
Human Immunodeficiency Virus Type-1 (HIV-1) Transcriptional Regulation, Latency and Therapy in the Central Nervous System

 ,
,  ,
, 


Abstract
1. Introduction
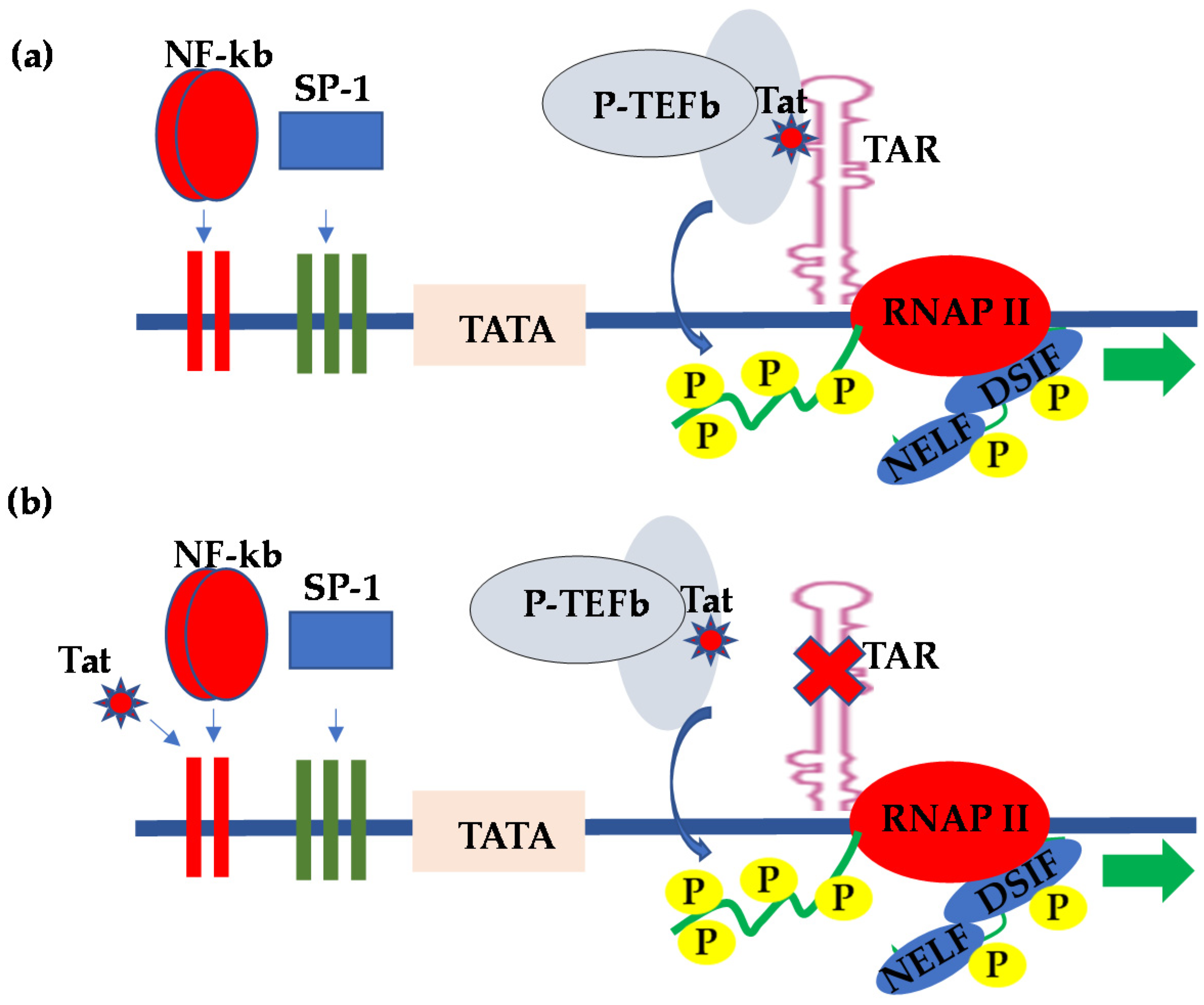
2. HIV Transcriptional Regulation in the CNS
3. HIV Latency in the CNS
4. Neuro-HIV Drugs and Therapy in the CNS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Tyagi, M. An Update on the HIV DNA Vaccine Strategy. Vaccines 2021, 9, 605. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–860. [Google Scholar] [CrossRef]
- Hokello, J.; Sharma, A.L.; Tyagi, M. Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome. Viruses 2020, 12, 868. [Google Scholar] [CrossRef]
- Fauci, A.S. Immunopathogenesis of HIV infection. J. Acquir. Immune Defic. Syndr. 1993, 6, 655–662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Poli, G.; Pantaleo, G.; Fauci, A.S. Immunopathogenesis of human immunodeficiency virus infection. Clin. Infect. Dis. 1993, 17 (Suppl. 1), S224–S229. [Google Scholar] [PubMed]
- Fauci, A.S. Multifactorial nature of human immunodeficiency virus disease: Implications for therapy. Science 1993, 262, 1011–1018. [Google Scholar] [CrossRef]
- Kramer-Hammerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef]
- Cosenza, M.A.; Zhao, M.L.; Si, Q.; Lee, S.C. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002, 12, 442–455. [Google Scholar] [CrossRef]
- Edara, V.V.; Ghorpade, A.; Borgmann, K. Insights into the Gene Expression Profiles of Active and Restricted Red/Green-HIV+ Human Astrocytes: Implications for Shock or Lock Therapies in the Brain. J. Virol. 2020, 94, e01563-19. [Google Scholar] [CrossRef]
- Li, G.H.; Anderson, C.; Jaeger, L.; Do, T.; Major, E.O.; Nath, A. Cell-to-cell contact facilitates HIV transmission from lymphocytes to astrocytes via CXCR4. AIDS 2015, 29, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.P.; Jorgen, G.G.H.; Pakkenberg, B. Preferential loss of large neocortical neurons during HIV infection: A study of the size distribution of neocortical neurons in the human brain. Brain Res. 1999, 828, 119–126. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef]
- Kingsman, S.M.; Kingsman, A.J. The regulation of human immunodeficiency virus type-1 gene expression. Eur. J. Biochem. 1996, 240, 491–507. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bourgeois, C.F.; Pearson, R.; Tyagi, M.; West, M.J.; Wong, J.; Wu, S.Y.; Chiang, C.M.; Karn, J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006, 25, 3596–3604. [Google Scholar] [CrossRef]
- Kumar, K.P.; Akoulitchev, S.; Reinberg, D. Promoter-proximal stalling results from the inability to recruit transcription factor IIH to the transcription complex and is a regulated event. Proc. Natl. Acad. Sci. USA 1998, 95, 9767–9772. [Google Scholar] [CrossRef]
