
Delivering Two Tumour Antigens Survivin and Mucin-1 on Virus-Like Particles Enhances Anti-Tumour Immune Responses

 ,
,  , ,
, ,  , and
, and 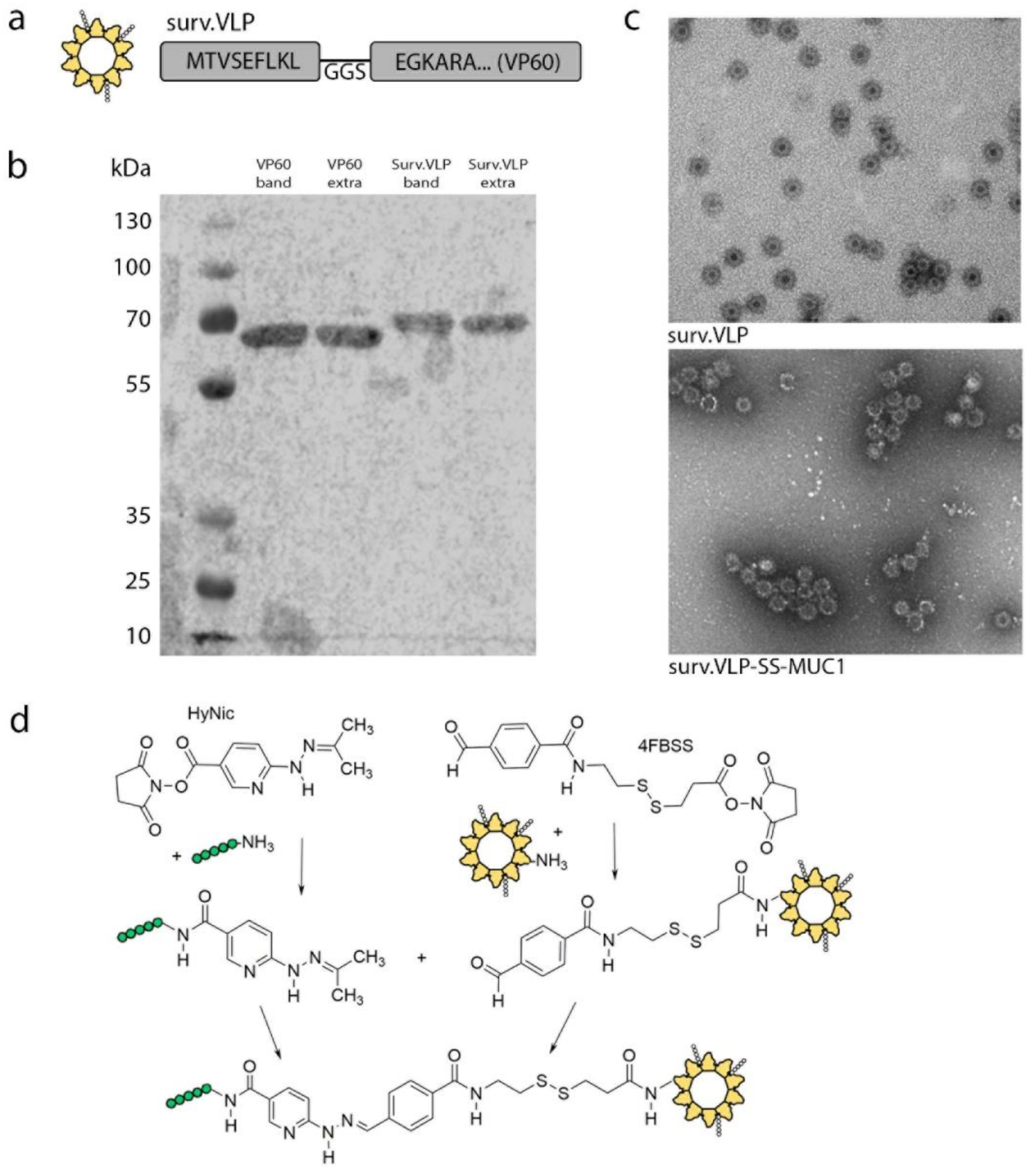{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of RHDV VLP
2.2. Confirmation of VLP Expression
2.3. Mass Spectrometry
2.4. Conjugation of MUC1 Peptide to VLP
2.5. Conjugate Cleavage Assay
2.6. Animals: Source and Ethics
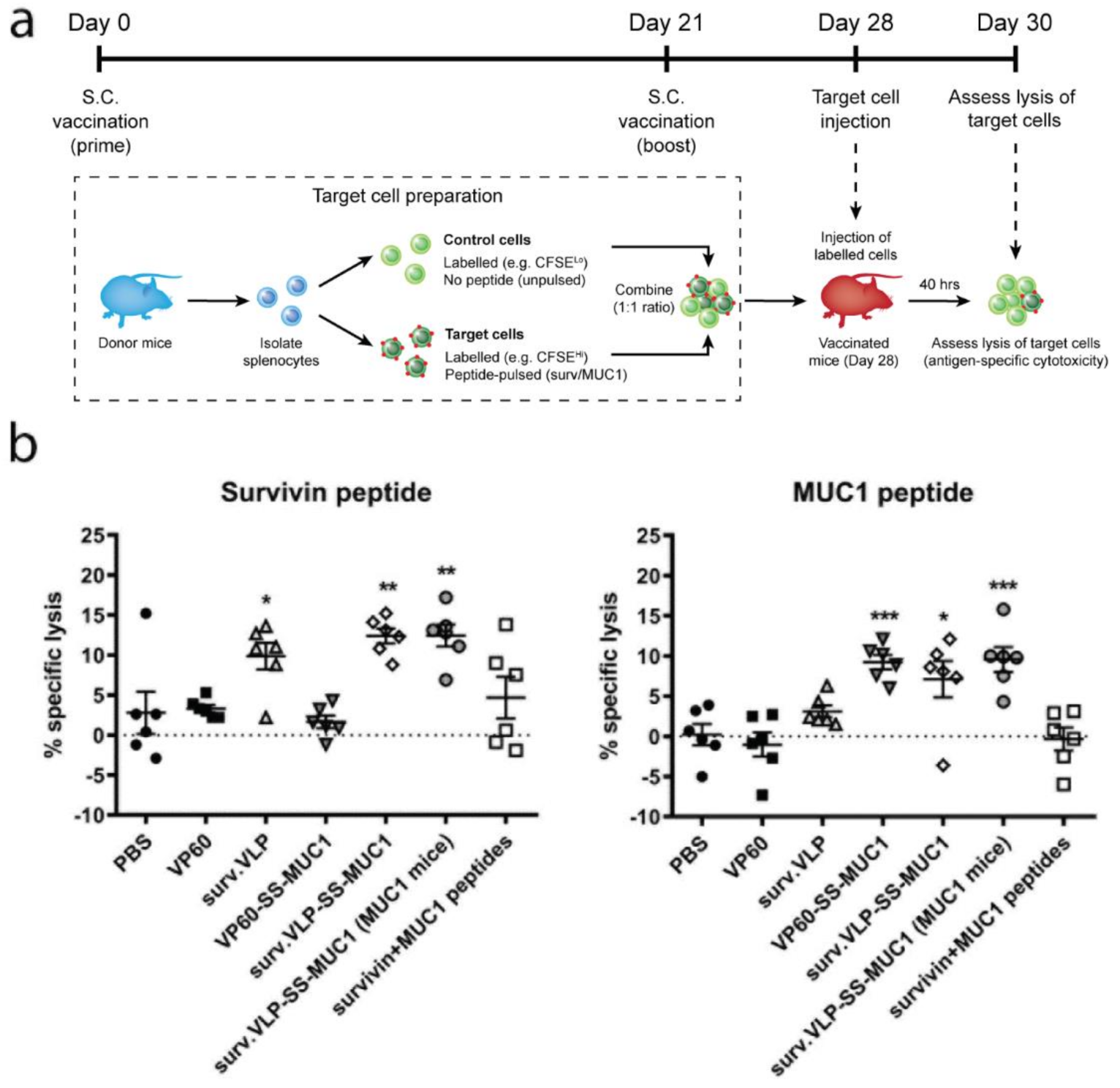
2.7. In Vivo Cytotoxicity
2.8. Tumour Cell Culture and Implantation
2.9. Tumour Cell Titration
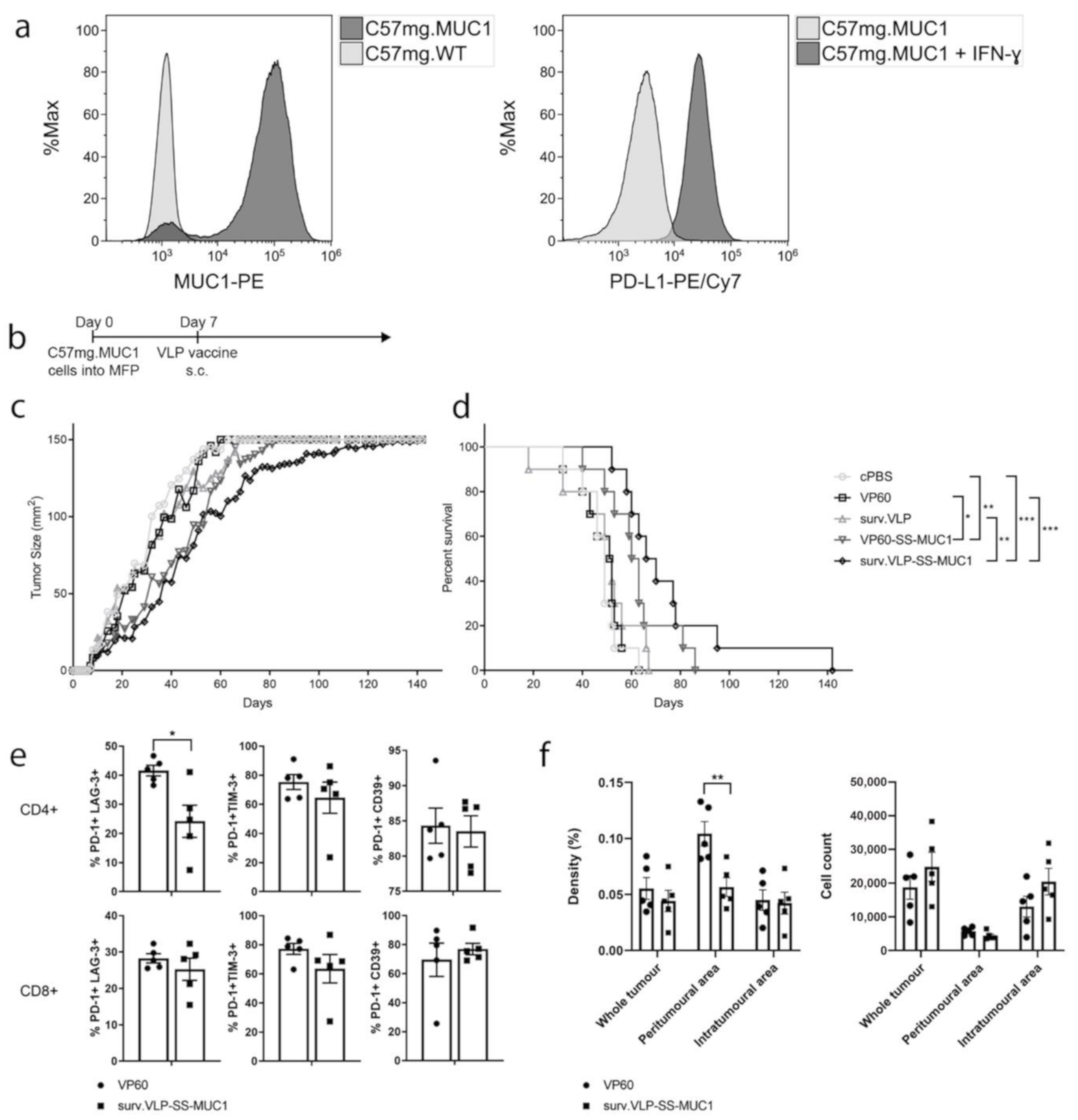
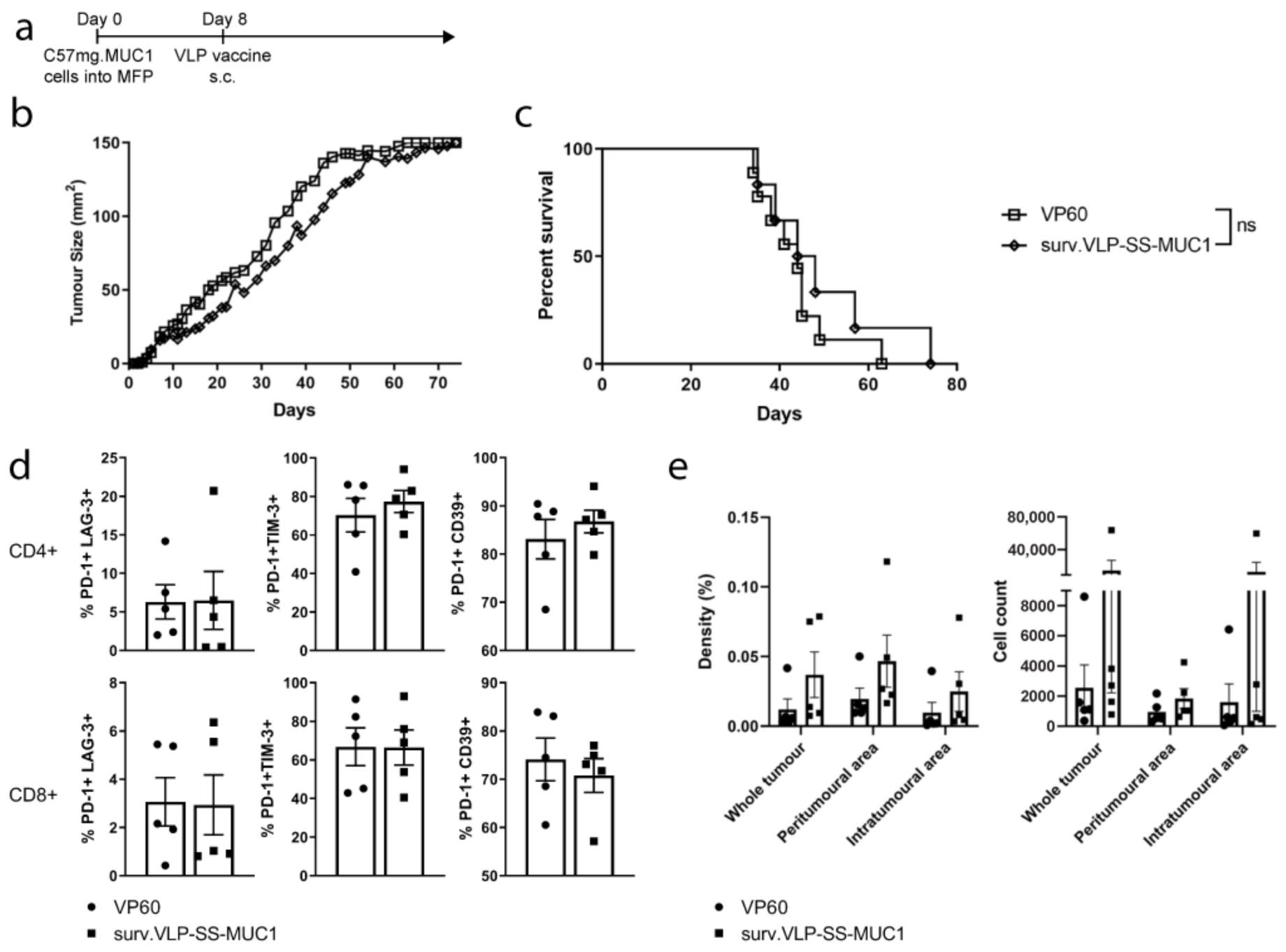
2.10. Therapeutic Tumour Trial
2.11. Tumour Infiltrating Lymphocytes
2.12. Statistical Analysis
3. Results
3.1. VLP Design and Conjugation
3.2. Cytotoxicity Assay
3.3. Therapeutic Tumour Trial in Wild-Type Mice
3.4. Therapeutic Tumour Trial in MUC1 Transgenic Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Mavratzas, A.; Seitz, J.; Smetanay, K.; Schneeweiss, A.; Jäger, D.; Fremd, C. Atezolizumab for use in PD-L1-positive unresectable, locally advanced or metastatic triple-negative breast cancer. Future Oncol. 2020, 16, 4439–4453. [Google Scholar] [CrossRef]
- Kramer, K.; Shields, N.J.; Poppe, V.; Young, S.L.; Walker, G.F. Intracellular Cleavable CpG Oligodeoxynucleotide-Antigen Conjugate Enhances Anti-tumor Immunity. Mol. Ther. 2017, 25, 62–70. [Google Scholar] [CrossRef][Green Version]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef]
