
The Impact of Mutations on the Pathogenic and Antigenic Activity of SARS-CoV-2 during the First Wave of the COVID-19 Pandemic: A Comprehensive Immunoinformatics Analysis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Sequence Alignment and Mutation Analysis
2.3. The Impact of Mutations on the Structural and Functional Properties of the Encoded Viral Proteins
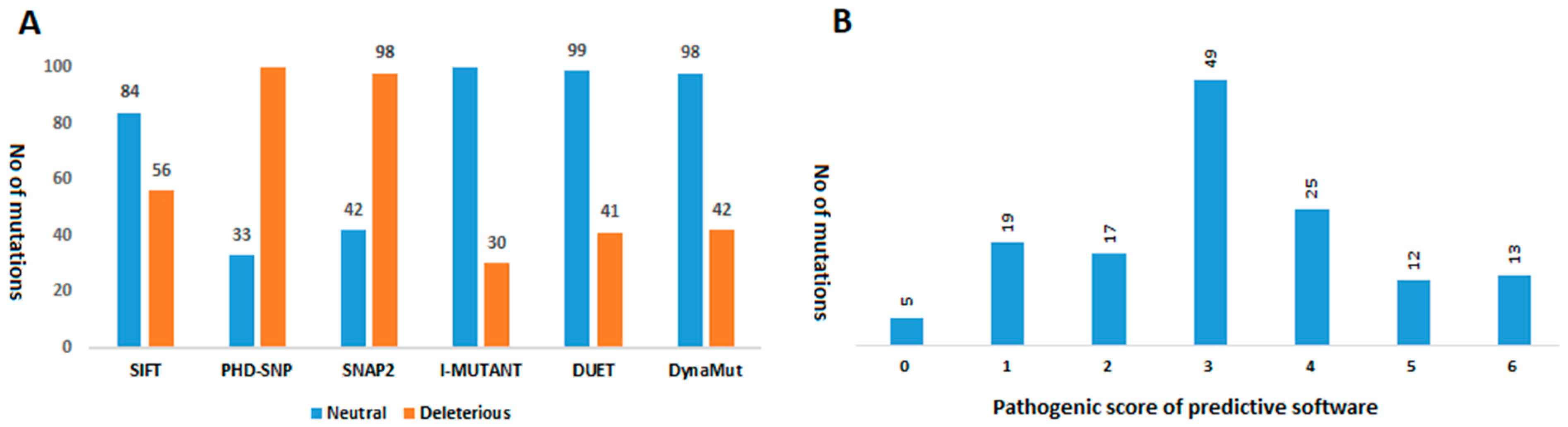
2.3.1. Predicting the Functional Impact of Mutations
2.3.2. Predicting Protein Stability Changes upon Mutations
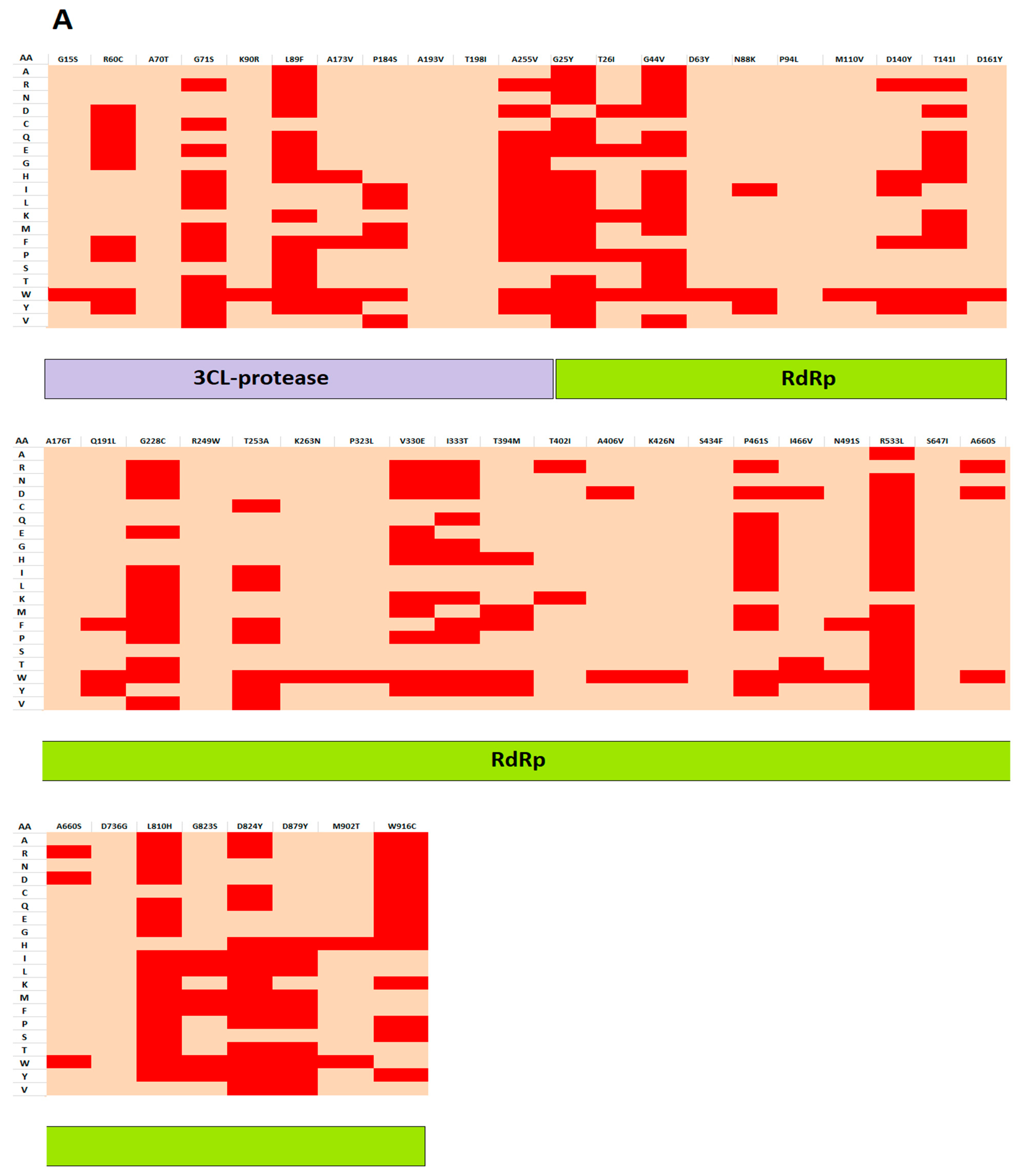
2.3.3. Mutation Screening
2.3.4. Normal Mode Analysis
2.3.5. Mapping the Ligand Binding Sites with Mutations
2.3.6. Epitope Mapping
2.3.7. Co-Occurring Mutations in Reported and Predicted Epitopes
3. Results
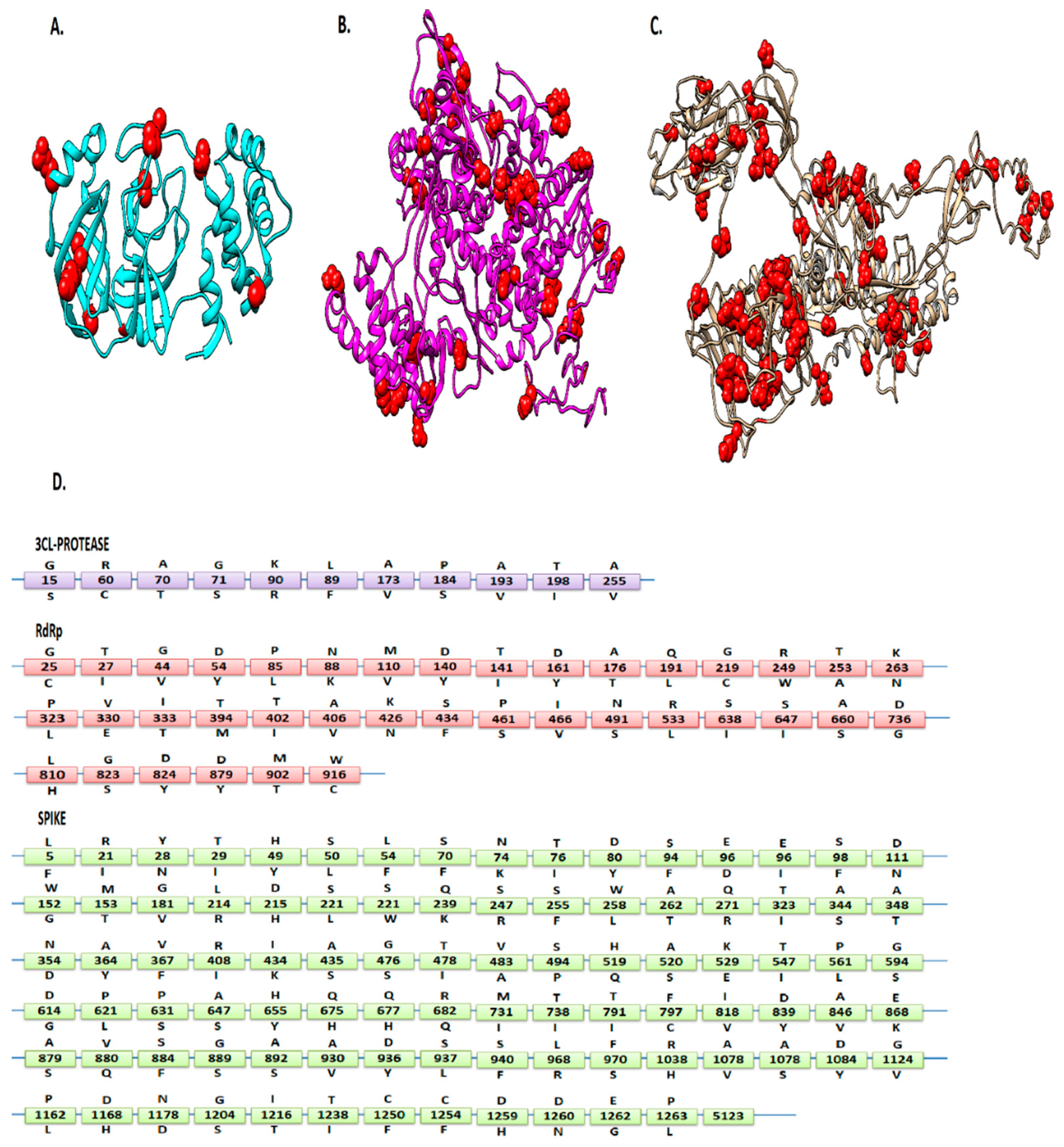
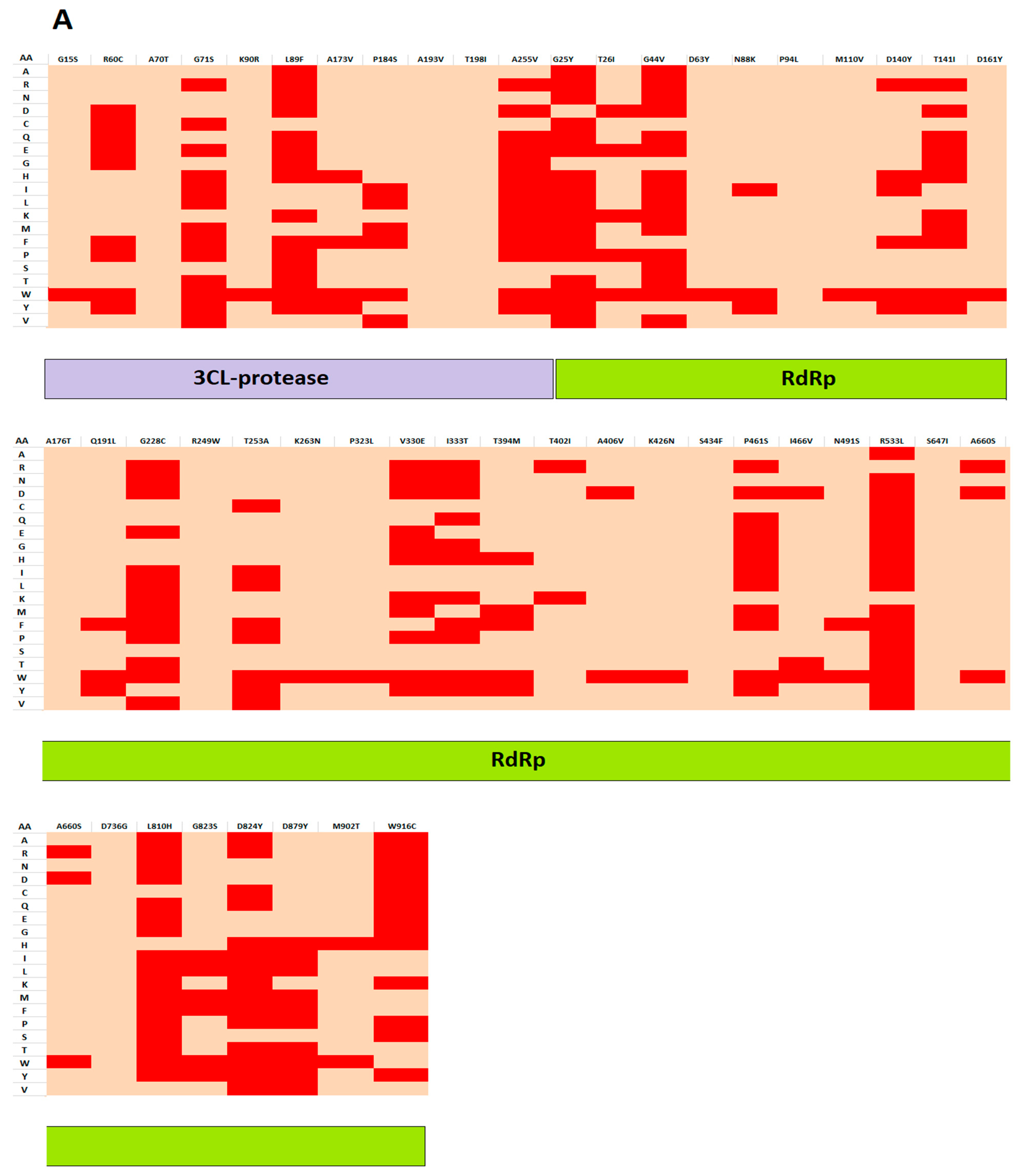
3.1. Mutations Residing in S, RdRp, and 3CLpro Sequences
3.2. Analyzing the Effect of Mutations on Structural and Functional Stability of the Respective Proteins
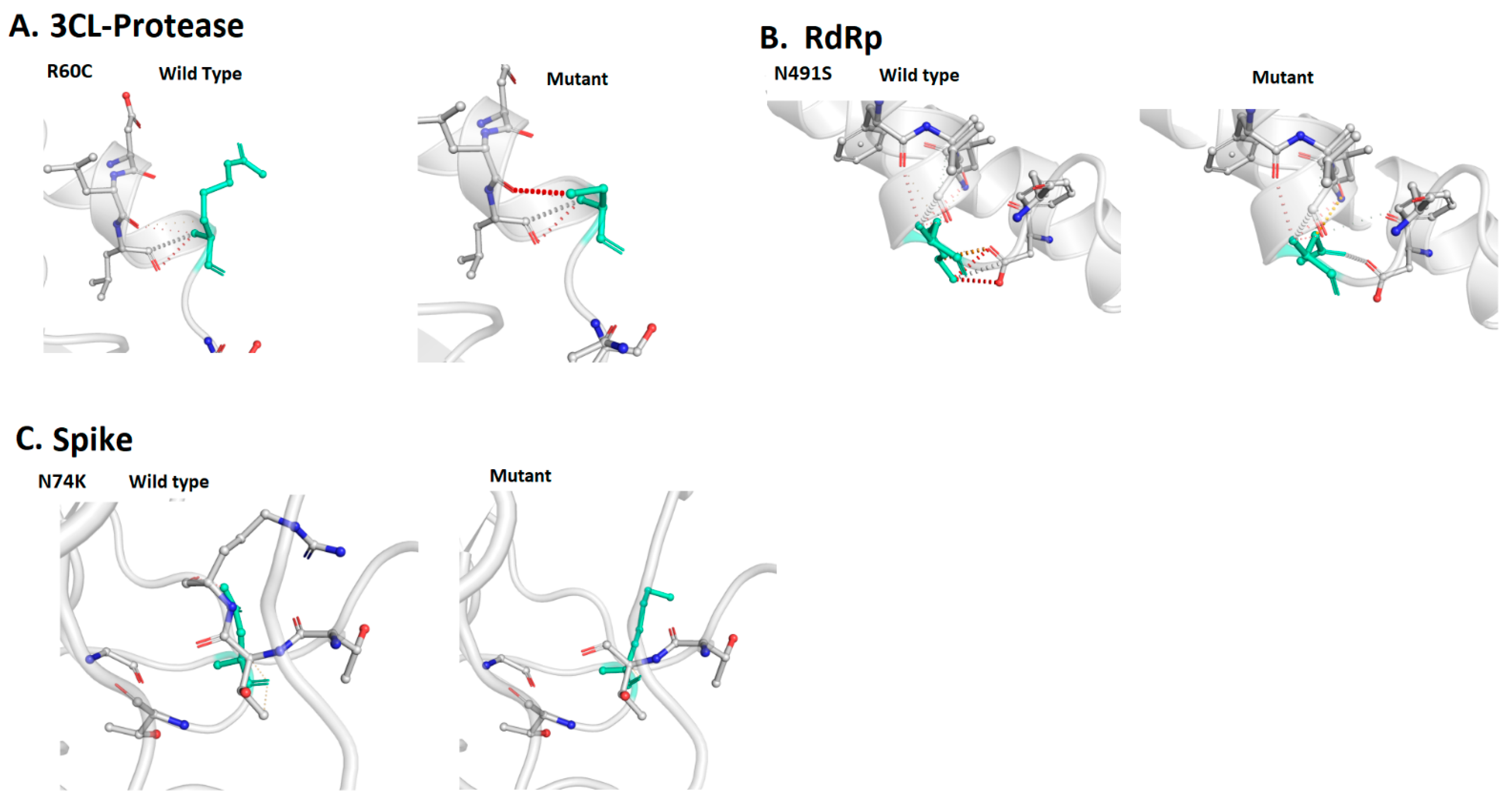
3.3. Localization of the Deleterious Mutations within the Binding Sites of Viral Proteins
3.4. Normal Mode Analysis of Highly Deleterious Mutations
3.5. Overlap of the Reported Mutations within the Predicted Epitopes
3.6. Estimating the Antigenicity of Epitopes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pachetti, M.; Marini, B.; Benedetti, F.; Ciudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. SARS-CoV-2: An emerging coronavirus that causes a global threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Petrone, M.E.; Holmes, E.C. We shouldn’t worry when a virus mutates during disease outbreaks. Nat. Microbiol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-R.; Liu, Y.