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol. Cell. Biol. 2002, 22, 4622–4637. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 10, 4189–4196. [Google Scholar] [CrossRef]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Hurley, J.H. Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex. Proc. Natl. Acad. Sci. USA 2018, 115, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Tyagi, M. AP-1 and NF-kappaB synergize to transcriptionally activate latent HIV upon T-cell receptor activation. FEBS Lett. 2021, 595, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Morris, G.F.; Lockyer, J.M.; Lu, M.; Wang, Z.; Morris, C.B. Distinct transcriptional pathways of TAR-dependent and TAR-independent human immunodeficiency virus type-1 transactivation by Tat. Virology 1997, 235, 48–64. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, J.P.; Pomerantz, R.; Bagasra, O.; Chowdhury, M.; Rappaport, J.; Khalili, K.; Amini, S. TAR-independent transactivation by Tat in cells derived from the CNS: A novel mechanism of HIV-1 gene regulation. EMBO J. 1992, 11, 3395–3403. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Pomerantz, R.J.; Oakes, J.W.; Khalili, K.; Amini, S. A CNS-enriched factor that binds to NF-kappa B and is required for interaction with HIV-1 tat. Oncogene 1995, 10, 395–400. [Google Scholar]
- Kundu, M.; Ansari, S.A.; Chepenik, L.G.; Pomerantz, R.J.; Khalili, K.; Rappaport, J.; Amini, S. HIV-1 regulatory protein tat induces RNA binding proteins in central nervous system cells that associate with the viral trans-acting-response regulatory motif. J. Hum. Virol. 1999, 2, 72–80. [Google Scholar]
- Taylor, J.P.; Pomerantz, R.J.; Raj, G.V.; Kashanchi, F.; Brady, J.N.; Amini, S.; Khalili, K. Central nervous system-derived cells express a kappa B-binding activity that enhances human immunodeficiency virus type 1 transcription in vitro and facilitates TAR-independent transactivation by Tat. J. Virol. 1994, 68, 3971–3981. [Google Scholar] [CrossRef]
- Cupp, C.; Taylor, J.P.; Khalili, K.; Amini, S. Evidence for stimulation of the transforming growth factor beta 1 promoter by HIV-1 Tat in cells derived from CNS. Oncogene 1993, 8, 2231–2236. [Google Scholar]
- Sawaya, B.E.; Thatikunta, P.; Denisova, L.; Brady, J.; Khalili, K.; Amini, S. Regulation of TNFalpha and TGFbeta-1 gene transcription by HIV-1 Tat in CNS cells. J. Neuroimmunol. 1998, 87, 33–42. [Google Scholar] [CrossRef]
- Gray, L.R.; Cowley, D.; Welsh, C.; Lu, H.K.; Brew, B.J.; Lewin, S.R.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol. Psychiatry 2016, 21, 574–584. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M.; Simon, G.L. Mechanisms of HIV Transcriptional Regulation by Drugs of Abuse. Curr. HIV Res. 2016, 14, 442–454. [Google Scholar] [CrossRef]
