
Targeted Delivery of BZLF1 to DEC205 Drives EBV-Protective Immunity in a Spontaneous Model of EBV-Driven Lymphoproliferative Disease


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Volunteers
2.2. Sample Collection and Storage
2.3. Cloning and Production of Anti-DEC205-BZLF1 Vaccine
2.4. Dendritic Cells and PBMC Co-Culture
2.5. Pentamer Quantitative Flow Cytometry
2.6. T-Cell Repertoire of EBV-Specific T-Cells
2.7. In Vivo Preclinical Model of EBV-LPD
2.8. ELISpot Assay
2.9. Mass Cytometry
2.10. Cytotoxicity Assay
2.11. Statistical Analyses
3. Results
3.1. αDEC205-BZLF1 Fusion Protein Promotes Expansion of EBV-Specific T-Cells
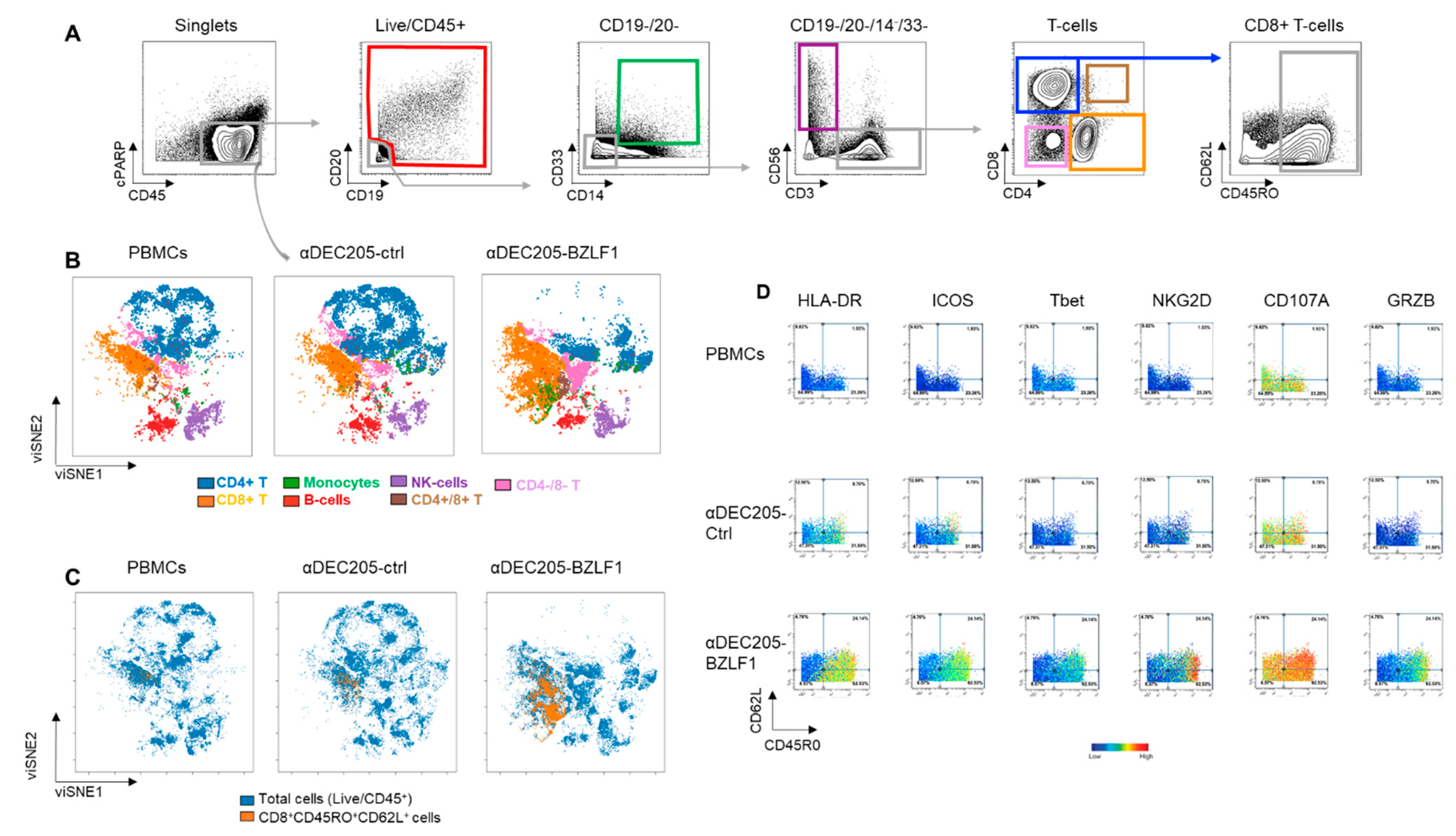
3.2. Effector Cells Expanded in αDEC205-BZLF1 CoCx Exhibited Activation and Cytotoxic Phenotype and Show Enhanced Anti-Tumor Effects In Vitro
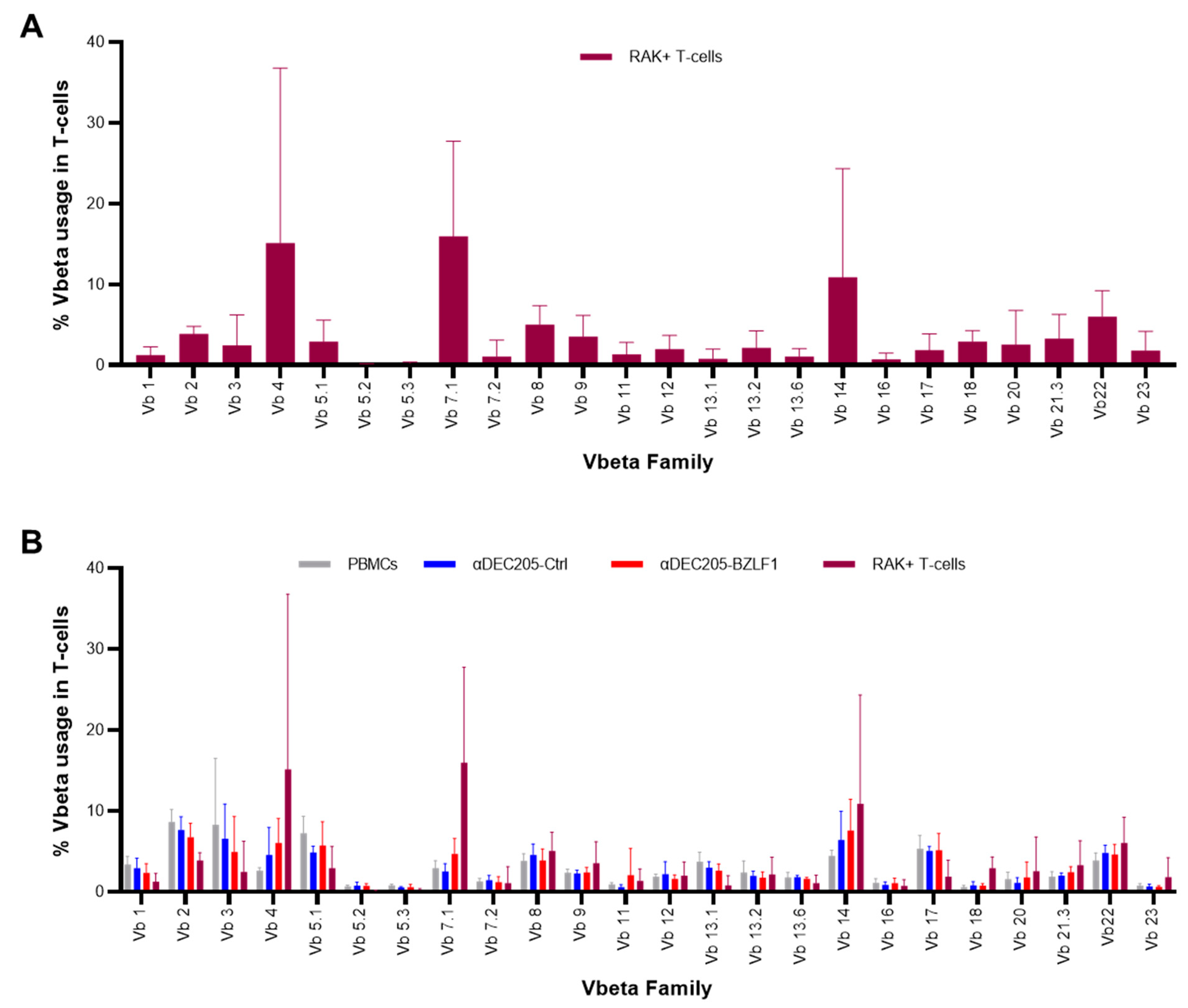
3.3. Clonal Expansion of the TCR Repertoire of RAK+ T-Cells
3.4. Vaccination of Hu-PBL-SCID Mice with Anti-DEC205-BZLF1 Drives BZLF1-Specific Immunity and Improves Survival from Fatal EBV-LPD Disease
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Epstein, M.A. Virus Particles. In Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Henle, W.; Henle, G. The sero-epidemiology of Epstein-Barr virus. Adv. Pathobiol. 1976, 5, 5–17. [Google Scholar]
- Rickinson, A.B.; Moss, D.J. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu. Rev. Immunol. 1997, 15, 405–431. [Google Scholar] [CrossRef]
- Miyashita, E.M.; Yang, B.; Babcock, G.J.; Thorley-Lawson, D.A. Identification of the site of Epstein-Barr virus persistence in vivo as a resting B cell. J. Virol. 1997, 71, 4882–4891. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV Persistence--Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar]
- Shindiapina, P.; Ahmed, E.H.; Mozhenkova, A.; Abebe, T.; Robert, A.B. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front. Oncol. 2020, 10, 1723. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Rooney, C.M.; Heslop, H.E. Post-transplant lymphoproliferative disorders. Annu. Rev. Med. 2005, 56, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Decker, L.L.; Freeman, R.B.; Thorley-Lawson, D.A. Epstein-barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J. Exp. Med. 1999, 190, 567–576. [Google Scholar] [CrossRef]
- Hosseini--Moghaddam, S.M.; Alhomayeed, B.; Soliman, N.; Weir, M.A.; House, A.A. Primary Epstein-Barr virus infection, seroconversion, and post-transplant lymphoproliferative disorder in seronegative renal allograft recipients: A prospective cohort study. Transpl. Infect. Dis. 2016, 18, 423–430. [Google Scholar] [CrossRef]
- Matas, A.J.; Smith, J.M.; Skeans, M.A.; Thompson, B.; Gustafson, S.K.; Stewart, D.E.; Cherikh, W.S.; Wainright, J.L.; Boyle, G.; Snyder, J.J.; et al. OPTN/SRTR 2013 Annual Data Report: Kidney. Am. J. Transpl. 2015, 15, 1–34. [Google Scholar] [CrossRef]
