
Epigenetic Mechanisms of HIV-1 Persistence


Abstract
1. Introduction
2. Epigenetic Mechanisms of HIV-1 Latency
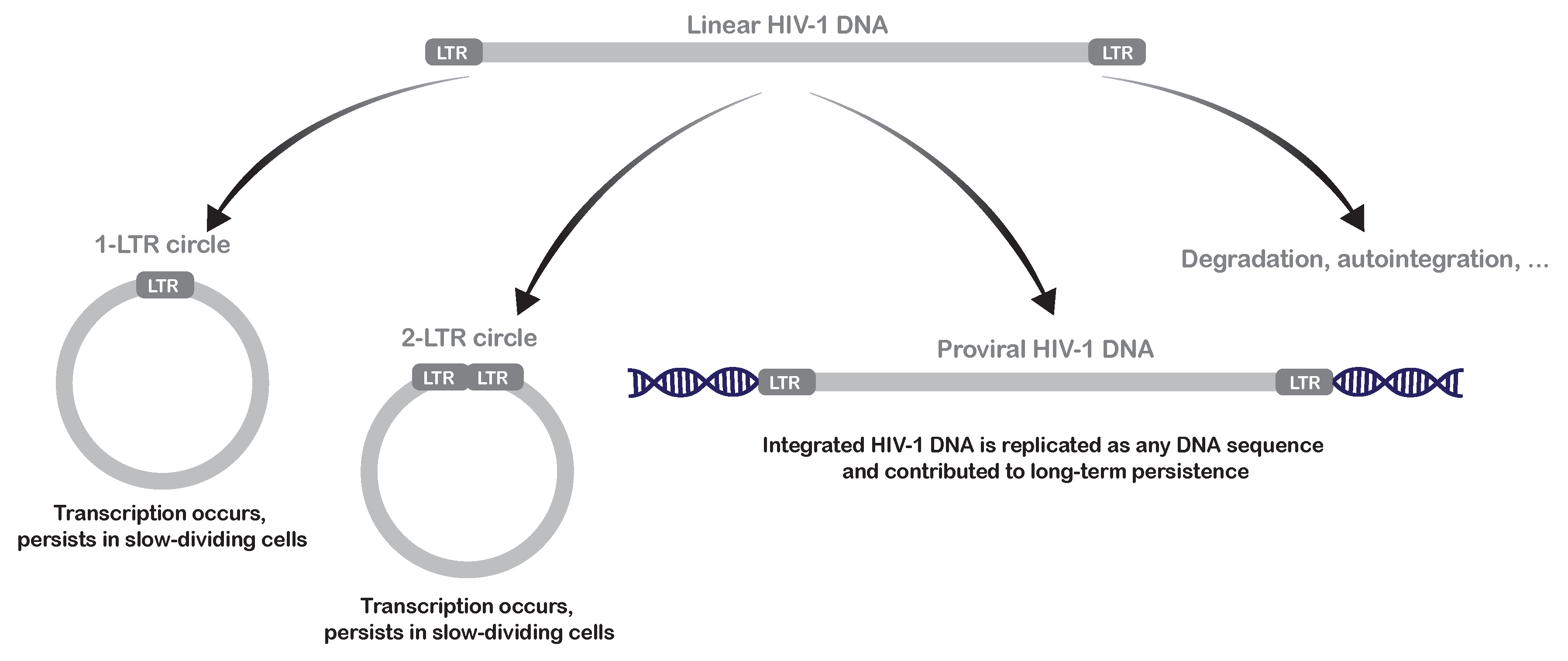
2.1. Mechanisms of Pre-Integration Latency
2.2. Mechanisms of Post-Integration Latency
2.2.1. Nucleosome Positioning on the HIV-1 Provirus
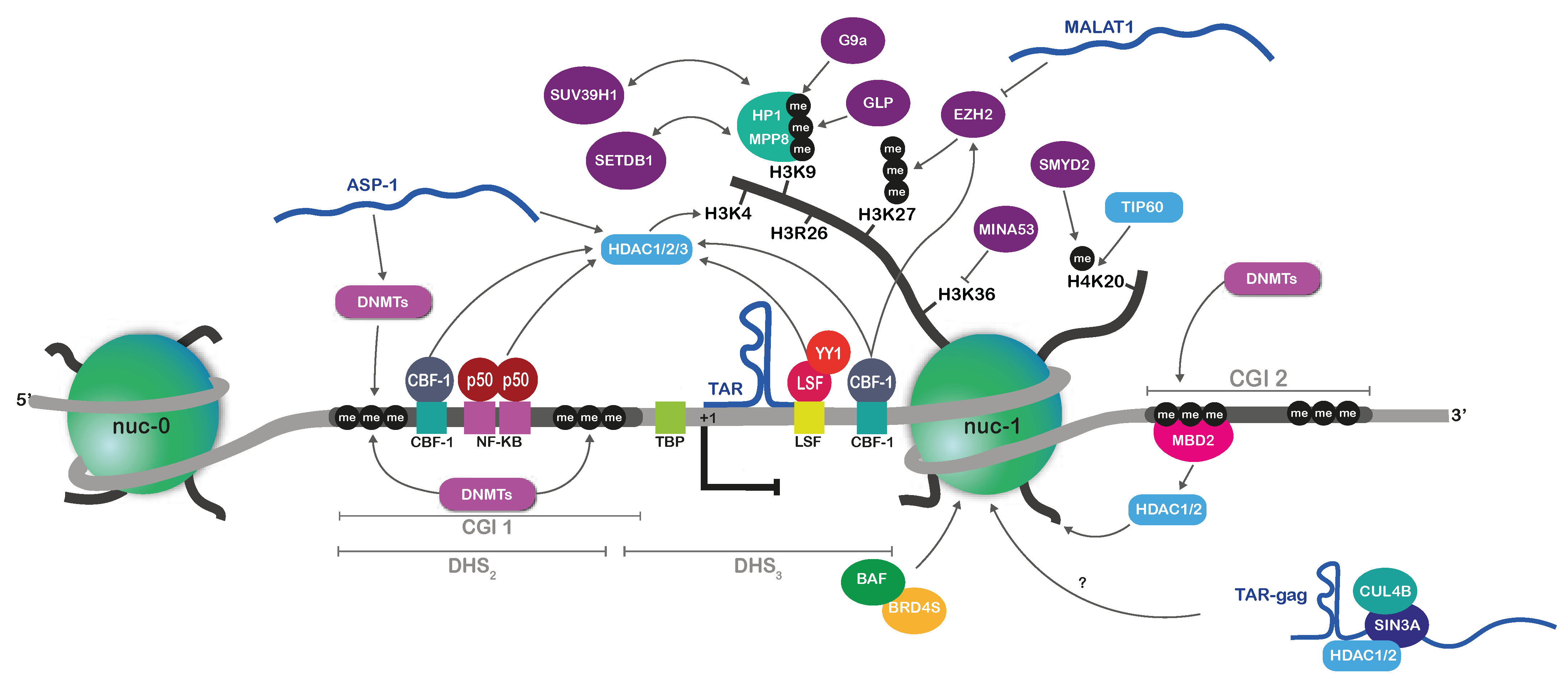
2.2.2. Repressive Histone Marks on the HIV-1 Promoter during Latency
2.2.3. Implication of DNA Methylation in HIV-1 Latency
2.2.4. ncRNA-Mediated Mechanisms of HIV-1 Epigenetic Silencing
2.2.5. Nuclear Position of the HIV-1 Provirus
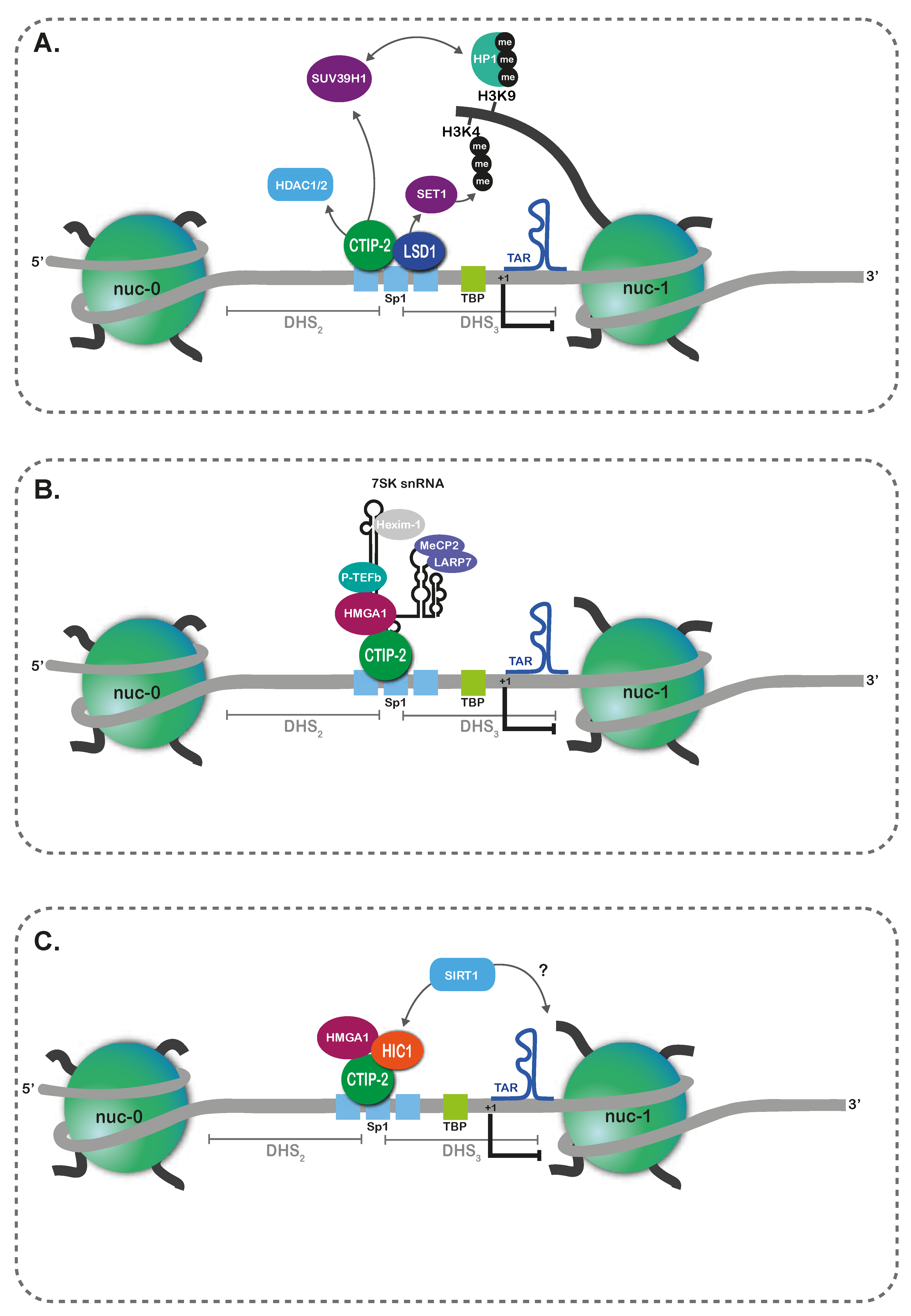
3. Epigenetic Persistence in Myeloid Lineages
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 5mC | 5-methylcytosine |
| ACSS2 | Acyl-coenzyme A synthetase short-chain family member 2 |
| AIDS | Acquired Immunodeficiency Syndrome |
| APOBEC3A | Apolipoprotein B mRNA editing enzyme catalytic subunit 3A |
| ASP-1 | HIV-1 antisense protein |
| ATP | Adenosine triphosphate |
| BAF/PBAF | BRG1- or HBRM-associated factor/polybromo-associated BAF |
| BLV | bovine leukemia virus |
| BRD4 | bromodomain-containing protein 4 |
| cART | combination antiretroviral therapy |
| CBF-1 | C-promoter binding factor-1 |
| cDNA | complementary DNA |
| CGI | CpG island |
| CpG | CpG dinucleotide |
| CTIP2 | COUP-TF interacting protein 2 |
| DHS | DNase I hypersensitive site |
| DNA | Deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| EHMT | Euchromatin histone methyltransferase |
| FACT | Facilitates Chromatin Transcription |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| HDACi | inhibitors of HDAC |
| HERVs | human endogenous retroviruses |
| HIC1 | Hypermethylated in cancer 1 |
| HIRA | Histone Cell Cycle Regulator |
| HIV | Human Immunodeficiency Virus |
| HKMTs | histone lysine methyltransferases |
| HMGA1 | High Mobility Group AT-hook 1 |
| HMT | Histone MethylTransferase |
| HP1 | heterochromatin protein 1 |
| HTLV | human T-cell leukemia virus |
| HUSH | Human Silencing Hub |
| INI-1 | Integrase Interactor 1 |
| INO80 | INO80 Complex ATPase |
| KAT | lysine acetyltransferase |
| LEDGF/p75 | lens epithelium-derived growth factor/transcription co-activator p75 |
| lncRNA | long non-coding RNA |
| LRA | Latency reversing agent |
| LSD1 | lysine-specific demethylase 1 |
| LSF | late SV40 factor |
| LTR | long terminal repeat |
| miRNA | microRNA |
| MLV | murine leukemia virus |
| Mnase | Micrococcal nuclease |
| NCR | non-coding region |
| ncRNA | non-coding RNA |
| NDR | nucleosome-depleted region |
| NKILA | NF-κB-interacting long non-coding RNA |
| nuc | nucleosome |
| NURD/Mi-2/CHD | Nucleosome Remodeling Deacetylase/Mi-2/Chromodomain Helicase DNA-binding |
| PBMC | peripheral blood mononuclear cell |
| PCAF | p300/CBP associated factor |
| PHD | Plant homeodomain |
| piRNA | piwi-interacting RNAs |
| PML | promyelocytic leukemia |
| PRC | Polycomb repressive complex |
| PRMTs | protein arginine methyltransferases |
| P-TEFb | positive transcription elongation factor B |
| PTM | post-translational modification |
| RNA | Ribonucleic acid |
| SETDB1 | SET domain, bifurcated 1 |
| SIRT1 | Sirtuin 1 |
| SUV39H1 | Suppressor of variegation 3-9 homolog 1 |
| SWI/SNF | Switch/Sucrose Non-Fermentable |
| SWR1 | SWI2/SNF2-Related 1 Chromatin Remodeling |
| TAF | TBP-associated factor |
| TAR | Trans-activating responsive elements |
| Tat | Trans-Activator of Transcription |
| TCM | central memory CD4+ T cell |
| TSS | transcription start site |
| YY1 | Yin Yang 1 |
References
- Deeks, S.; Lewin, S.; Havlir, D. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb. Perspect. Med. 2012, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Eisele, E.; Siliciano, R.F. Redefining the Viral Reservoirs that Prevent HIV-1 Eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.W.; Moir, S.; Fauci, A.S. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 2015, 16, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immun. Rev. 2018, 48, 872–895. [Google Scholar] [CrossRef]