-M.; Tseng, Y.-C.; Ma, C. Better influenza vaccines: An industry perspective. J. Biomed. Sci. 2020, 27, 33. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene. Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta. Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Ristevski, B.; Chen, M. Big data analytics in medicine and healthcare. J. Integr. Bioinf. 2018, 15, 20170030. [Google Scholar] [CrossRef]
- Samad, F.A.; Suliman, B.A.; Basha, S.H.; Manivasagam, T.; Essa, M.M. A comprehensive In Silico analysis on the structural and functional impact of SNPs in the congenital heart defects associated with NKX2-5 gene—A molecular dynamic simulation approach. PLoS ONE 2016, 11, e0153999. [Google Scholar]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.J.; Ribeiro, D.; Moreira, L.; Amaral, O. In silico analysis of missense mutations as a first step in functional studies: Examples from two sphingolipidoses. Int. J. Mol. Sci. 2018, 19, 3409. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.F.; Chien, C.S.; Yarmishyn, A.A.; Lin, Y.Y.; Luo, Y.H.; Lin, Y.T.; Lai, W.Y.; Yang, D.M.; Chou, S.J.; Yang, Y.P.; et al. A review of SARS-CoV-2 and the ongoing clinical trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||||
| Mutations | SIFT | PHD-SNP | SNAP2 | i-Mutant | DUET | DynaMut | SCORE | |
