacillus Calmette-Guerin infection on transplanted tumours in the mouse. Nature 1959, 184 (Suppl. S5), 291–292. [Google Scholar] [CrossRef]
- Morales, A.; Eidinger, D.; Bruce, A.W. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J. Urol 1976, 116, 180–183. [Google Scholar] [CrossRef]
- Wood, L.M.; Paterson, Y. Attenuated Listeria monocytogenes: A powerful and versatile vector for the future of tumor immunotherapy. Front. Cell Infect. Microbiol. 2014, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Compans, R.W. Host responses from innate to adaptive immunity after vaccination: Molecular and cellular events. Mol. Cells 2009, 27, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Arunachalam, P.S.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.W.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef]
- Nunberg, J.H.; Doyle, M.V.; York, S.M.; York, C.J. Interleukin 2 acts as an adjuvant to increase the potency of inactivated rabies virus vaccine. Proc. Natl. Acad. Sci. USA 1989, 86, 4240–4243. [Google Scholar] [CrossRef]
- Tovey, M.G.; Lallemand, C. Adjuvant activity of cytokines. Methods Mol. Biol. 2010, 626, 287–309. [Google Scholar] [CrossRef]
- Luckey, A.M.; Anderson, T.; Silverman, M.H.; Webster, G. Safety, tolerability and pharmacodynamics of a novel immunomodulator, MIS416, in patients with chronic progressive multiple sclerosis. Mult Scler. J. Exp. Transl. Clin. 2015, 1, 2055217315583385. [Google Scholar] [CrossRef]
- Martins, K.A.; Bavari, S.; Salazar, A.M. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev. Vaccines 2015, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, E.; Liu, H.; Huckriede, A.; Hak, E. Safety and tolerability evaluation of the use of Montanide ISA51 as vaccine adjuvant: A systematic review. Hum. Vaccin Immunother. 2016, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Nagatomo, D.; Taniai, M.; Ariyasu, H.; Taniguchi, M.; Aga, M.; Ariyasu, T.; Ohta, T.; Fukuda, S. Cholesteryl Pullulan Encapsulated TNF-alpha Nanoparticles Are an Effective Mucosal Vaccine Adjuvant against Influenza Virus. Biomed. Res. Int. 2015, 2015, 471468. [Google Scholar] [CrossRef]
- Baz Morelli, A.; Becher, D.; Koernig, S.; Silva, A.; Drane, D.; Maraskovsky, E. ISCOMATRIX: A novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. J. Med. Microbiol. 2012, 61, 935–943. [Google Scholar] [CrossRef]
- Cooper, C.L.; Davis, H.L.; Morris, M.L.; Efler, S.M.; Adhami, M.A.; Krieg, A.M.; Cameron, D.W.; Heathcote, J. CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: A double-blind phase I/II study. J. Clin. Immunol. 2004, 24, 693–701. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Bazin-Lee, H.; Evans, J.T.; Casella, C.R.; Mitchell, T.C. MPL Adjuvant Contains Competitive Antagonists of Human TLR4. Front. Immunol. 2020, 11, 577823. [Google Scholar] [CrossRef]
- Zhu, D.; Tuo, W. QS-21: A Potent Vaccine Adjuvant. Nat. Prod. Chem. Res. 2016, 3. [Google Scholar] [CrossRef]
- Slingluff, C.L., Jr.; Petroni, G.R.; Olson, W.C.; Smolkin, M.E.; Chianese-Bullock, K.A.; Mauldin, I.S.; Smith, K.T.; Deacon, D.H.; Varhegyi, N.E.; Donnelly, S.B.; et al. A randomized pilot trial testing the safety and immunologic effects of a MAGE-A3 protein plus AS15 immunostimulant administered into muscle or into dermal/subcutaneous sites. Cancer Immunol. Immunother. 2016, 65, 25–36. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/news-room/facts-in-pictures/detail/immunization (accessed on 5 December 2019).
- CDC. Available online: https://www.statista.com/chart/24658/fully-vaccinated-americans-who-became-infected-with-covid-19/ (accessed on 19 April 2021).