- Chun, T.-W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.; Margolick, J.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.; Kajdas, J.; Finzi, D.; Quinn, T.; Chadwick, K.; Margolick, J.; Kovacs, C.; Gange, S.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Perelson, A.S.; Neumann, A.U.; Markowitz, M.; Leonard, J.M.; Ho, D.D. HIV-1 Dynamics in vivo: Virion Clearance Rate, Infected Cell Life-Span, and Viral Generation Time. Science 1996, 271, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.M.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S.Y. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Sarabia, N.; Bateson, R.E.; Dahl, N.P.; Crooks, A.M.; Kuruc, J.D.; Margolis, D.M.; Archin, N.M. Quantitation of Replication-Competent HIV-1 in Populations of Resting CD4+ T Cells. J. Virol. Methods 2014, 88, 14070–14077. [Google Scholar] [CrossRef]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef]
- Hiener, B.; Horsburgh, B.A.; Eden, J.S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef]
- Zerbato, J.M.; McMahon, D.K.; Sobolewski, M.D.; Mellors, J.W.; Sluis-Cremer, N. Naïve CD4+ T Cells Harbor a Large Inducible Reservoir of Latent, Replication-Competent HIV-1. Clin. Infect. Dis. 2019, 69, 1919–1925. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Chomont, N. HIV persistence in the setting of antiretroviral therapy: When, where and how does HIV hide? J. Virus Erad. 2015, 1, 59–66. [Google Scholar] [CrossRef]
- Le Douce, V.; Herbein, G.; Rohr, O.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010, 7, 32. [Google Scholar] [CrossRef]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.-A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Victor Garcia, J. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef]
- Zhang, J.; Perelson, A.S. Contribution of Follicular Dendritic Cells to persistent HIV viremia. J. Virol. 2013, 87, 3–4. [Google Scholar] [CrossRef]
- Kandathil, A.J.; Sugawara, S.; Balagopal, A. Are T cells the only HIV-1 reservoir? Retrovirology 2016, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.; Winckelmann, A.; Palmer, S. HIV-1 Reservoirs During Suppressive Therapy. Trends Microbiol. 2016, 24, 345–355. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Li, G. Structural insights of nucleosome and the 30-nm chromatin fiber. Curr. Opin. Struct. Biol. 2016, 36, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Reinberg, D. Facultative Heterochromatin: Is There a Distinctive Molecular Signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef]
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Murr, R. Interplay between different epigenetic modifications and mechanisms. Adv. Genet. 2010, 70, 101–141. [Google Scholar]
- Spivak, A.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2019, 10, 3060. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Koyanagi, Y.; Miles, S.; Wiley, C.; Vinters, H.V.; Chen, I.S.Y. High levels of unintegrated HIV -1 DNA in brain tissue of AIDS dementia patients. Nature 1990, 395, 367–370. [Google Scholar] [CrossRef]
- Sharkey, M.E.; Teo, I.; Greenough, T.; Sharova, N.; Luzuriaga, K.; Sullivan, J.L.; Bucy, R.P.; Kostrikis, L.G.; Haase, A.; Veryard, C.; et al. Persistence of episomal HIV-1 infection intermediates in patients onhighly active anti-retroviral therapy. Nat. Med. 2000, 6, 76–81. [Google Scholar] [CrossRef]
- Sloan, R.D.; Wainberg, M.A. The role of unintegrated DNA in HIV infection. Retrovirology 2011, 8, 1–15. [Google Scholar] [CrossRef]
- Brussel, A.; Sonigo, P. Evidence for Gene Expression by Unintegrated Human Immunodeficiency Virus Type 1 DNA Species. J. Virol. 2004, 78, 11263–11271. [Google Scholar] [CrossRef]
- Thierry, S.; Thierry, E.; Subra, F.; Deprez, E.; Leh, H.; Bury-Moné, S.; Delelis, O. Opposite transcriptional regulation of integrated vs unintegrated HIV genomes by the NF-κB pathway. Sci. Rep. 2016, 6, 1–12. [Google Scholar]