| 3CL-protease | G15S | - | - | √ | √ | √ | √ | 4 |
| R60C | √ | √ | √ | √ | √ | √ | 6 | |
| A70T | - | - | - | √ | √ | √ | 3 | |
| G71S | - | - | - | √ | √ | √ | 3 | |
| K90R | - | - | - | √ | √ | √ | 3 | |
| L89F | √ | √ | √ | √ | √ | √ | 6 | |
| A173V | - | - | - | - | - | - | 0 | |
| P184S | - | - | - | √ | √ | √ | 3 | |
| A193V | - | - | - | - | - | - | 0 | |
| T198I | - | - | - | - | - | - | 0 | |
| A255V | - | - | - | √ | √ | √ | 3 | |
| (B) | ||||||||
| RdRp | G25Y | √ | √ | √ | √ | √ | √ | 6 |
| T26I | - | - | - | √ | √ | √ | 3 | |
| G44V | - | - | √ | √ | √ | √ | 4 | |
| D63Y | √ | - | - | √ | √ | √ | 4 | |
| N88K | - | - | - | √ | √ | √ | 3 | |
| P94L | - | - | - | √ | √ | √ | 3 | |
| M110V | √ | - | - | √ | √ | √ | 4 | |
| D140Y | √ | √ | - | - | - | √ | 3 | |
| T141I | √ | - | - | √ | - | - | 2 | |
| D161Y | - | √ | - | - | - | - | 1 | |
| A176T | - | - | - | √ | √ | √ | 3 | |
| Q191L | √ | - | - | √ | - | - | 2 | |
| G228C | √ | √ | - | √ | √ | √ | 5 | |
| R249W | √ | - | √ | √ | √ | √ | 5 | |
| T262A | - | - | - | √ | - | - | 1 | |
| K263N | - | - | - | - | √ | √ | 2 | |
| P323L | - | - | √ | √ | - | - | 2 | |
| V330E | √ | √ | √ | √ | √ | √ | 6 | |
| I333T | √ | √ | - | √ | √ | √ | 5 | |
| T394M | - | - | √ | √ | - | - | 2 | |
| T402I | √ | - | - | √ | - | - | 2 | |
| A406V | - | - | - | √ | √ | √ | 3 | |
| K426N | √ | - | - | √ | √ | √ | 4 | |
| S434F | √ | - | - | - | √ | √ | 3 | |
| P461S | - | - | - | √ | √ | √ | 3 | |
| I466V | - | √ | √ | √ | √ | 4 | ||