- Smith, J.M.; Rudser, K.; Gillen, D.; Kestenbaum, B.; Seliger, S.; Weiss, N.; McDonald, R.A.; Davis, C.L.; Stehmen-Breen, C. Risk of Lymphoma after Renal Transplantation Varies with Time: An Analysis of the United States Renal Data System. Transplant 2006, 81, 175–180. [Google Scholar] [CrossRef]
- Opelz, G.; Döhler, B. Lymphomas After Solid Organ Transplantation: A Collaborative Transplant Study Report. Arab. Archaeol. Epigr. 2004, 4, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Loren, A.W.; Porter, D.L.; Stadtmauer, A.E.; Tsai, E.D. Post-transplant lymphoproliferative disorder: A review. Bone Marrow Transp. 2003, 31, 145–155. [Google Scholar] [CrossRef]
- Heslop, H.E. How I treat EBV lymphoproliferation. Blood 2009, 114, 4002–4008. [Google Scholar] [CrossRef] [PubMed]
- Abbas, F.; El Kossi, M.; Shaheen, I.S.; Sharma, A.; Halawa, A. Post-transplantation lymphoproliferative disorders: Current concepts and future therapeutic approaches. World J. Transpl. 2020, 10, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Nourse, J.P.; Jones, K.; Gandhi, M.K. Epstein-Barr Virus-related post-transplant lymphoproliferative disorders: Pathogenetic insights for targeted therapy. Am. J. Transpl. 2011, 11, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Trappe, R.U.; Dierickx, D.; Zimmermann, H.; Morschhauser, F.; Mollee, P.; Zaucha, J.M.; Dreyling, M.H.; Dührsen, U.; Reinke, P.; Verhoef, G.; et al. Response to Rituximab Induction Is a Predictive Marker in B-Cell Post-Transplant Lymphoproliferative Disorder and Allows Successful Stratification Into Rituximab or R-CHOP Consolidation in an International, Prospective, Multicenter Phase II Trial. J. Clin. Oncol. 2017, 35, 536–543. [Google Scholar] [CrossRef]
- Bollard, C.M.; Aguilar, L.; Straathof, K.C.; Gahn, B.; Huls, M.H.; Rousseau, A.; Sixbey, J.; Gresik, M.V.; Carrum, G.; Hudson, M.; et al. Cytotoxic T lymphocyte therapy for Epstein-Barr virus+ Hodgkin’s disease. J. Exp. Med. 2004, 200, 1623–1633. [Google Scholar] [CrossRef]
- Haque, T.; Taylor, C.; Wilkie, G.M.; Murad, P.; Amlot, P.L.; Beath, S.; McKiernan, P.J.; Crawford, D.H. Complete regression of posttransplant lymphoproliferative disease using partially HLA-matched Epstein Barr virus-specific cytotoxic T cells. Transplantation 2001, 72, 1399–1402. [Google Scholar] [CrossRef]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef]
- Bollard, C.M.; Savoldo, B.; Rooney, C.M.; Heslop, H.E. Adoptive T-cell therapy for EBV-associated post-transplant lymphoproliferative disease. Acta Haematol. 2003, 110, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.-F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, A.S. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Balfour, H.H., Jr. Progress, prospects, and problems in Epstein-Barr virus vaccine development. Curr. Opin. Virol. 2014, 6, 1–5. [Google Scholar] [CrossRef][Green Version]
- Cohen, J.I. Epstein-barr virus vaccines. Clin. Transl. Immunol. 2015, 4, e32. [Google Scholar] [CrossRef]