- Zardavas, D.; Irrthum, A.; Swanton, C.; Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 2015, 12, 381–394. [Google Scholar] [CrossRef]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Vaca, L. Interaction Between Virus-Like Particles (VLPs) and Pattern Recognition Receptors (PRRs) From Dendritic Cells (DCs): Toward Better Engineering of VLPs. Front. Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef]
- Zeltins, A. Construction and Characterization of Virus-Like Particles: A Review. Mol. Biotechnol. 2013, 53, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Romanowski, B. Long term protection against cervical infection with the human papillomavirus: Review of currently available vaccines. Hum. Vaccines 2011, 7, 161–169. [Google Scholar] [CrossRef][Green Version]
- Peacey, M.; Wilson, S.; Baird, M.A.; Ward, V.K. Versatile RHDV virus-like particles: Incorporation of antigens by genetic modification and chemical conjugation. Biotechnol. Bioeng. 2007, 98, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.; Wall, K. Immunological Evaluation of Recent MUC1 Glycopeptide Cancer Vaccines. Vaccines 2016, 4, 25. [Google Scholar] [CrossRef]
- Domínguez-Romero, A.N.; Martínez-Cortés, F.; Munguía, M.E.; Odales, J.; Gevorkian, G.; Manoutcharian, K. Generation of multiepitope cancer vaccines based on large combinatorial libraries of survivin-derived mutant epitopes. Immunology 2020, 161, 123–138. [Google Scholar] [CrossRef]
- Antonilli, M.; Rahimi, H.; Visconti, V.; Napoletano, C.; Ruscito, I.; Zizzari, I.G.; Caponnetto, S.; Barchiesi, G.; Iadarola, R.; Pierelli, L.; et al. Triple peptide vaccination as consolidation treatment in women affected by ovarian and breast cancer: Clinical and immunological data of a phase I/II clinical trial. Int. J. Oncol. 2016, 48, 1369–1378. [Google Scholar] [CrossRef]