| N491S | √ | - | - | √ | √ | √ | 4 | |
| R533L | √ | √ | - | √ | - | - | 3 | |
| S647I | √ | - | - | - | - | - | 1 | |
| A660S | √ | - | - | √ | √ | √ | 4 | |
| D736G | - | - | - | √ | - | - | 1 | |
| L810H | √ | √ | √ | √ | √ | √ | 6 | |
| G823S | - | √ | √ | √ | 3 | |||
| D824Y | √ | √ | √ | √ | √ | √ | 6 | |
| D879Y | - | - | √ | √ | - | - | 2 | |
| M902T | - | - | √ | √ | - | - | 2 | |
| W916C | √ | √ | - | √ | √ | √ | 5 | |
| (C) | ||||||||
| Spike | L5F | - | - | - | √ | √ | √ | 3 |
| P9L | - | - | √ | √ | √ | 3 | ||
| R21I | - | - | - | √ | - | - | 1 | |
| Y28N | - | - | √ | √ | √ | √ | 4 | |
| T29I | √ | - | - | - | - | - | 1 | |
| H49Y | √ | - | - | - | - | - | 1 | |
| S50L | - | - | - | √ | - | - | 1 | |
| L54F | - | - | - | √ | √ | √ | 3 | |
| S71F | √ | √ | - | √ | √ | √ | 5 | |
| N74K | √ | √ | √ | √ | √ | √ | 6 | |
| T76I | - | - | - | √ | - | - | 1 | |
| D80Y | - | - | √ | √ | - | - | 2 | |
| S94F | √ | - | - | - | √ | √ | 3 | |
| E96D | √ | - | - | √ | √ | √ | 4 | |
| E96I | √ | - | - | √ | √ | √ | 4 | |
| S98F | - | - | - | √ | √ | √ | 3 | |
| D111N | - | - | - | √ | √ | √ | 3 | |
| W152G | - | - | √ | √ | √ | √ | 4 | |
| M153T | - | - | - | √ | √ | √ | 3 | |
| G181V | - | - | - | √ | √ | √ | 3 | |
| R214L | - | - | √ | √ | - | - | 2 | |
| D215H | - | - | - | √ | - | - | 1 | |
| S221L | - | - | - | - | √ | - | 1 | |
| S221W | √ | - | - | - | - | - | 1 | |
| Q239K | - | - | - | - | √ | √ | 2 | |
| S247R | √ | - | - | - | - | - | 1 | |
| S255F | - | - | - | - | √ | √ | 2 | |
| W258L | - | - | - | √ | √ | √ | 3 | |