- Orzalli, M.H.; Knipe, D.M. Cellular Sensing of Viral DNA and Viral Evasion Mechanisms. Annu. Rev. Microbiol. 2014, 68, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Kantor, B.; Hong, M.; Webster-Cyriaque, J.; Monahan, P.E.; Kafri, T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc. Natl. Acad. Sci. USA 2009, 106, 18786–18791. [Google Scholar] [CrossRef] [PubMed]
- Machida, S.; Depierre, D.; Chen, H.C.; Thenin-Houssier, S.; Petitjean, G.; Doyen, C.M.; Takaku, M.; Cuvier, O.; Benkirane, M. Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome. Proc. Natl. Acad. Sci. USA 2020, 117, 6822–6830. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, G.Z.; Cingöz, O.; Goff, S.P. NP220 mediates silencing of unintegrated retroviral DNA. Nature 2018, 564, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chougui, G.; Margottin-Goguet, F. HUSH, a Link Between Intrinsic Immunity and HIV Latency. Front. Microbiol. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.; Morel, M.; Lahouassa, H.; Leduc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef]
- Sharkey, M.; Triques, K.; Kuritzkes, D.R.; Stevenson, M. In vivo evidence for instability of episomal human immunodeficiency virus type 1 cDNA. J. Virol. 2005, 79, 5203–5210. [Google Scholar] [CrossRef]
- Pierson, T.C.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Gange, S.J.; Siliciano, R.F. Intrinsic Stability of Episomal Circles Formed during Human Immunodeficiency Virus Type 1 Replication. J. Virol. 2002, 76, 4138–4144. [Google Scholar] [CrossRef]
- Darcis, G.; Van Driessche, B.; Bouchat, S.; Kirchhoff, F.; Van Lint, C. Molecular Control of HIV and SIV Latency. In HIV-1 Latency. Current Topics in Microbiology and Immunology; Silvestri, G., Lichterfeld, M., Eds.; Springer: Berlin, Germany, 2017; Volume 417, pp. 1–22. [Google Scholar]
- Sarracino, A.; Marcello, A. The Relevance of Post-Transcriptional Mechanisms in HIV Latency Reversal. Curr. Pharm. Des. 2017, 23, 4103–4111. [Google Scholar] [CrossRef]
- Struhl, K.; Segal, E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Clark, D.J. Major Determinants of Nucleosome Positioning. Biophys. J. 2018, 114, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef]
- Gross, D.S.; Garrard, W.T. Nuclease Hypersensitive Sites in Chromatin. Annu. Rev. Biochem. 1988, 57, 159–197. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.; Thurman, R.; Song, L.; Safi, A.; Stamatoyannopoulos, J.; Lenhard, B.; Crawford, G.; Furey, T. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 2013, 23, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991, 65, 6790–6799. [Google Scholar] [CrossRef]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol. Cell 2017, 67, 1001–1012.e6. [Google Scholar] [CrossRef]
- Van Duyne, R.; Guendel, I.; Narayanan, A.; Gregg, E.; Shafagati, N.; Tyagi, M.; Easley, R.; Klase, Z.; Nekhai, S.; Kehn-Hall, K.; et al. Varying modulation of HIV-1 LTR activity by BAF complexes. J. Mol. Biol. 2011, 411, 581–596. [Google Scholar] [CrossRef]
- Verdin, E.; Paras, P.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Marian, C.A.; Stoszko, M.; Wang, L.; Leighty, M.W.; de Crignis, E.; Maschinot, C.A.; Gatchalian, J.; Carter, B.C.; Chowdhury, B.; Hargreaves, D.C.; et al. Small Molecule Targeting of Specific BAF (mSWI/SNF) Complexes for HIV Latency Reversal. Cell Chem. Biol. 2018, 25, 1443–1455.e14. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Parra, M.; Vries, R.G.J.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar] [CrossRef] [PubMed]
- Tréand, C.; Du Chéné, I.; Brès, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Ishizaka, A.; Tomizawa, M.; Okazaki, T.; Yamamichi, N.; Kawana-Tachikawa, A.; Iwamoto, A.; Iba, H. Loss of the Brm-Type SWI/SNF Chromatin Remodeling Complex Is a Strong Barrier to the Tat-Independent Transcriptional Elongation of Human Immunodeficiency Virus Type 1 Transcripts. J. Virol. 2009, 83, 11569–11580. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lesbats, P.; Botbol, Y.; Chevereau, G.; Vaillant, C.; Calmels, C.; Arneodo, A.; Andreola, M.L.; Lavigne, M.; Parissi, V. Functional coupling between HIV-1 integrase and the SWI/SNF chromatin remodeling complex for efficient in vitro integration into stable nucleosomes. PLoS Pathog. 2011, 7, e1001280. [Google Scholar] [CrossRef] [PubMed]