- Porcu, P.; Eisenbeis, C.F.; Pelletier, R.P.; Davies, E.A.; Baiocchi, R.A.; Roychowdhury, S.; Vourganti, S.; Nuovo, G.J.; Marsh, W.L.; Ferketich, A.K.; et al. Successful treatment of posttransplantation lymphoproliferative disorder (PTLD) following renal allografting is associated with sustained CD8+ T-cell restoration. Blood 2002, 100, 2341–2348. [Google Scholar] [CrossRef]
- Brooks, J.M.; Long, H.M.; Tierney, R.J.; Shannon-Lowe, C.; Leese, A.M.; Fitzpatrick, M.; Taylor, G.S.; Rickinson, A.B. Early T Cell Recognition of B Cells following Epstein-Barr Virus Infection: Identifying Potential Targets for Prophylactic Vaccination. PLoS Pathog. 2016, 12, e1005549. [Google Scholar] [CrossRef]
- Baiocchi, R.A.; Ward, J.S.; Carrodeguas, L.; Eisenbeis, C.F.; Peng, R.; Roychowdhury, S.; Vourganti, S.; Sekula, T.; O’Brien, M.; Moeschberger, M.; et al. GM-CSF and IL-2 induce specific cellular immunity and provide protection against Epstein-Barr virus lymphoproliferative disorder. J. Clin. Investig. 2001, 108, 887–894. [Google Scholar] [CrossRef]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr Virus BZLF1 Gene, a Switch from Latency to Lytic Infection, Is Expressed as an Immediate-Early Gene after Primary Infection of B Lymphocytes. J. Virol. 2006, 81, 1037–1042. [Google Scholar] [CrossRef]
- Miller, G.; Lipman, M. Release of Infectious Epstein-Barr Virus by Transformed Marmoset Leukocytes. Proc. Natl. Acad. Sci. USA 1973, 70, 190–194. [Google Scholar] [CrossRef]
- Hui-Yuen, J.; McAllister, S.; Koganti, S.; Hill, E.; Bhaduri-McIntosh, S. Establishment of Epstein-Barr Virus Growth-transformed Lymphoblastoid Cell Lines. J. Vis. Exp. 2011, 2011, e3321. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gong, S.; Maric, S.; Misulovin, Z.; Pack, M.; Mahnke, K.; Nussenzweig, M.C.; Steinman, R.M. A monoclonal antibody to the DEC-205 endocytosis receptor on human dendritic cells. Hum. Immunol. 2000, 61, 729–738. [Google Scholar] [CrossRef]
- Lehmann, C.H.K.; Heger, L.; Heidkamp, G.F.; Baranska, A.; Luehr, J.J.; Hoffmann, A.; Dudziak, D. Direct Delivery of Antigens to Dendritic Cells via Antibodies Specific for Endocytic Receptors as a Promising Strategy for Future Therapies. Vaccines 2016, 4, 8. [Google Scholar] [CrossRef]
- Tsuji, T.; Matsuzaki, J.; Kelly, M.P.; Ramakrishna, V.; Vitale, L.; He, L.Z.; Keler, T.; Odunsi, K.; Old, L.J.; Ritter, G.; et al. Antibody-Targeted NY-ESO-1 to Mannose Receptor or DEC-205 In Vitro Elicits Dual Human CD8+ and CD4+ T Cell Responses with Broad Antigen Specificity. J. Immunol. 2011, 186, 1218–1227. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nat. Cell Biol. 1983, 301, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Kasai, M.; Yoneda, T.; Habu, S.; Maruyama, Y.; Okumura, K.; Tokunaga, T. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature 1981, 291, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Leipold, M.D.; Newell, E.W.; Maecker, H.T. Multiparameter Phenotyping of Human PBMCs Using Mass Cytometry. Methods Mol. Biol. 2015, 1343, 81–95. [Google Scholar] [PubMed]
- Finck, R.; Simonds, E.F.; Jager, A.; Krishnaswamy, S.; Sachs, K.; Fantl, W.; Pe’Er, D.; Nolan, G.P.; Bendall, S.C. Normalization of mass cytometry data with bead standards. Cytom. Part A 2013, 83, 483–494. [Google Scholar] [CrossRef]