| A262T | - | - | - | √ | √ | √ | 3 | |
| Q271R | - | - | - | √ | √ | √ | 3 | |
| T323I | - | - | - | √ | - | 1 | ||
| A344S | - | - | - | √ | √ | √ | 3 | |
| A348T | √ | - | - | √ | √ | √ | 4 | |
| N354D | - | - | - | √ | √ | √ | 3 | |
| D364Y | √ | - | - | √ | - | - | 1 | |
| V367F | - | - | - | √ | √ | √ | 3 | |
| R408I | - | - | - | √ | - | - | 1 | |
| I434K | - | - | √ | √ | √ | √ | 4 | |
| A435S | √ | - | - | √ | √ | √ | 4 | |
| G476S | - | - | - | √ | √ | - | 2 | |
| T478I | - | - | - | √ | √ | √ | 3 | |
| V483A | - | - | - | √ | √ | √ | 3 | |
| S494P | - | - | - | - | √ | √ | 2 | |
| H519Q | - | - | - | - | - | - | 0 | |
| A520S | - | - | - | - | - | - | 0 | |
| K529E | - | - | - | √ | - | - | 1 | |
| T547I | - | - | √ | √ | - | - | 2 | |
| P561L | - | - | - | √ | - | - | 1 | |
| G594S | - | - | - | √ | √ | √ | 3 | |
| D614G | - | - | √ | √ | √ | √ | 3 | |
| P621S | - | - | - | √ | √ | √ | 3 | |
| P631S | - | - | - | √ | √ | √ | 3 | |
| A647S | - | - | - | √ | √ | √ | 3 | |
| H655Y | √ | √ | √ | - | - | - | 3 | |
| Q675H | - | - | √ | √ | √ | √ | 4 | |
| Q677H | - | - | - | √ | √ | √ | 3 | |
| R682Q | - | - | √ | √ | √ | √ | 4 | |
| M731I | - | - | - | √ | √ | √ | 3 | |
| T739I | √ | √ | √ | √ | - | - | 4 | |
| T791I | - | - | - | √ | - | - | 1 | |
| F797C | √ | √ | √ | √ | √ | √ | 6 | |
| I818V | - | - | - | √ | √ | √ | 3 | |
| D839Y | √ | √ | √ | - | √ | √ | 5 | |
| A846V | - | - | √ | √ | √ | √ | 4 | |
| V860Q | √ | √ | √ | √ | √ | √ | 6 | |
| E868K | - | - | - | √ | √ | √ | 3 | |