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef]
- Brisse, M.; Vrba, S.M.; Kirk, N.; Liang, Y.; Ly, H. Emerging Concepts and Technologies in Vaccine Development. Front. Immunol. 2020, 11, 583077. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Antigen | Vaccine Name | Vaccine Type | Indication | Clinical Trial # | Clinical Outcome | Ref # |
|---|---|---|---|---|---|---|
| MUC1 | Tecemotide/ L-BLP25 | Peptide | Stage III non-small-cell lung cancer, completed chemoradiotherapy | NCT00409188 (Phase III) | Median OS: 25.6 months to 22.3 months (treatment to control) | [14] |
| STn-KLH | Glycopeptide | Metastatic Breast Cancer | Unlisted (Phase III) | Median OS: 23.1 months to 22.3 months (treatment to control) Median PFS: 3.4 months to 3.0 months (treatment to control) | [15] | |
| PANVAC-V/F | Viral | Stage IV pancreatic cancer | NCT00088660 (Phase III) | Cancelled due to lack of clinical efficacy | [16,17,18] | |
| HER2/neu | E75 | Peptide | High risk node negative breast cancer | NCT00841399 NCT00584789 (Phase I/II) | 5-year DFS: 89.7 % to 80.2 % (treatment to control) | [19] |
| E75 | Peptide | High risk node negative breast cancer | NCT01479244 (Phase III) | 3-year Kaplan–Meier estimated DFS: 77.1 % to 77.5 % (treatment to control) | [20] | |
| p53 | SLP-p53® | Peptide | Epithelial ovarian cancer, with observed elevated levels of CA-125 | Unlisted- Trial approved by Medical Ethical Committee of the University Medical Center Groningen (Phase II) | Stable Disease observed in 2/20 patients | [21] |
| SLP-p53® | Peptide | Epithelial ovarian cancer, with observed elevated levels of CA-125 | NCT00844506 (Phase II) | Stable Disease observed in 2/10 patients | [22] | |
| SLP-p53® | Peptide | Colorectal Cancer | ISRCTN43704292 (Phase I/II) | N/A | [23] | |
| SLP-p53® | Peptide | Platinum-resistant ovarian cancer | NTC01639885 (Phase I/II) | Partial Response observed in 2 patients and stable disease observed in 4 patients | [24] | |
| ALVAC | Cellular/Viral | Colorectal Cancer | Unlisted- Trial approved by local and national medical ethics/ biological safety committee and the Dutch Ministry of Health and Environment (Phase I) | N/A | [25] | |
| MVAp53 | Viral | Recurrent epithelial ovarian/peritoneal/fallopian tube cancer | NCT02275039 (Phase I) | Median PFS: 3.0 months (treatment) | [26] | |
| hTERT | GV1001 | Peptide | Advanced / Metastatic Pancreatic Cancer | ISRCTN4382138 (Phase III) | Median OS: 6.4 months to 6.6 months to 4.5 months (control to concurrent treatment to sequential treatment) Median PFS: 7.89 months to 6.94 months to 8.36 months (control to concurrent treatment to sequential treatment) | [27] |
| UVI | Peptide | Hormone-naïve prostate cancer | NCT01784913 (Phase I/IIa) | Stable disease observed in 17 of 21 treated patients | [28] | |
| VX-001 | Peptide | Stage IV non-small cell lung cancer | NCT01935154 (Phase II) | Median TTF: 3.6 months to 3.5 months (treatment to control) Median OS: 14.3 months to 11.3 months (treatment to control) | [29] | |
| Survivin | SurVaxM | Peptide | Recurring malignant glioma | NCT01250470 (Phase I) | Median OS: 86.6 weeks Median DFS: 17.6 weeks | [30] |
| SurVaxM | Peptide | Newly diagnosed glioblastoma | NCT02455557 (Phase II) | Median PFS: 13.9 months | [31] | |
| SurVaxM | Peptide | Recurring malignant glioblastoma | NCT04013672 (Phase II) | Active Trial | N/A | |
| EMD640744 | Peptide | Solid Mass tumours (metastatic or locally advanced) | NCT01012102 (Phase I) | Stable disease observed in 28% of treated patients | [32] | |