- Tyagi, M.; Weber, J.; Bukrinsky, M.; Simon, G.L. The effects of cocaine on HIV transcription. J. Neurovirol. 2016, 22, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Swepson, C.; Ranjan, A.; Balasubramaniam, M.; Pandhare, J.; Dash, C. Cocaine Enhances HIV-1 Transcription in Macrophages by Inducing p38 MAPK Phosphorylation. Front. Microbiol. 2016, 7, 823. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Mahajan, S.D.; Bindukumar, B.; Sykes, D.; Schwartz, S.A.; Nair, M.P. Proteomic analysis of the effects of cocaine on the enhancement of HIV-1 replication in normal human astrocytes (NHA). Brain Res. 2006, 1123, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.; Farley, K.; El-Hage, N.; Aiamkitsumrit, B.; Fassnacht, R.; Kashanchi, F.; Ochem, A.; Simon, G.L.; Karn, J.; Hauser, K.F.; et al. Cocaine promotes both initiation and elongation phase of HIV-1 transcription by activating NF-kappaB and MSK1 and inducing selective epigenetic modifications at HIV-1 LTR. Virology 2015, 483, 185–202. [Google Scholar] [CrossRef]
- Sonti, S.; Sharma, A.L.; Tyagi, M. HIV-1 persistence in the CNS: Mechanisms of latency, pathogenesis and an update on eradication strategies. Virus Res. 2021, 303, 198523. [Google Scholar] [CrossRef]
- Sharma, A.L.; Hokello, J.; Sonti, S.; Zicari, S.; Sun, L.; Alqatawni, A.; Bukrinsky, M.; Simon, G.; Chauhan, A.; Daniel, R.; et al. CBF-1 Promotes the Establishment and Maintenance of HIV Latency by Recruiting Polycomb Repressive Complexes, PRC1 and PRC2, at HIV LTR. Viruses 2020, 12, 1040. [Google Scholar] [CrossRef]
- Sharma, A.L.; Hokello, J.; Tyagi, M. Circumcision as an Intervening Strategy against HIV Acquisition in the Male Genital Tract. Pathogens 2021, 10, 806. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef]
- Zicari, S.; Sharma, A.L.; Sahu, G.; Dubrovsky, L.; Sun, L.; Yue, H.; Jada, T.; Ochem, A.; Simon, G.; Bukrinsky, M.; et al. DNA dependent protein kinase (DNA-PK) enhances HIV transcription by promoting RNA polymerase II activity and recruitment of transcription machinery at HIV LTR. Oncotarget 2020, 11, 699–726. [Google Scholar] [CrossRef]
- Alqatawni, A.; Sharma, A.L.; Attilus, B.; Tyagi, M.; Daniel, R. Shedding Light on the Role of Extracellular Vesicles in HIV Infection and Wound Healing. Viruses 2020, 12, 584. [Google Scholar] [CrossRef]
- Hokello, J.; Sharma, A.L.; Dimri, M.; Tyagi, M. Insights into the HIV Latency and the Role of Cytokines. Pathogens 2019, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Tyagi, M. Combinatorial Use of Both Epigenetic and Non-Epigenetic Mechanisms to Efficiently Reactivate HIV Latency. Int. J. Mol. Sci. 2021, 22, 3697. [Google Scholar] [CrossRef] [PubMed]
- North, T.W.; Higgins, J.; Deere, J.D.; Hayes, T.L.; Villalobos, A.; Adamson, L.; Shacklett, B.L.; Schinazi, R.F.; Luciw, P.A. Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. J. Virol. 2010, 84, 2913–2922. [Google Scholar] [CrossRef][Green Version]