| A879S | - | - | - | √ | √ | √ | 3 | |
| S884F | √ | √ | √ | - | √ | √ | 5 | |
| G889S | √ | - | - | √ | √ | √ | 4 | |
| A892S | - | - | - | √ | √ | √ | 3 | |
| A930V | √ | √ | √ | √ | √ | √ | 6 | |
| D936Y | √ | √ | √ | √ | √ | √ | 6 | |
| S937L | √ | - | √ | - | √ | √ | 4 | |
| S940F | √ | √ | √ | - | √ | √ | 5 | |
| L966R | √ | √ | √ | - | √ | √ | 5 | |
| F970S | √ | √ | √ | √ | √ | √ | 6 | |
| A1078V | √ | √ | √ | - | - | - | 3 | |
| A1078S | - | - | - | - | √ | √ | 2 | |
| D1084Y | - | √ | √ | - | √ | √ | 4 | |
| G1124V | - | - | - | √ | √ | √ | 3 | |
| P1162L | - | √ | - | √ | √ | √ | 4 | |
| D1168H | √ | √ | √ | √ | √ | √ | 6 | |
| N1178D | √ | - | - | √ | √ | √ | 4 | |
| G1204S | - | - | - | √ | √ | √ | 3 | |
| I1216T | √ | √ | - | √ | √ | √ | 5 | |
| T1238I | √ | - | √ | - | - | - | 2 | |
| C1250F | √ | √ | √ | √ | √ | - | 5 | |
| C1254F | √ | √ | √ | √ | - | √ | 5 | |
| D1259H | √ | - | - | √ | √ | √ | 4 | |
| D1260N | - | - | - | √ | √ | √ | 3 | |
| E1262G | - | - | - | √ | √ | √ | 3 | |
| P1263L | √ | - | - | √ | √ | √ | 4 | |
| Protein | Epitope Position | Mutation Position | Name | Predicted Epitopes | Antigenicity (without Mutations) | Predicted Epitopes with Mutations | Antigenicity (with Mutations) |
|---|---|---|---|---|---|---|---|
| Spike MHCI | 69 | S71F | T1 | HVSGTNGTK | 1 | HVS/FGTNGTK | 0.6 |
| 515 | H519Q | T2 | FELLHAPAT | 0.5 | FELLH/QAPAT | 0.1 | |
| 515 | A520S | T3 | FELLHAPAT | 0.5 | FELLHA/SPAT | 0.2 | |
| 545 | T547I | T4 | GLTGTGVLT | 1 | GLT/IGTGVLT | 0.8 | |