- Gallastegui, E.; Millan-Zambrano, G.; Terme, J.-M.J.-M.; Chavez, S.; Jordan, A.; Millán-Zambrano, G.; Terme, J.-M.J.-M.; Chávez, S.; Jordan, A. Chromatin reassembly factors are involved in transcriptional interference promoting HIV latency. J. Virol. 2011, 85, 3187–3202. [Google Scholar] [CrossRef]
- Nakamura, M.; Basavarajaiah, P.; Rousset, E.; Beraud, C.; Latreille, D.; Henaoui, I.S.; Lassot, I.; Mari, B.; Kiernan, R. Spt6 levels are modulated by PAAF1 and proteasome to regulate the HIV-1 LTR. Retrovirology 2012, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gérard, A.; Ségéral, E.; Naughtin, M.; Abdouni, A.; Charmeteau, B.; Cheynier, R.; Rain, J.-C.C.; Emiliani, S. The Integrase Cofactor LEDGF/p75 Associates with Iws1 and Spt6 for Postintegration Silencing of HIV-1 Gene Expression in Latently Infected Cells. Cell Host Microbe 2015, 17, 107–117. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Rodgers, M.J.; Banks, D.J.; Bradley, K.A.; Young, J.A.T. CHD1 and CHD2 are positive regulators of HIV-1 gene expression. Virol. J. 2014, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, D.C. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Romerio, F.; Gabriel, M.N.; Margolis, D.M. Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. J. Virol. 1997, 71, 9375–9382. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A Limited Group of Class I Histone Deacetylases Acts To Repress Human Immunodeficiency Virus Type 1 Expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar] [CrossRef]
- Huber, K.; Doyon, G.; Plaks, J.; Fyne, E.; Mellors, J.W.; Sluis-Cremer, N. Inhibitors of histone deacetylases: Correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011, 286, 22211–22218. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- He, G.; Margolis, D.M. Counterregulation of Chromatin Deacetylation and Histone Deacetylase Occupancy at the Integrated Promoter of Human Immunodeficiency Virus Type 1 (HIV-1) by the HIV-1 Repressor YY1 and HIV-1 Activator Tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, I.; Della Chiara, G.; D’Ambrosio, R.L.; Huichalaf, C.; Brambilla, P.; Corbetta, S.; Riba, M.; Piccirillo, R.; Valente, S.; Casari, G.; et al. Amino acid starvation induces reactivation of silenced transgenes and latent HIV-1 provirus via down-regulation of histone deacetylase 4 (HDAC4). Proc. Natl. Acad. Sci. USA 2012, 109, E2284–E2293. [Google Scholar] [CrossRef]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV/AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Li, Z.; Mbonye, U.; Feng, Z.; Wang, X.; Gao, X.; Karn, J.; Zhou, Q. The KAT5-Acetyl-Histone4-Brd4 axis silences HIV-1 transcription and promotes viral latency. PLoS Pathog. 2018, 14, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Cho, W.-K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of Histone H3 Lysine 9 (H3K) Methyltransferase G9a in the Maintenance of HIV-1 Latency and Its Reactivation. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Qu, X.; Li, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, P.; Liu, S.; Kong, C.; Wang, X.; et al. Involvement of histone methyltransferase GLP in HIV-1 latency through catalysis of H3K9 dimethylation. Virology 2013, 440, 182–189. [Google Scholar] [CrossRef]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple histone lysine methyltransferases are required for the establishment and maintenance of HIV-1 latency. MBio 2017, 8, 1–15. [Google Scholar] [CrossRef]
- du Chéné, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host Microbe 2017, 21, 569–579. [Google Scholar] [CrossRef]