- Spudich, S.; Gisslen, M.; Hagberg, L.; Lee, E.; Liegler, T.; Brew, B.; Fuchs, D.; Tambussi, G.; Cinque, P.; Hecht, F.M.; et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J. Infect. Dis. 2011, 204, 753–760. [Google Scholar] [CrossRef]
- Lustig, G.; Cele, S.; Karim, F.; Derache, A.; Ngoepe, A.; Khan, K.; Gosnell, B.I.; Moosa, M.S.; Ntshuba, N.; Marais, S.; et al. T cell derived HIV-1 is present in the CSF in the face of suppressive antiretroviral therapy. PLoS Pathog. 2021, 17, e1009871. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Ferretti, F.; Peterson, J.; Lee, E.; Fuchs, D.; Boschini, A.; Gisslen, M.; Angoff, N.; Price, R.W.; Cinque, P.; et al. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well controlled plasma viral load. AIDS 2012, 26, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.K. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J. Neuroimmune Pharmacol. 2019, 14, 110–119. [Google Scholar] [CrossRef]
- Reu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, M.; Leon-Rivera, R.; Li, M.; Gama, L.; Clements, J.E.; Berman, J.W. Mechanisms of CNS Viral Seeding by HIV+ CD14+ CD16+ Monocytes: Establishment and Reseeding of Viral Reservoirs Contributing to HIV-Associated Neurocognitive Disorders. mBio 2017, 8, e01280-17. [Google Scholar] [CrossRef]
- Leon-Rivera, R.; Veenstra, M.; Donoso, M.; Tell, E.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Central Nervous System (CNS) Viral Seeding by Mature Monocytes and Potential Therapies To Reduce CNS Viral Reservoirs in the cART Era. mBio 2021, 12, e03633-20. [Google Scholar] [CrossRef] [PubMed]
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, G.N.; Alvarez-Carbonell, D.; Chateau, M.; Karn, J.; Cannon, P.M. HIV-1 infection of microglial cells in a reconstituted humanized mouse model and identification of compounds that selectively reverse HIV latency. J. Neurovirol. 2018, 24, 192–203. [Google Scholar] [CrossRef]
- Li, G.H.; Henderson, L.; Nath, A. Astrocytes as an HIV Reservoir: Mechanism of HIV Infection. Curr. HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef]
- Chiodi, F.; Fuerstenberg, S.; Gidlund, M.; Asjo, B.; Fenyo, E.M. Infection of brain-derived cells with the human immunodeficiency virus. J. Virol. 1987, 61, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, C.; Nath, A.; Amemiya, K.; Major, E.O. Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J. Virol. 1991, 65, 6094–6100. [Google Scholar] [CrossRef]
- Atwood, W.J.; Tornatore, C.S.; Traub, R.; Conant, K.; Drew, P.D.; Major, E.O. Stimulation of HIV type 1 gene expression and induction of NF-kappa B (p50/p65)-binding activity in tumor necrosis factor alpha-treated human fetal glial cells. AIDS Res. Hum. Retrovir. 1994, 10, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.M.; Durham, L.C.; Schwartz, L.; Seth, P.; Maric, D.; Major, E.O. Human immunodeficiency virus type 1 infection of human brain-derived progenitor cells. J. Virol. 2004, 78, 7319–7328. [Google Scholar] [CrossRef] [PubMed]