| 612 | D614G | T5 | YQDVNCTEV | 1.6 | YQD/GVNCTEV | 1.3 | |
| 654 | H655Y | T6 | EHVNNSYEC | 1 | EH/YVNNSYEC | 0.9 | |
| 1210 | I1216T | T7 | IKWPWYIWL | 0.9 | IKWPWYI/TWL | 0.6 | |
| 1257 | E1262G | T8 | DEDDSEPVL | 0.5 | DEDDSE/GPVL | 0.33 | |
| Spike MHCII | 231 | Q239K | T9 | IGINITRFQ | 1.33 | IGINITRFQ/K | 1.2 |
| 318 | T323I | T10 | FRVQPTESI | 0.9 | FRVQPT/IESI | 1 | |
| 353 | N354D | T11 | WNRKRISNC | 0.5 | WN/DRKRISNC | 0.4 | |
| 512 | H519Q | T12 | VLSFELLHA | 1 | VLSFELLH/QA | 0.77 | |
| 512 | A520S | T13 | VLSFELLHA | 1 | VLSFELLHA/S | 0.8 | |
| 3CL-protease MHCI | 68 | A70T | T14 | VQAGNVQLR | 1.9 | VQA/TGNVQLR | 1.8 |
| 68 | G71S | T15 | VQAGNVQLR | 1.9 | VQAG/SNVQLR | 1.4 | |
| 3CL-protease MHCII | 57 | R60C | T16 | LLIRKSNHN | 0.7 | LLIR/CKSNHN | 0.3 |
| 67 | G71S | T17 | FLVQAGNVQ | 0.8 | FLVQAG/SNVQ | 0.7 | |
| RdRp MHCI | 18, 24 | G25Y | T18 | RLTPCGTGT | 1.1 | RLTPCGTG/YT | 0.6 |
| TGTSTDVVY | TG/YTSTDVVY | ||||||
| 18, 24 | T26I | T19 | RLTPCGTGT | 1.1 | RLTPCGTGT/I | 0.9 | |
| TGTSTDVVY | 0.7 | TGT/ISTDVVY | 0.3 | ||||
| 37 | G44V | T20 | IYNDKVAGF | 0.5 | IYNDKVAG/VF | 0.1 | |
| 90 | P94L | T21 | LKDCPAVAK | 0.6 | LKDCP/LAVAK | 0.5 | |
| 155 | D161Y | T22 | DYFNKKDWY | 1.2 | DYFNKKD/YWY | 0.3 | |
| 174 | A176T | T23 | VYANLGERV | 0.8 | VYA/TNLGERV | 0.1 | |
| 184 | Q191L | T24 | QALLKTVQF | 0.5 | QALLKTVQ/LF | 0.2 | |
| 400 | T402I | T25 | ALTNNVAFQ | 1.2 | ALT/INNVAFQ | 0.4 | |
| 429 | S434F | T26 | FKEGSSVEL | 0.6 | FKEGS/FSVEL | 0.2 | |
| 527 | R533L | T27 | LFAYTKRNV | 1 | LFAYTKR/LNV | 0.9 | |