- Garg, H.; Hada, R.S.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Combination immunotherapy with Survivin and luteinizing hormone-releasing hormone fusion protein in murine breast cancer model. World J. Clin. Oncol. 2018, 9, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Xu, L.; Huang, W.; Tonn, T. Epitopes of MUC1 Tandem Repeats in Cancer as Revealed by Antibody Crystallography: Toward Glycopeptide Signature-Guided Therapy. Molecules 2018, 23, 1326. [Google Scholar] [CrossRef] [PubMed]
- Stergiou, N.; Glaffig, M.; Jonuleit, H.; Schmitt, E.; Kunz, H. Immunization with a synthetic human MUC1 glycopeptide vaccine against tumor-associated MUC1 breaks tolerance in human MUC1 transgenic mice. Chem. Med. Chem. 2017, 12, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, V.; Supekar, N.T.; Wei, J.; McCurry, D.B.; Dueck, A.C.; Kosiorek, H.E.; Trivedi, P.P.; Bradley, J.M.; Madsen, C.S.; Pathangey, L.B.; et al. MUC1 Vaccines, Comprised of Glycosylated or Non-Glycosylated Peptides or Tumor-Derived MUC1, Can Circumvent Immunoediting to Control Tumor Growth in MUC1 Transgenic Mice. PLoS ONE 2016, 11, e0145920. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef]
- Pathangey, L.B.; Lakshminarayanan, V.; Suman, V.J.; Pockaj, B.A.; Mukherjee, P.; Gendler, S.J. Aberrant Glycosylation of Anchor-Optimized MUC1 Peptides Can Enhance Antigen Binding Affinity and Reverse Tolerance to Cytotoxic T Lymphocytes. Biomolecules 2016, 6, 31. [Google Scholar] [CrossRef]