- Zhang, Z.; Nikolai, B.C.; Gates, L.A.; Jung, S.Y.; Siwak, E.B.; He, B.; Rice, A.P.; O’Malley, B.W.; Feng, Q. Crosstalk between histone modifications indicates that inhibition of arginine methyltransferase CARM1 activity reverses HIV latency. Nucleic Acids Res. 2017, 45, 9348–9360. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kong, W.; Jean, M.; Fiches, G.; Zhou, D.; Hayashi, T.; Que, J.; Santoso, N.; Zhu, J. A CRISPR/Cas9 screen identifies the histone demethylase MINA53 as a novel HIV-1 latency-promoting gene (LPG). Nucleic Acids Res. 2019, 47, 7333–7347. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Lakhikumar Sharma, A.; Hokello, J.; Sonti, S.; Zicari, S.; Sun, L.; Alqatawni, A.; Bukrinsky, M.; Simon, G.; Chauhan, A.; Daniel, R.; et al. CBF-1 promotes the establishment and maintenance of HIV latency by recruiting Polycomb repressive complexes, PRC1 and PRC2, at HIV LTR. Viruses 2020, 12, 1–22. [Google Scholar]
- Taura, M.; Song, E.; Ho, Y.C.; Iwasaki, A. Apobec3A maintains HIV-1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc. Natl. Acad. Sci. USA 2019, 116, 2282–2289. [Google Scholar] [CrossRef]
- Jiang, G.; Nguyen, D.; Archin, N.M.; Yukl, S.A.; Méndez-Lagares, G.; Tang, Y.; Elsheikh, M.M.; Thompson, G.R.; Hartigan-O’Connor, D.J.; Margolis, D.M.; et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J. Clin. Investig. 2018, 128, 1190–1198. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef]
- Verdikt, R.; Darcis, G.; Ait-Ammar, A.; Van Lint, C. Applications of CRISPR/Cas9 tools in deciphering the mechanisms of HIV-1 persistence. Curr. Opin. Virol. 2019, 38, 63–69. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic Cytosine Methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Wolffe, A.P. DNA Methylation in Health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA methylation in cancer and aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed]
- Pierard, V.; Guiguen, A.; Colin, L.; Wijmeersch, G.; Vanhulle, C.; Van Driessche, B.; Dekoninck, A.; Blazkova, J.; Cardona, C.; Merimi, M.; et al. DNA cytosine methylation in the bovine leukemia virus promoter is associated with latency in a lymphoma-derived B-cell line: Potential involvement of direct inhibition of cAMP-responsive element (CRE)-binding protein/CRE modulator/activation transcription. J. Biol. Chem. 2010, 285, 19434–19449. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Hamano, A.; Koiwa, T.; Watanabe, T. 5’ long terminal repeat (LTR)-selective methylation of latently infected HIV-1 provirus that is demethylated by reactivation signals. Retrovirology 2006, 3, 69. [Google Scholar] [CrossRef]
- Leung, D.C.; Lorincz, M.C. Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem. Sci. 2012, 37, 127–133. [Google Scholar] [CrossRef]
- Chavez, L.; Kauder, S.; Verdin, E. In vivo, in vitro, and in silico analysis of methylation of the HIV-1 provirus. Methods 2011, 53, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bednarik, D.P.; Mosca, J.D.; Raj, N.B. Methylation as a modulator of expression of human immunodeficiency virus. J. Virol. 1987, 61, 1253–1257. [Google Scholar] [CrossRef]
- Bednarik, D.; Cook, J.; Pitha, P. Inactivation of the HIV LTR by DNA CpG methylation: Evidence for a role in latency. EMBO J. 1990, 9, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kauder, S.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.; Nelson, A.; Moir, S.; Chun, T.-W.; Fauci, A. Paucity of HIV DNA Methylation in Latently Infected, Resting CD4+ T Cells from Infected Individuals Receiving Antiretroviral Therapy. J. Virol. 2012, 86, 1–10. [Google Scholar] [CrossRef]
- Ho, Y.-C.; Shan, L.; Hosmane, N.; Wang, J.; Laskey, S.; Rosenbloom, D.; Lai, J.; Blankson, J.; Siliciano, J.; Siliciano, R. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Weber, S.; Weiser, B.; Kemal, K.S.; Burger, H.; Ramirez, C.M.; Korn, K.; Anastos, K.; Kaul, R.; Kovacs, C.; Doerfler, W. Epigenetic analysis of HIV-1 proviral genomes from infected individuals: Predominance of unmethylated CpG’s. Virology 2014, 449, 181–189. [Google Scholar] [CrossRef][Green Version]