| 897 | M902T | T28 | GHMLDMYSV | 0.4 | GHMLDM/TYSV | 0.1 | |
| RdRpMHCII | 37 | G44V | T29 | IYNDKVAGF | 0.5 | IYNDKVAG/VF | 0.1 |
| 241 | R249W | T30 | LMPILTLTR | 0.9 | LMPILTLTR/W | 1.1 | |
| 387 | T394M | T31 | LLLDKRTTC | 1.33 | LLLDKRTT/MC | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baloch, Z.; Ikram, A.; Hakim, M.S.; Awan, F.M. The Impact of Mutations on the Pathogenic and Antigenic Activity of SARS-CoV-2 during the First Wave of the COVID-19 Pandemic: A Comprehensive Immunoinformatics Analysis. Vaccines 2021, 9, 1410. https://doi.org/10.3390/vaccines9121410
Baloch Z, Ikram A, Hakim MS, Awan FM. The Impact of Mutations on the Pathogenic and Antigenic Activity of SARS-CoV-2 during the First Wave of the COVID-19 Pandemic: A Comprehensive Immunoinformatics Analysis. Vaccines. 2021; 9(12):1410. https://doi.org/10.3390/vaccines9121410
Chicago/Turabian StyleBaloch, Zulqarnain, Aqsa Ikram, Mohamad S. Hakim, and Faryal Mehwish Awan. 2021. "The Impact of Mutations on the Pathogenic and Antigenic Activity of SARS-CoV-2 during the First Wave of the COVID-19 Pandemic: A Comprehensive Immunoinformatics Analysis" Vaccines 9, no. 12: 1410. https://doi.org/10.3390/vaccines9121410
APA StyleBaloch, Z., Ikram, A., Hakim, M. S., & Awan, F. M. (2021). The Impact of Mutations on the Pathogenic and Antigenic Activity of SARS-CoV-2 during the First Wave of the COVID-19 Pandemic: A Comprehensive Immunoinformatics Analysis. Vaccines, 9(12), 1410. https://doi.org/10.3390/vaccines9121410