- Jha, K.; Shukla, M.; Pandey, M. Survivin expression and targeting in breast cancer. Surg. Oncol. 2012, 21, 125–131. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Zhang, H.-H.; Liu, C.-L.; Wu, H.; Wang, P.; Xia, Q.; Zhang, L.-X.; Li, B.; Wu, J.-X.; Yu, B.; et al. Enhancement of survivin-specific anti-tumor immunity by adenovirus prime protein-boost immunity strategy with DDA/MPL adjuvant in a murine melanoma model. Int. Immunopharmacol. 2013, 17, 9–17. [Google Scholar] [CrossRef]
- Donaldson, B.; Al-Barwani, F.; Pelham, S.J.; Young, K.; Ward, V.K.; Young, S.L. Multi-target chimaeric VLP as a therapeutic vaccine in a model of colorectal cancer. J. Immunother. Cancer 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- McKee, S.J.; Young, V.L.; Clow, F.; Hayman, C.M.; Baird, M.A.; Hermans, I.F.; Young, S.L.; Ward, V.K. Virus-like particles and α-galactosylceramide form a self-adjuvanting composite particle that elicits anti-tumor responses. J. Control Release 2012, 159, 338–345. [Google Scholar] [CrossRef]
- Rowse, G.J.; Tempero, R.M.; VanLith, M.L.; Hollingsworth, M.A.; Gendler, S.J. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998, 58, 315–321. [Google Scholar] [PubMed]
- Hofmann, U.B.; Voigt, H.; Andersen, M.H.; Straten, P.T.; Becker, J.C.; Eggert, A.O. Identification and characterization of survivin-derived H-2Kb-restricted CTL epitopes. Eur. J. Immunol. 2009, 39, 1419–1424. [Google Scholar] [CrossRef]
- Cai, H.; Sun, Z.-Y.; Chen, M.-S.; Zhao, Y.-F.; Kunz, H.; Li, Y.-M. Synthetic Multivalent Glycopeptide-Lipopeptide Antitumor Vaccines: Impact of the Cluster Effect on the Killing of Tumor Cells. Angew. Chem. Int. Ed. 2014, 53, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aal, A.-B.M.; Lakshminarayanan, V.; Thompson, P.; Supekar, N.; Bradley, J.M.; Wolfert, M.A.; Cohen, P.A.; Gendler, S.J.; Boons, G. Immune and anticancer responses elicited by fully synthetic aberrantly glycosylated MUC1 tripartite vaccines modified by a TLR2 or TLR9 agonist. Chembiochem 2014, 15, 1508–1513. [Google Scholar] [CrossRef]