- Amir, E.A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Jedema, I.; Van Der Werff, N.M.; Barge, R.M.Y.; Willemze, R.; Falkenburg, J.H.F. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood 2004, 103, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Gudgeon, N.H.; Callan, M.F.; Fazou, C.; Hasegawa, H.; Salmon, M.; Rickinson, A.B. EBV-specific CD8+ T cell memory: Relationships between epitope specificity, cell phenotype, and immediate effector function. J. Immunol. 2001, 167, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Frahm, M.A.; Picking, R.A.; Kuruc, J.D.; McGee, K.S.; Gay, C.; Eron, J.J.; Hicks, C.B.; Tomaras, G.D.; Ferrari, G. CD4+CD8+T Cells Represent a Significant Portion of the Anti-HIV T Cell Response to Acute HIV Infection. J. Immunol. 2012, 188, 4289–4296. [Google Scholar] [CrossRef] [PubMed]
- Ortolani, C. Cytofluorimetric identification of two populations of double positive (CD4+,CD8+) T lymphocytes in human peripheral blood. Biochem. Biophys. Res. Commun. 1993, 191, 601–609. [Google Scholar] [CrossRef]
- Ortolani, C.; Forti, E.; Radin, E.L.I.G.I.O.; Cibin, R.; Cossarizza, A. Association of Epstein-Barr virus reactivation with the recovery of CD4/CD8 double-negative T lymphocytes after haploidentical hematopoietic stem cell transplantation. Bone Marrow Transpl. 2017, 52, 264–269. [Google Scholar]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The Role of the NKG2D Immunoreceptor in Immune Cell Activation and Natural Killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef]
- Tary-Lehmann, M.; Saxon, A. Human mature T cells that are anergic in vivo prevail in SCID mice reconstituted with human peripheral blood. J. Exp. Med. 1992, 175, 503–516. [Google Scholar] [CrossRef]
- Tary-Lehmann, M.; Saxon, A.; Lehmann, P.V. The human immune system in hu-PBL-SCID mice. Immunol. Today 1995, 16, 529–533. [Google Scholar] [CrossRef]
- DHHS, U.S. Organdonor.gov. Available online: https://www.organdonor.gov/statistics-stories/statistics.html (accessed on 4 August 2020).
- Curtis, E.R.; Travis, L.B.; Rowlings, A.P.; Socié, G.; Kingma, D.W.; Banks, P.M.; Jaffe, E.S.; Sale, E.G.; Horowitz, M.M.; Witherspoon, R.P.; et al. Risk of lymphoproliferative disorders after bone marrow transplantation: A multi-institutional study. Blood 1999, 94, 2208–2216. [Google Scholar]
- Murray, J.E.; Wilson, R.E.; Tilney, N.L.; Merrill, J.P.; Cooper, W.C.; Birtch, A.G.; Carpenter, C.B.; Hager, E.B.; Dammin, G.J.; Harrison, J.H. Five years’ experience in renal transplantation with immunosuppressive drugs: Survival, function, complications, and the role of lymphocyte depletion by thoracic duct fistula. Ann. Surg. 1968, 168, 416–435. [Google Scholar] [CrossRef]
- Opelz, G.; Henderson, R. Incidence of non-Hodgkin lymphoma in kidney and heart transplant recipients. Lancet 1993, 342, 1514–1516. [Google Scholar] [CrossRef]