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder-pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 309. [Google Scholar] [CrossRef]
- Levine, A.J.; Service, S.; Miller, E.N.; Reynolds, S.M.; Singer, E.J.; Shapshak, P.; Martin, E.M.; Sacktor, N.; Becker, J.T.; Jacobson, L.P.; et al. Genome-wide association study of neurocognitive impairment and dementia in HIV-infected adults. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159B, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Mothobi, N.Z.; Brew, B.J. Neurocognitive dysfunction in the highly active antiretroviral therapy era. Curr. Opin. Infect. Dis. 2012, 25, 4–9. [Google Scholar] [CrossRef]
- Thomas, S.A. Anti-HIV drug distribution to the central nervous system. Curr. Pharm. Des. 2004, 10, 1313–1324. [Google Scholar] [CrossRef]
- Mdanda, S.; Ntshangase, S.; Singh, S.D.; Naicker, T.; Kruger, H.G.; Baijnath, S.; Govender, T. Zidovudine and Lamivudine as Potential Agents to Combat HIV-Associated Neurocognitive Disorder. Assay Drug Dev. Technol. 2019, 17, 322–329. [Google Scholar] [CrossRef]
- Mdanda, S.; Ntshangase, S.; Singh, S.D.; Naicker, T.; Kruger, H.G.; Baijnath, S.; Govender, T. Mass spectrometric investigations into the brain delivery of abacavir, stavudine and didanosine in a rodent model. Xenobiotica 2020, 50, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Salcedo-Gomez, P.M.; Yedidi, R.S.; Zhao, R.; Hayashi, H.; Hasegawa, K.; Nakamura, T.; Martyr, C.D.; Ghosh, A.K.; Mitsuya, H. Novel Central Nervous System (CNS)-Targeting Protease Inhibitors for Drug-Resistant HIV Infection and HIV-Associated CNS Complications. Antimicrob. Agents Chemother. 2019, 63, e00466-19. [Google Scholar] [CrossRef]
- Beck, S.E.; Queen, S.E.; Metcalf Pate, K.A.; Mangus, L.M.; Abreu, C.M.; Gama, L.; Witwer, K.W.; Adams, R.J.; Zink, M.C.; Clements, J.E.; et al. An SIV/macaque model targeted to study HIV-associated neurocognitive disorders. J. Neurovirol. 2018, 24, 204–212. [Google Scholar] [CrossRef]
- Dental, C.; Proust, A.; Ouellet, M.; Barat, C.; Tremblay, M.J. HIV-1 Latency-Reversing Agents Prostratin and Bryostatin-1 Induce Blood-Brain Barrier Disruption/Inflammation and Modulate Leukocyte Adhesion/Transmigration. J. Immunol. 2017, 198, 1229–1241. [Google Scholar] [CrossRef]
- Nair, M.; Jayant, R.D.; Kaushik, A.; Sagar, V. Getting into the brain: Potential of nanotechnology in the management of NeuroAIDS. Adv. Drug Deliv. Rev. 2016, 103, 202–217. [Google Scholar] [CrossRef]
- Jayant, R.D.; Atluri, V.S.; Agudelo, M.; Sagar, V.; Kaushik, A.; Nair, M. Sustained-release nanoART formulation for the treatment of neuroAIDS. Int. J. Nanomed. 2015, 10, 1077–1093. [Google Scholar] [CrossRef]