- Kobukai, S.; Kremers, G.-J.; Cobb, J.G.; Baheza, R.; Xie, J.; Kuley, A.; Zhu, M.; Pham, W. Induction of Antitumor Immunity by Dendritic Cells Loaded with Membrane-Translocating Mucin 1 Peptide Antigen. Transl. Oncol. 2011, 4, 1–8. [Google Scholar] [CrossRef][Green Version]
- Lakshminarayanan, V.; Thompson, P.; Wolfert, M.A.; Buskas, T.; Bradley, J.M.; Pathangey, L.B.; Madsen, C.S.; Cohen, P.A.; Gendler, S.J.; Boons, G.-J. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc. Natl. Acad. Sci. USA 2012, 109, 261–266. [Google Scholar] [CrossRef]
- Win, S.J.; Ward, V.K.; Dunbar, P.R.; Young, S.L.; Baird, M.A. Cross-presentation of epitopes on virus-like particles via the MHC I receptor recycling pathway. Immunol. Cell Biol. 2011, 89, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Peers-Adams, A.; Win, S.J.; Scullion, S.; Wilson, M.; Young, V.L.; Jennings, P.; Ward, V.K.; Baird, M.A.; Young, S.L. Antigen Incorporated In Virus-like Particles Is Delivered to Specific Dendritic Cell Subsets That Induce An Effective Antitumor Immune Response In Vivo. J. Immunother. 2013, 36, 11–19. [Google Scholar] [CrossRef]
- Peacey, M.; Wilson, S.; Perret, R.; Ronchese, F.; Ward, V.K.; Young, V.; Young, S.L.; Baird, M.A. Virus-like particles from rabbit hemorrhagic disease virus can induce an anti-tumor response. Vaccine 2008, 26, 5334–5337. [Google Scholar] [CrossRef]