- Faull, R.J.; Hollett, P.; McDonald, S.P. Lymphoproliferative disease after renal transplantation in Australia and New Zealand. Transplantation 2005, 80, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Cockfield, S.M. Identifying the patient at risk for post-transplant lymphoproliferative disorder. Transpl. Infect. Dis. 2001, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lewin, K. Post-transplant lymphoproliferative disorders. Pathol. Oncol. Res. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Lucas, K.G.; Small, T.N.; Heller, G.; Dupont, B.; O’Reilly, R.J. The development of cellular immunity to Epstein-Barr virus after allogeneic bone marrow transplantation. Blood 1996, 87, 2594–2603. [Google Scholar] [CrossRef]
- Leblond, V.; Sutton, L.; Dorent, R.; Davi, F.; Bitker, O.M.; Gabarre, J.; Charlotte, F.; Ghoussoub, J.J.; Fourcade, C.; Fischer, A. Lymphoproliferative disorders after organ transplantation: A report of 24 cases observed in a single center. J. Clin. Oncol. 1995, 13, 961–968. [Google Scholar] [CrossRef]
- McDonald, R.A.; Smith, J.M.; Ho, M.; Lindblad, R.; Ikle, D.; Grimm, P.; Wyatt, R.; Arar, M.; Liereman, D.; Bridges, N.; et al. Incidence of PTLD in Pediatric Renal Transplant Recipients Receiving Basiliximab, Calcineurin Inhibitor, Sirolimus and Steroids. Arab. Archaeol. Epigr. 2008, 8, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Walti, L.N.; Mugglin, C.; Sidler, D.; Mombelli, M.; Manuel, O.; Hirsch, H.H.; Khanna, N.; Mueller, N.; Berger, C.; Boggian, K.; et al. Association of Antiviral Prophylaxis and Rituximab Use with Post-transplant Lymphoproliferative Disorders (PTLD): A Nationwide Cohort Study. Am. J. Transpl. 2020. [Google Scholar] [CrossRef]
- Aldabbagh, M.A.; Gitman, M.R.; Kumar, D.; Humar, A.; Rotstein, C.; Husain, S. The Role of Antiviral Prophylaxis for the Prevention of Epstein-Barr Virus-Associated Posttransplant Lymphoproliferative Disease in Solid Organ Transplant Recipients: A Systematic Review. Arab. Archaeol. Epigr. 2017, 17, 770–781. [Google Scholar] [CrossRef]
- Martin, S.I.; Dodson, B.; Wheeler, C.; Davis, J.; Pesavento, T.; Bumgardner, G.L. Monitoring Infection with Epstein-Barr Virus among Seromismatch Adult Renal Transplant Recipients. Arab. Archaeol. Epigr. 2011, 11, 1058–1063. [Google Scholar] [CrossRef]
- Savoldo, B.; Goss, J.A.; Hammer, M.M.; Zhang, L.; Lopez, T.; Gee, A.P.; Lin, Y.-F.; Quiros-Tejeira, R.E.; Reinke, P.; Schubert, S.; et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus–specific cytotoxic T lymphocytes (CTLs). Blood 2006, 108, 2942–2949. [Google Scholar] [CrossRef]
- van Zyl, D.G.; Mautner, J.; Delecluse, H.-J. Progress in EBV Vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef]