| Gp100 | MDX-1379 | Peptide | Metastatic, unresectable Stage III/IV Melanoma | NCT00094653 (Phase III) | Median OS: 10.0 months to 6.4 months to 10.1 months (Treatment to monoclonal antibody monotherapy to vaccine monotherapy) Median PFS: 2.76 months to 2.86 months to 2.76 months (Treatment to monoclonal antibody monotherapy to vaccine monotherapy) | [33] |
| gp100:209–217 (210V) | Peptide | Advanced Stage III cutaneous melanoma/IV melanoma | NCT00019682 (Phase III) | Median OS: 17.8 months to 11.1 months (treatment to IL-2 monotherapy) Median PFS: 2.2 months to 1.6 months (treatment to IL-2 monotherapy) | [34] | |
| PAP | Sipuleucel-T (Provenge®) | Cellular- Dendritic Cell | Hormone-refractory prostate cancer | Unlisted- Trial approved by local institutional review boards at each study center and all patients signed institutional review board approved informed consent (Phase II) | N/A 38% of patients developed immune responses to PAP. Decline in PSA level by >50% was observed in 3 patients, and 25–49% in another 3 | [35] |
| Sipuleucel-T (Provenge®) | Cellular- Dendritic Cell | Metastatic, asymptomatic hormone-refractory prostate cancer | Unlisted- Trial approved by local institutional review boards at each study center and all patients signed institutional review board approved informed consent (Phase III) | Median OS: 25.9 months to 21.4 months (treatment to control) Median PFS: 11.7 months to 10.0 months (treatment to control) | [36] | |
| Sipuleucel-T (Provenge®) | Cellular- Dendritic Cell | Advanced prostate cancer | NCT00005947 NCT01133704 (Phase III) | Median OS: 23.2 months to 18.9 months (treatment to control) A 33% reduction in risk of death was observed for treated patients | [37] | |
| Sipuleucel-T (Provenge®) | Cellular- Dendritic Cell | Castration resistant prostate cancer | NCT00065442 (Phase III) | Median OS: 25.8 months to 21.7 months (treatment to control) A 22% reduction in risk of death was observed for treated patients | [38] | |
| MAGEA3 | recMAGE-A3 | Protein | Stage IB, II, IIIA MAGE-A3-positive non-small cell lung cancer | NCT00480025 (Phase III) | Median DFS: 60.5 months to 57.9 months (treatment to control) | [39] |
| recMAGE-A3 | Protein | Stage IIIB/IIIC MAGE-A3-positive melanoma | NCT00796445 (Phase III) | Median DFS: 11.0 months to 11.2 months (treatment to control) | [40] | |
| NY-ESO-1 | CHP-NY-ESO-1 | Peptide | Urothelial cancer, Prostate Cancer, Malignant solid tumours | UMIN000005246 UMIN000008006 (Phase I) | N/A | [41] |
| NY-ESO-1/ iscomatrix | Peptide | Resected Stage IIc, IIIb, IIIc and IV melanoma | LUD2003-009 (Phase II) | Median DFS: 4.67 months to 5.79 months (treatment to control) | [42] | |
| Neoantigen | Personalized Neoantigen Vaccine | Peptide | Stage IIB/C and IVM1a/b melanoma | NCT01970358 (Phase I) | 4 of 6 treated patients had no disease recurrence at 2-years follow up. Other 2 patients experienced total regression post anti-PD-1 therapy | [43] |
| IVAC Mutanome/ RBL001/002 | mRNA | Stage IIIA-C/IV NY-ESO-1 and/or tyrosinase positive melanoma | NCT02035956 (Phase I) | 8 of 13 treated patients had no disease recurrence at 1-2 years follow up. 2 out of 5 patients with recurrent disease showed objective response to vaccination with delayed relapse | [44] | |
| Personalized Neoantigen Vaccine | Peptide | Newly diagnosed (MGMT)-unmethylated glioblastoma | NCT02287428 (Phase I) | Median OS: 16.8 months Median PFS: 7.6 months | [45] | |