- Lacunza, E.; Baudis, M.; Colussi, A.G.; Segal-Eiras, A.; Croce, M.V.; Abba, M.C. MUC1 oncogene amplification correlates with protein overexpression in invasive breast carcinoma cells. Cancer Genet. Cytogenet. 2010, 201, 102–110. [Google Scholar] [CrossRef]
- Mukherjee, P.; Madsen, C.S.; Ginardi, A.R.; Tinder, T.L.; Jacobs, F.; Parker, J.; Agrawal, B.; Longenecker, B.M.; Gendler, S.J. Mucin 1-specific immunotherapy in a mouse model of spontaneous breast cancer. J. Immunother. 2003, 26, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Al-Barwani, F.; Donaldson, B.; Pelham, S.J.; Young, S.L.; Ward, V.K. Antigen delivery by virus-like particles for immunotherapeutic vaccination. Ther. Deliv. 2014, 5, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jarvis, D. Protein N-Glycosylation in the Baculovirus-Insect Cell System. Curr. Drug Targets 2007, 8, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Redman, J.M.; Gulley, J.L. Combining vaccines and immune checkpoint inhibitors to prime, expand, and facilitate effective tumor immunotherapy. Expert Rev. Vaccines 2018, 17, 697–705. [Google Scholar] [CrossRef]
- Madan, R.A.; Mohebtash, M.; Arlen, P.M.; Vergati, M.; Rauckhorst, M.; Steinberg, S.M.; Tsang, K.Y.; Poole, D.J.; Parnes, H.L.; Wright, J.J.; et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 501–508. [Google Scholar] [CrossRef]
- Duperret, E.K.; Wise, M.C.; Trautz, A.; Villarreal, D.O.; Ferraro, B.; Walters, J.; Yan, J.; Khan, A.; Masteller, E.; Humeau, L.; et al. Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol. Ther. 2018, 26, 435–445. [Google Scholar] [CrossRef]