- Nemerow, G.R.; Mold, C.; Schwend, V.K.; Tollefson, V.; Cooper, N.R. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: Sequence homology of gp350 and C3 complement fragment C3d. J. Virol. 1987, 61, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Finerty, S.; Tarlton, J.; Mackett, M.; Conway, M.; Arrand, J.R.; Watkins, P.E.; Morgan, A.J. Protective immunization against Epstein-Barr virus-induced disease in cottontop tamarins using the virus envelope glycoprotein gp340 produced from a bovine papillomavirus expression vector. J. Gen. Virol. 1992, 73, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.Y.; Huang, T.M.; Ruan, L.; Miao, Y.H.; Lu, H.; Chu, C.M.; Motz, M.; Wolf, H. First EBV vaccine trial in humans using recombinant vaccinia virus expressing the major membrane antigen. Dev. Boil. Stand. 1995, 84, 171–177. [Google Scholar]
- Moutschen, M.; Léonard, P.; Sokal, E.M.; Smets, F.; Haumont, M.; Mazzu, P.; Bollen, A.; Denamur, F.; Peeters, P.; Dubin, G.; et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein–Barr virus vaccine in healthy adults. Vaccine 2007, 25, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Léonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 vaccine for infectious mononucleosis: A phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef]
- Rees, L.; Tizard, E.J.; Morgan, A.J.; Cubitt, W.D.; Finerty, S.; Oyewole-Eletu, T.A.; Owen, K.; Royed, C.; Stevens, S.J.; Shroff, R.C.; et al. A phase I trial of epstein-barr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. Transplantation 2009, 88, 1025–1029. [Google Scholar] [CrossRef]
- Elliott, S.L.; Suhrbier, A.; Miles, J.J.; Lawrence, G.; Pye, S.J.; Le, T.T.; Rosenstengel, A.; Nguyen, T.; Allworth, A.; Burrows, S.R.; et al. Phase I Trial of a CD8+ T-Cell Peptide Epitope-Based Vaccine for Infectious Mononucleosis. J. Virol. 2007, 82, 1448–1457. [Google Scholar] [CrossRef]
- Roskrow, M.A.; Suzuki, N.; Gan, Y.J.; Sixbey, J.W.; Ng, C.Y.; Kimbrough, S.; Hudson, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Epstein-Barr Virus (EBV)-Specific Cytotoxic T Lymphocytes for the Treatment of Patients With EBV-Positive Relapsed Hodgkin’s Disease. Blood 1998, 91, 2925–2934. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained Complete Responses in Patients With Lymphoma Receiving Autologous Cytotoxic T Lymphocytes Targeting Epstein-Barr Virus Latent Membrane Proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef]
- Straathof, K.C.M.; Bollard, C.M.; Popat, U.; Huls, M.H.; Lopez, T.; Morriss, M.C.; Gresik, M.V.; Gee, A.P.; Russell, H.V.; Brenner, M.K.; et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus–specific T lymphocytes. Blood 2005, 105, 1898–1904. [Google Scholar] [CrossRef]
- Chia, W.-K.; Teo, M.; Wang, W.-W.; Lee, B.; Ang, S.-F.; Tai, W.-M.; Chee, C.-L.; Ng, J.; Kan, R.; Lim, W.-T.; et al. Adoptive T-cell Transfer and Chemotherapy in the First-line Treatment of Metastatic and/or Locally Recurrent Nasopharyngeal Carcinoma. Mol. Ther. 2014, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Smith, A.C.; Ng, C.Y.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.K.; Bowman, L.C.; Krance, A.R.; Brenner, M.K.; et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood 1998, 92, 1549–1555. [Google Scholar] [CrossRef]
- Hislop, A.D.; Kuo, M.; Drake-Lee, A.B.; Akbar, A.N.; Bergler, W.; Hammerschmitt, N.; Khan, N.; Palendira, U.; Leese, A.M.; Timms, J.M.; et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J. Clin. Investig. 2020, 130, 733–747. [Google Scholar]