| APVAC2 | Peptide (TAA + neoantigen) | Newly diagnosed glioblastoma | NCT02149225 (Phase I) | Median OS: 29.0 months Median PFS: 14.2 months | [46] | |
| mRNA-4157 | mRNA | Melanoma Bladder carcinoma HPV-negative head & neck squamous cell carcinoma Non-small cell lung cancer Small cell lung cancer Microsatellite colon cancer | NCT03313778 | Active Trial CPI-naïve HPV-negative HNSCC patient median PFS: 9.8 months | [47] | |
| HPV (E6/E7) | VGX-3100 | DNA | Cervical intraepithelial neoplasia grade 2/3 | NCT00685412 (Phase I) | N/A | [48] |
| VGX-3100 | DNA | Cervical intraepithelial neoplasia grade 2/3 | NCT01304524, EudraCT2012-001334-33 (Phase II) | Histopathological regression observed in 49.5% of treated patients to 30.6% in the control subgroup | [49] | |
| MVA E2 | Viral | HPV intraepithelial lesions | Unlisted- Trial approved by Ethics and Scientific Committee of hospitals and corresponding health authorities from Estado de Mexico, (Phase III) | Complete regression observed in 94.82% (825/870) and 73.33% (220/300) of female patients with low-grade and high-grade lesions. Complete regression observed in 100% of male patients enrolled | [50] | |
| HPV16-SLP | Peptide | HPV16-positive cervical carcinoma | Unlisted- Trial approved by Medical Ethical Committee of the Leiden University Medical Center (Phase II) | N/A | [51] | |
| HPV16-SLP | Peptide | HPV16-induced advanced or recurrent gynecological carcinoma | Unlisted- Trial approved by Medical Ethical Committee of the Leiden University Medical Center (Phase II) | Median OS: 12.6 months | [52] | |
| MEDI0457 | DNA | HPV associated head and neck squamous cell carcinoma | NCT02163057 (Phase Ib/II) | 12-months DFS: 89.4% of treated patients | [53] | |
| AMV002 | DNA | HPV-associated oropharyngeal squamous cell carcinoma | ACTRN12618000140257 (Phase I) | N/A | [54] | |
| SQZ-PBMC-HPV | Cellular- Whole Cell | HPV16+ Recurrent, Locally Advanced or Metastatic Solid Tumors | NCT04084951 (Phase I) | Active Trial- Recruiting | N/A | |
| EBV (LMP1/2) | MVA-EL | Viral | Nasopharyngeal Carcinoma | NCT01256853, NCT01147991 (Phase I trials) | N/A | [55,56] |
| Ad-ΔLMP1-LMP2 transduced DCs | Cellular—Dendritic Cell | Epstein–Barr virus (EBV)-positive nasopharyngeal carcinoma | Unlisted- Trial approved by Institutional Review Board of the National Cancer Centre, Singapore (Phase II) | Median OS: 6.0 months Median PFS: 1.92 months Of 3 out of 12 treated patients, 1 patient exhibited partial responses to the vaccine for 7.5 months. The other 2 patients maintained stable disease for 6.5 and 7.5 months | [57] | |
| HCV (HCV Core) | C-35 peptide vaccine | Peptide | HCV-positive advanced hepatocellular carcinoma | UMIN000003520, UMIN000005634 (Phase II) | Median OS: 6.05 months | [58] |
| PSA | PROSTVAC-V/F-Tricom | Viral | Metastatic castration resistant prostate cancer | NCT00078585 (Phase II) | Median OS: 26.2 months to 16.3 months (treatment to control) | [59] |
| PROSTVAC-V/F-Tricom | Viral | Metastatic castration resistant prostate cancer | NCT01322490 (Phase III) | Median OS: 34.4 months to 32.2 months to 34.3 months (PROSTVAC-VF monotherapy to PROSTVAC-VF + GM-CSF to control) | [60] | |
| Multiple Antigens | ||||||
| MAGE-A3, MAGE-C2, tyrosinase, gp100 | TriMix-DC | mRNA | Stage III/IV Melanoma | NCT01302496 (Phase II) | Tumor response observed in 38% of treated patients, 8 complete and 7 partial responses were observed. 6 patients displayed stable disease. In 5-years follow-up, 7 complete and 1 partial response observed (n = 15) Median PFS: 6.21 months Median OS: 13.57 months | [61] |