- Palacios, J.A.; Pérez-Piñar, T.; Toro, C.; Sanz-Minguela, B.; Moreno, V.; Valencia, E.; Gómez-Hernando, C.; Rodés, B. Long-term nonprogressor and elite controller patients who control viremia have a higher percentage of methylation in their HIV-1 proviral promoters than aviremic patients receiving highly active antiretroviral therapy. J. Virol. 2012, 86, 13081–13084. [Google Scholar] [CrossRef][Green Version]
- Trejbalova, K.; Kovarova, D.; Blazkova, J.; Machala, L.; Jilich, D.; Weber, J.; Kucerova, D.; Vencálek, O.; Hirsch, I.; Hejnar, J. Development of 5’ LTR DNA methylation of latent HIV-1 provirus in cell line models and in long-term-infected individuals. Clin. Epigenet. 2016, 8, 1–20. [Google Scholar] [CrossRef]
- Cortés-Rubio, C.N.; Salgado-Montes de Oca, G.; Prado-Galbarro, F.J.; Matías-Florentino, M.; Murakami-Ogasawara, A.; Kuri-Cervantes, L.; Carranco-Arenas, A.P.; Ormsby, C.E.; Cortés-Rubio, I.K.; Reyes-Terán, G.; et al. Longitudinal variation in human immunodeficiency virus long terminal repeat methylation in individuals on suppressive antiretroviral therapy. Clin. Epigenet. 2019, 11, 1–17. [Google Scholar] [CrossRef]
- Csankovszki, G.; Nagy, A.; Jaenisch, R. Synergism of Xist Rna, DNA Methylation, and Histone Hypoacetylation in Maintaining X Chromosome Inactivation. J. Cell Biol. 2001, 153, 773–784. [Google Scholar] [CrossRef]
- Clark, S.J.; Melki, J. DNA methylation and gene silencing in cancer: Which is the guilty party? Oncogene 2002, 21, 5380–5387. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase Methylates DNA Processively with High Preference for Hemimethylated Target Sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Song, J.; Wang, Y.; Zhao, Y.; Guda, K.; Yang, S.; Kao, H.-Y.Y.; Xu, Y.; Willis, J.; Markowitz, S.D.; et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 2010, 3, 1–11. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.Y.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Triboulet, R.; Mari, B.; Lin, Y.-L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Sun, G.; Li, H.; Wu, X.; Covarrubias, M.; Scherer, L.; Meinking, K.; Luk, B.; Chomchan, P.; Alluin, J.; Gombart, A.F.; et al. Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res. 2012, 40, 2181–2196. [Google Scholar] [CrossRef]
- Moyano, M.; Stefani, G. piRNA involvement in genome stability and human cancer. J. Hematol. Oncol. 2015, 8, 1–10. [Google Scholar] [CrossRef]
- He, Z.; Jing, S.; Yang, T.; Chen, J.; Huang, F.; Zhang, W.; Peng, Z.; Liu, B.; Ma, X.; Wu, L.; et al. PIWIL4 Maintains HIV-1 Latency by Enforcing Epigenetically Suppressive Modifications on the 5′ Long Terminal Repeat. J. Virol. 2020, 94, 1–22. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Huan, C.; Yang, J.; Li, Z.; Zheng, B.; Wang, Y. NF-KB-Interacting Long Noncoding RNA Regulates HIV-1 Replication and Latency by Repressing NF-KB Signaling. J. Virol. 2020, 94, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Sun, W.W.; Li, L.; Ma, L.; Sun, L.; Jin, X.; Li, T.; Hou, W.; Wang, J.H. Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter. Nucleic Acids Res. 2019, 47, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Houzet, L.; Yeung, M.L.; de Lame, V.; Desai, D.; Smith, S.M.; Jeang, K.-T. MicroRNA profile changes in human immunodeficiency virus type 1 (HIV-1) seropositive individuals. Retrovirology 2008, 5, 118. [Google Scholar] [CrossRef] [PubMed]
- Bignami, F.; Pilotti, E.; Bertoncelli, L.; Ronzi, P.; Gulli, M.; Marmiroli, N.; Magnani, G.; Pinti, M.; Lopalco, L.; Mussini, C.; et al. Stable changes in CD4+ T lymphocyte miRNA expression after exposure to HIV-1. Blood 2012, 119, 6259–6267. [Google Scholar] [CrossRef] [PubMed]
- Barichievy, S.; Naidoo, J.; Mhlanga, M.M. Non-coding RNAs and HIV: Viral manipulation of host dark matter to shape the cellular environment. Front. Genet. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Saayman, S.; Ackley, A.; Turner, A.-M.W.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-Encoded Antisense Long Noncoding RNA Epigenetically Regulates Viral Transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef]