- Hislop, A.D.; Kuo, M.; Drake-Lee, A.B.; Akbar, A.N.; Bergler, W.; Hammerschmitt, N.; Khan, N.; Palendira, U.; Leese, A.M.; Timms, J.M.; et al. Tonsillar homing of Epstein-Barr virus-specific CD8+ T cells and the virus-host balance. J. Clin. Investig. 2005, 115, 2546–2555. [Google Scholar] [CrossRef]
- Gurer, C.; Strowig, T.; Brilot, F.; Pack, M.; Trumpfheller, C.; Arrey, F.; Park, C.G.; Steinman, R.M.; Münz, C. Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood 2008, 112, 1231–1239. [Google Scholar] [CrossRef]
- Krueger, K.M.; Ison, M.G.; Ghossein, C. Practical Guide to Vaccination in All Stages of CKD, Including Patients Treated by Dialysis or Kidney Transplantation. Am. J. Kidney Dis. 2020, 75, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.H.; Baiocchi, R.A. Murine Models of Epstein-Barr Virus-Associated Lymphomagenesis. Ilar J. 2016, 57, 55–62. [Google Scholar] [CrossRef]
- Baiocchi, R.A.; Caligiuri, M.A. Low-dose interleukin 2 prevents the development of Epstein-Barr virus (EBV)-associated lymphoproliferative disease in scid/scid mice reconstituted i.p. with EBV-seropositive human peripheral blood lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 5577–5581. [Google Scholar] [CrossRef]
- Baiocchi, R.A.; Ross, A.M.; Tan, J.C.; Chou, C.C.; Sullivan, L.; Haldar, S.; Monne, M.; Seiden, M.V.; Narula, S.K.; Sklar, J. Lymphomagenesis in the SCID-hu mouse involves abundant production of human interleukin-10. Blood 1995, 85, 1063–1074. [Google Scholar] [CrossRef]
- Hartlage, A.S.; Liu, T.; Patton, J.T.; Garman, S.L.; Zhang, X.; Kurt, H.; Lozanski, G.; Lustberg, M.E.; Caligiuri, M.A.; Baiocchi, R.A. The Epstein–Barr Virus Lytic Protein BZLF1 as a Candidate Target Antigen for Vaccine Development. Cancer Immunol. Res. 2015, 3, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Habib, M.; Buisson, M.; Lupo, J.; Agbalika, F.; Socié, G.; Germi, R.; Baccard, M.; Imbert-Marcille, B.-M.; Dantal, J.; Morand, P.; et al. Lytic EBV infection investigated by detection of Soluble Epstein-Barr virus ZEBRA in the serum of patients with PTLD. Sci. Rep. 2017, 7, 10479. [Google Scholar] [CrossRef]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; et al. Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: Expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020, 33, 2407–2421. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Suzuki, R.; Oguchi, M. Advances in the treatment of extranodal NK/T-cell lymphoma, nasal type. Blood 2018, 131, 2528–2540. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.; Zell, J.; Ziogas, A.; Anton-Culver, H. Epidemiology of nasopharyngeal carcinoma in the United States: Improved survival of Chinese patients within the keratinizing squamous cell carcinoma histology. Ann. Oncol. 2007, 18, 29–35. [Google Scholar] [CrossRef] [PubMed]