- Jayant, R.D.; Atluri, V.S.R.; Tiwari, S.; Pilakka-Kanthikeel, S.; Kaushik, A.; Yndart, A.; Nair, M. Novel nanoformulation to mitigate co-effects of drugs of abuse and HIV-1 infection: Towards the treatment of NeuroAIDS. J. Neurovirol. 2017, 23, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.S.; Reddy, M.K.; Horning, J.L.; Labhasetwar, V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials 2008, 29, 4429–4438. [Google Scholar] [CrossRef] [PubMed]
- Asahchop, E.L.; Meziane, O.; Mamik, M.K.; Chan, W.F.; Branton, W.G.; Resch, L.; Gill, M.J.; Haddad, E.; Guimond, J.V.; Wainberg, M.A.; et al. Reduced antiretroviral drug efficacy and concentration in HIV-infected microglia contributes to viral persistence in brain. Retrovirology 2017, 14, 47. [Google Scholar] [CrossRef]


{kind=link}
| Anatomic Sites | Specific Sites |
|---|---|
| Brain | |
| Primary Lymphatic tissue/organ | Thymus and Bone marrow |
| Non-Lymphoid tissues | Liver, Kidney, Adipose, reproductive tract and other |
| Secondary lymphatic tissue/organs | Tonsils, Adenoids, spleen, Mucosa-associated lymphatic tissues and lymph nodes |
| Peripheral Blood |
| Target | Name of Drugs |
|---|---|
| Entry inhibitors | Maraviroc (Celsentri), Enfuvirtide |
| Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) | Abacavir, Emtricitabine, Lamivudine, Tenofovir disoproxil, Tenofovir alafenamide, Zidovudine |
| Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | Doravirine, Efavirenz, Etravirine, Nevirapine, Rilpivirine |
| Integrase inhibitors | Bictegravir, Dolutegravir, Elvitegravir, Raltegravir, Cabotegravir |
| Protease inhibitors (PIs) | Atazanavir, Darunavir, Lopinavir, Ritonavir (Norvir), Cobicistat (Tybost) |
| Attachment and post-attachment inhibitors | Fostemsavir (Rukobia), Ibalizumab (Trogarzo) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hokello, J.; Sharma, A.L.; Tyagi, P.; Bhushan, A.; Tyagi, M. Human Immunodeficiency Virus Type-1 (HIV-1) Transcriptional Regulation, Latency and Therapy in the Central Nervous System. Vaccines 2021, 9, 1272. https://doi.org/10.3390/vaccines9111272
Hokello J, Sharma AL, Tyagi P, Bhushan A, Tyagi M. Human Immunodeficiency Virus Type-1 (HIV-1) Transcriptional Regulation, Latency and Therapy in the Central Nervous System. Vaccines. 2021; 9(11):1272. https://doi.org/10.3390/vaccines9111272
Chicago/Turabian StyleHokello, Joseph, Adhikarimayum Lakhikumar Sharma, Priya Tyagi, Alok Bhushan, and Mudit Tyagi. 2021. "Human Immunodeficiency Virus Type-1 (HIV-1) Transcriptional Regulation, Latency and Therapy in the Central Nervous System" Vaccines 9, no. 11: 1272. https://doi.org/10.3390/vaccines9111272
APA StyleHokello, J., Sharma, A. L., Tyagi, P., Bhushan, A., & Tyagi, M. (2021). Human Immunodeficiency Virus Type-1 (HIV-1) Transcriptional Regulation, Latency and Therapy in the Central Nervous System. Vaccines, 9(11), 1272. https://doi.org/10.3390/vaccines9111272