- Lopes, A.; Vanvarenberg, K.; Kos, Š.; Lucas, S.; Colau, D.; van den Eynde, B.; Préat, V.; Vandermeulen, G. Combination of immune checkpoint blockade with DNA cancer vaccine induces potent antitumor immunity against P815 mastocytoma. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.A.; Lewin, S.A.; Dranoff, G.; Mooney, D.J. Vaccines combined with immune checkpoint antibodies promote cytotoxic T-cell activity and tumor eradication. Cancer Immunol. Res. 2016, 4, 95–100. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. Npj Vaccines 2019, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, K.; Young, V.L.; Donaldson, B.C.; Woodall, M.J.; Shields, N.J.; Walker, G.F.; Ward, V.K.; Young, S.L. Delivering Two Tumour Antigens Survivin and Mucin-1 on Virus-Like Particles Enhances Anti-Tumour Immune Responses. Vaccines 2021, 9, 463. https://doi.org/10.3390/vaccines9050463
Campbell K, Young VL, Donaldson BC, Woodall MJ, Shields NJ, Walker GF, Ward VK, Young SL. Delivering Two Tumour Antigens Survivin and Mucin-1 on Virus-Like Particles Enhances Anti-Tumour Immune Responses. Vaccines. 2021; 9(5):463. https://doi.org/10.3390/vaccines9050463
Chicago/Turabian StyleCampbell, Katrin, Vivienne L. Young, Braeden C. Donaldson, Matthew J. Woodall, Nicholas J. Shields, Greg F. Walker, Vernon K. Ward, and Sarah L. Young. 2021. "Delivering Two Tumour Antigens Survivin and Mucin-1 on Virus-Like Particles Enhances Anti-Tumour Immune Responses" Vaccines 9, no. 5: 463. https://doi.org/10.3390/vaccines9050463
APA StyleCampbell, K., Young, V. L., Donaldson, B. C., Woodall, M. J., Shields, N. J., Walker, G. F., Ward, V. K., & Young, S. L. (2021). Delivering Two Tumour Antigens Survivin and Mucin-1 on Virus-Like Particles Enhances Anti-Tumour Immune Responses. Vaccines, 9(5), 463. https://doi.org/10.3390/vaccines9050463