- Zapata, J.C.; Campilongo, F.; Barclay, R.A.; DeMarino, C.; Iglesias-Ussel, M.D.; Kashanchi, F.; Romerio, F. The Human Immunodeficiency Virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology 2017, 506, 34–44. [Google Scholar] [CrossRef]
- Barclay, R.A.; Schwab, A.; Demarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from uninfected cells activate transcription of latent HIV-1. J. Biol. Chem. 2017, 292, 11682–11701. [Google Scholar] [CrossRef]
- Pinto, D.O.; Scott, T.A.; Demarino, C.; Pleet, M.L.; Vo, T.T.; Saifuddin, M.; Kovalskyy, D.; Erickson, J.; Cowen, M.; Barclay, R.A.; et al. Effect of transcription inhibition and generation of suppressive viral non-coding RNAs. Retrovirology 2019, 16, 1–17. [Google Scholar] [CrossRef]
- McDonel, P.; Costello, I.; Hendrich, B. Keeping things quiet: Roles of NuRD and Sin3 co-repressor complexes during mammalian development. Int. J. Biochem. Cell Biol. 2009, 41, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Pombo, A.; Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015, 16, 245–257. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Casartelli, N.; Pellin, D.; Rizzi, E.; Souque, P.; Severgnini, M.; Di Serio, C.; Fricke, T.; Diaz-Griffero, F.; Zimmer, C.; et al. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Lucic, B.; Chen, H.-C.; Kuzman, M.; Zorita, E.; Wegner, J.; Minneker, V.; Wang, W.; Fronza, R.; Laufs, S.; Schmidt, M.; et al. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Lusic, M.; Marini, B.; Ali, H.; Lucic, B.; Luzzati, R.; Giacca, M. Proximity to PML nuclear bodies regulates HIV-1 latency in CD4+ T cells. Cell Host Microbe 2013, 13, 665–677. [Google Scholar] [CrossRef]
- Raices, M.; D’Angelo, M.A. Nuclear pore complexes and regulation of gene expression. Curr. Opin. Cell Biol. 2017, 46, 26–32. [Google Scholar] [CrossRef]
- Sun, W.W.; Jiao, S.; Sun, L.; Zhou, Z.; Jin, X.; Wang, J.H. SUN2 modulates HIV-1 infection and latency through association with lamin A/C to maintain the repressive Chromatin. MBio 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An Unhealthy Constellation. Cell Host Microbe Rev. 2016, 19, 304–310. [Google Scholar] [CrossRef]
- Wong, J.K.; Yukl, S.A. Tissue Reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Herskovitz, J.; Gendelman, H.E. HIV and the Macrophage: From Cell Reservoirs to Drug Delivery to Viral Eradication. J. Neuroimmune Pharmacol. 2019, 14, 52–67. [Google Scholar] [CrossRef]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Beddall, M.H.; Yu, D.; Iyer, S.R.; Marsh, J.W.; Wu, Y. Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology 2008, 372, 300–312. [Google Scholar] [CrossRef]
- Marban, C.; Suzanne, S.; Dequiedt, F.; De Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Rohr, O.; Marban, C.; Aunis, D.; Schaeffer, E. Regulation of HIV-1 gene transcription: From lymphocytes to microglial cells. J. Leukoc. Biol. 2003, 74, 736–749. [Google Scholar] [CrossRef]
- Marban, C.; Redel, L.; Suzanne, S.; Van Lint, C.; Lecestre, D.; Chasserot-Golaz, S.; Leid, M.; Aunis, D.; Schaeffer, E.; Rohr, O. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res. 2005, 33, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; Van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Cherrier, T.; Le Douce, V.; Eilebrecht, S.; Riclet, R.; Marban, C.; Dequiedt, F.; Goumon, Y.; Paillart, J.-C.; Mericskay, M.; Parlakian, A.; et al. CTIP2 is a negative regulator of P-TEFb. Proc. Natl. Acad. Sci. USA 2013, 110, 12655–12660. [Google Scholar] [CrossRef]