- van Beek, J.; Zur Hausen, A.; Klein Kranenbarg, E.; van de Velde, C.J.; Middeldorp, J.M.; van den Brule, A.J.; Meijer, C.J.; Bloemena, E. EBV-positive gastric adenocarcinomas: A distinct clinicopathologic entity with a low frequency of lymph node involvement. J. Clin. Oncol. 2004, 22, 664–670. [Google Scholar] [CrossRef]
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, A.L. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCR | Normal Range | EBV-Specific CTLs | |||
|---|---|---|---|---|---|
| D-9 | D-77 | D-78 | D-81 | ||
| Vb 1 | 1.89–11.7 | 0.6 | 2.8 | 0.2 | 0.97 |
| Vb 2 | 4.03–23.48 | 4.4 | 6.2 | 2.4 | 2.3 |
| Vb 3 | 0.52–15.71 | 0.6 | 7.6 | 0.1 | 0.6 |
| Vb 4 | 0.79–3.26 | 47.5 | 2.2 | 6.4 | 0.8 |
| Vb 5.1 | 3.19–14.93 | 0.4 | 1.5 | 2.3 | 4.9 |
| Vb 5.2 | 0.49–4.98 | 0.1 | 0 | 0.2 | 0.1 |
| Vb 5.3 | 0.37–2.98 | 0.1 | 0 | 0.3 | 0.4 |
| Vb 7.1 | 0.64–20.01 | 6.5 | 21.7 | 5.1 | 26.1 |
| Vb 7.2 | 0.05–5.45 | 0.1 | 0 | 0 | 8.8 |
| Vb 8 | 2.26–29.47 | 4.1 | 5.5 | 2.3 | 6.4 |
| Vb 9 | 1.1–9.3 | 1.9 | 1.8 | 3 | 7 |
| Vb 11 | 0.25–5.11 | 1.3 | 0 | 0.7 | 0.3 |
| Vb 12 | 1–4.76 | 1.9 | 0.3 | 0.6 | 4.4 |
| Vb 13.1 | 1.62–8.16 | 0.1 | 0.1 | 2.6 | 0.3 |
| Vb 13.2 | 0.80–5.28 | 0.1 | 2 | 1.3 | 1.5 |
| Vb 13.6 | 0.84–8.8 | 0.2 | 1 | 0.4 | 1.3 |
| Vb 14 | 1.33–8.03 | 0.4 | 25.6 | 0.1 | 12.7 |
| Vb 16 | 0.42–1.9 | 0.7 | 0.3 | 0.1 | 0.5 |
| Vb 17 | 2.28–12.61 | 0.6 | 1 | 0.6 | 2.2 |
| Vb 18 | 0.58–5.23 | 3 | 4.5 | 2.2 | 1.4 |
| Vb 20 | 0–9.73 | 0.2 | 2.2 | 0.1 | 5.4 |
| Vb 21.3 | 1.08–5.97 | 1.8 | 7.6 | 1.6 | 1.9 |
| Vb22 | 1.99–9.89 | 9.1 | 2 | 5.4 | 7.2 |
| Vb 23 | 0.26–4.76 | 0.4 | 1 | 0.6 | 1.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, E.H.; Brooks, E.; Sloan, S.; Schlotter, S.; Jeney, F.; Hale, C.; Mao, C.; Zhang, X.; McLaughlin, E.; Shindiapina, P.; et al. Targeted Delivery of BZLF1 to DEC205 Drives EBV-Protective Immunity in a Spontaneous Model of EBV-Driven Lymphoproliferative Disease. Vaccines 2021, 9, 555. https://doi.org/10.3390/vaccines9060555
Ahmed EH, Brooks E, Sloan S, Schlotter S, Jeney F, Hale C, Mao C, Zhang X, McLaughlin E, Shindiapina P, et al. Targeted Delivery of BZLF1 to DEC205 Drives EBV-Protective Immunity in a Spontaneous Model of EBV-Driven Lymphoproliferative Disease. Vaccines. 2021; 9(6):555. https://doi.org/10.3390/vaccines9060555
Chicago/Turabian StyleAhmed, Elshafa Hassan, Eric Brooks, Shelby Sloan, Sarah Schlotter, Frankie Jeney, Claire Hale, Charlene Mao, Xiaoli Zhang, Eric McLaughlin, Polina Shindiapina, and et al. 2021. "Targeted Delivery of BZLF1 to DEC205 Drives EBV-Protective Immunity in a Spontaneous Model of EBV-Driven Lymphoproliferative Disease" Vaccines 9, no. 6: 555. https://doi.org/10.3390/vaccines9060555
APA StyleAhmed, E. H., Brooks, E., Sloan, S., Schlotter, S., Jeney, F., Hale, C., Mao, C., Zhang, X., McLaughlin, E., Shindiapina, P., Shire, S., Das, M., Prouty, A., Lozanski, G., Mamuye, A. T., Abebe, T., Alinari, L., Caligiuri, M. A., & Baiocchi, R. A. (2021). Targeted Delivery of BZLF1 to DEC205 Drives EBV-Protective Immunity in a Spontaneous Model of EBV-Driven Lymphoproliferative Disease. Vaccines, 9(6), 555. https://doi.org/10.3390/vaccines9060555