| NY-ESO-1, MAGE-A3, Tyrosinase TPTE | BNT-111 | mRNA | Advanced unresectable melanoma | NCT02410733 (Phase I) | Active Trial | [62] |
| PSA PSMA PSCA | VBIR | Viral | Prostate Cancer | NCT02616185 (Phase I) | Trial Completed as of 9 March 2021 | [63] |
| Undefined Antigens | GVAX® | Cellular- Whole Cell | Asymptomatic prostate cancer | NCT00089856 (Phase III) | Trial terminated based on IDMC recommendation, with 30% chance of meeting primary endpoint of improving OS. OS reported post-study revealed median OS: 20.7 months to 21.7 months (treatment to standard care) | [64,65] |
| GVAX® | Cellular- Whole Cell | Metastatic hormone refractory prostate cancer | NCT00133224 (Phase III) | Trial terminated following increased deaths in treatment arm to control | [65,66] | |
| Melacine | Cellular- Whole Cell | Resected primary cutaneous melanoma | Unlisted | 5-years DFS: 77% for treated patients | [67] | |
| Canvaxin | Cellular- Whole Cell | Stage III Melanoma | Unlisted- Trial approved by UCLA/ JWCI–Saint John’s Health Center Institutional Review Boards (Phase II) | Median OS: 56.4 months to 31.9 months (treatment to control) 5-years OS: 49% to 37% (treatment to control) | [68] | |
| Canvaxin | Cellular- Whole Cell | Stage III/IV Melanoma | Unlisted (Phase III) | Study was terminated as a result of an interim analysis, concluding low probability of demonstrating significant improvement in survival | [69] | |
| OncoVax | Cellular- Whole Cell | Colon Cancer | Unlisted- Trial approved by participating hospital boards in the Netherlands, (Phase III) | 61% risk reduction associated with longer recurrence-free period was observed in Stage II colon patients | [70] | |
| Unnamed Vaccine | Cellular- Whole Cell | Stage II/III Metastatic Melanoma | Unlisted (Phase I) | 5 of 40 assessable, treated patients reported a median PFS of 10.0 months | [71] | |
| GVAX + CRS-207 | Cellular- Whole Cell/Viral | Metastatic pancreatic adenocarcinoma | NCT01417000 (Phase II) | Median OS: 6.2 months to 3.9 months (CRS-207 co-administration with GVAX/Cyclophosphamide to GVAX/Cyclophosphamide monotherapy) | [72] | |
| GVAX + CRS-207 | Cellular- Whole Cell/Viral | Metastatic pancreatic adenocarcinoma | NCT02004262 (Phase IIb) | Median OS: 3.7 months to 5.4 months to 4.6 months (CRS-207 co-administration with GVAX/Cyclophosphamide to GVAX/Cyclophosphamide monotherapy to control) | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tay, B.Q.; Wright, Q.; Ladwa, R.; Perry, C.; Leggatt, G.; Simpson, F.; Wells, J.W.; Panizza, B.J.; Frazer, I.H.; Cruz, J.L.G. Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions. Vaccines 2021, 9, 535. https://doi.org/10.3390/vaccines9050535
Tay BQ, Wright Q, Ladwa R, Perry C, Leggatt G, Simpson F, Wells JW, Panizza BJ, Frazer IH, Cruz JLG. Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions. Vaccines. 2021; 9(5):535. https://doi.org/10.3390/vaccines9050535
Chicago/Turabian StyleTay, Ban Qi, Quentin Wright, Rahul Ladwa, Christopher Perry, Graham Leggatt, Fiona Simpson, James W. Wells, Benedict J. Panizza, Ian H. Frazer, and Jazmina L. G. Cruz. 2021. "Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions" Vaccines 9, no. 5: 535. https://doi.org/10.3390/vaccines9050535
APA StyleTay, B. Q., Wright, Q., Ladwa, R., Perry, C., Leggatt, G., Simpson, F., Wells, J. W., Panizza, B. J., Frazer, I. H., & Cruz, J. L. G. (2021). Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions. Vaccines, 9(5), 535. https://doi.org/10.3390/vaccines9050535