- Eilebrecht, S.; Le Douce, V.; Riclet, R.; Targat, B.; Hallay, H.; Van Driessche, B.; Schwartz, C.; Robette, G.; Van Lint, C.; Rohr, O.; et al. HMGA1 recruits CTIP2-repressed P-TEFb to the HIV-1 and cellular target promoters. Nucleic Acids Res. 2014, 42, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Le Douce, V.; Forouzanfar, F.; Eilebrecht, S.; Van Driessche, B.; Ait-Ammar, A.; Verdikt, R.; Kurashige, Y.; Marban, C.; Gautier, V.; Candolfi, E.; et al. HIC1 controls cellular- and HIV-1- gene transcription via interactions with CTIP2 and HMGA1. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Chen, W.Y.; Wang, D.H.; Chiu Yen, R.; Luo, J.; Gu, W.; Baylin, S.B. Tumor Suppressor HIC1 Directly Regulates SIRT1 to Modulate p53-Dependent DNA-Damage Responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Bellefroid, M.; Daouad, F.; Martinelli, V.; Van Assche, J.; Wallet, C.; Rodari, A.; De Rovere, M.; Fahrenkrog, B.; Schwartz, C.; et al. Inhibition of HIV-1 gene transcription by KAP1 in myeloid lineage. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Rouzine, I.M.; Weinberger, A.D.; Weinberger, L.S. An evolutionary role for HIV latency in enhancing viral transmission. Cell 2015, 160, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.-R.; et al. International AIDS Society global scientific strategy: Towards an HIV cure 2016. Nat. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J.-S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV- 1. EMBO Mol. Med. 2015, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Gagne, M.; Michaels, D.; Schiralli Lester, G.M.; Gummuluru, S.; Wong, W.W.; Henderson, A.J. Strength of T cell signaling regulates HIV-1 replication and establishment of latency. PLoS Pathog. 2019, 15, 1–21. [Google Scholar] [CrossRef]
- Lange, U.C.; Verdikt, R.; Ait-Ammar, A.; Van Lint, C. Epigenetic crosstalk in chronic infection with HIV-1. Semin. Immunopathol. 2020, 42, 187–200. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Latent Reservoirs | Persistent Reservoirs |
|---|---|---|
| Cell type | CD4+ T cells (e.g., TCM [6], TN [19], etc.) | Myeloid cells (e.g., macrophages [22,23]), Follicular dendritic cells [24], Epithelial cells? [25] Tissue-resident memory CD4+ T cells (TRM)? [26] |
| Causes for cART inefficiency | Low or no replication | Tissue localization and poor drug penetration |
| Time of establishment | Early during the infection | “Mature” reservoirs |
| Epigenetic mechanisms of HIV-1 gene regulation | Extensively studied | Poorly studied |
| Histone Deacetylases | ||
| Family | Members † | References |
| Class I | HDAC1, HDAC2, HDAC3, HDAC8 | [77,78,79,80,81,82] |
| Class IIa | HDAC4, HDAC5, HDAC7, HDAC9 | [83] |
| Class IIb | HDAC6, HDAC10 | |
| Class III (Sirtuins) | SIRT1-7 | [84] |
| Class IV | HDAC11 | |
| Histone Acetyltransferases | ||
| Family | Members † | References |
| GNAT | KAT2A/GCN5, KAT2B/PCAF | |
| MYST | KAT5/TIP60, KAT6A/MOZ/MYST3, KAT6B/MORF/MYST4, KAT7/HBO1/MYST2, KAT8/MOF/MYST1 | [86] |
| p300/CBP | KAT3B/p300, KAT3A/CBP | |
| Histone Methyltransferases | ||
| Family | Members † | References |
| HKMTs | ASH1L, DOT1L, EHMT1-2, EZH1, EZH2, MLL1-4, NSD1-3, SETD1A, SETD1B, SETD2, SETD7, SMYD2-3, SUV39H1-2, SUV420H1-2 | [87,88,89,90] [46,47] [91,92] |
| PRMTs | CARM1/PRMT4, PRMT1, PRMT5-7 | [93] |
| Histone Demethylases | ||
| Family | Members † | References |
| KDM | LSD1/KDM1A, LSD2/KDM1B | |
| JMJD | KDM2-8 classes that contain over 30 members, including MINA53/JMJD10 | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdikt, R.; Hernalsteens, O.; Van Lint, C. Epigenetic Mechanisms of HIV-1 Persistence. Vaccines 2021, 9, 514. https://doi.org/10.3390/vaccines9050514
Verdikt R, Hernalsteens O, Van Lint C. Epigenetic Mechanisms of HIV-1 Persistence. Vaccines. 2021; 9(5):514. https://doi.org/10.3390/vaccines9050514
Chicago/Turabian StyleVerdikt, Roxane, Olivier Hernalsteens, and Carine Van Lint. 2021. "Epigenetic Mechanisms of HIV-1 Persistence" Vaccines 9, no. 5: 514. https://doi.org/10.3390/vaccines9050514
APA StyleVerdikt, R., Hernalsteens, O., & Van Lint, C. (2021). Epigenetic Mechanisms of HIV-1 Persistence. Vaccines, 9(5), 514. https://doi.org/10.3390/vaccines9050514