
Identification of HLA-A2-Restricted Mutant Epitopes from Neoantigens of Esophageal Squamous Cell Carcinoma

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptides
2.2. Cell Lines, Peripheral Blood Mononuclear CELLS (PBMCs), and Animal
2.3. Antibodies
2.4. T2A2 Cell-Binding Assay and Stabilization Assay
2.5. Generation of Peptide-Specific T Cells by Autologous DCs
2.6. Induction of Peptide-Specific T Cells from HLA-A2.1/Kb Tg Mice
2.7. The Construction of Minigene and Transfection of Tumor Cell Lines
2.8. Intracellular Cytokine Staining Assay
2.9. Cytotoxicity Assay
2.10. Analyze the Possible Structural Models of the Candidate Peptide and HLA-A*0201 Molecule
2.11. Statistical Analysis
3. Results
3.1. Prediction, Synthesis, and Binding Capacity of MUT Peptide Derived from Mutant Antigen of ESCC
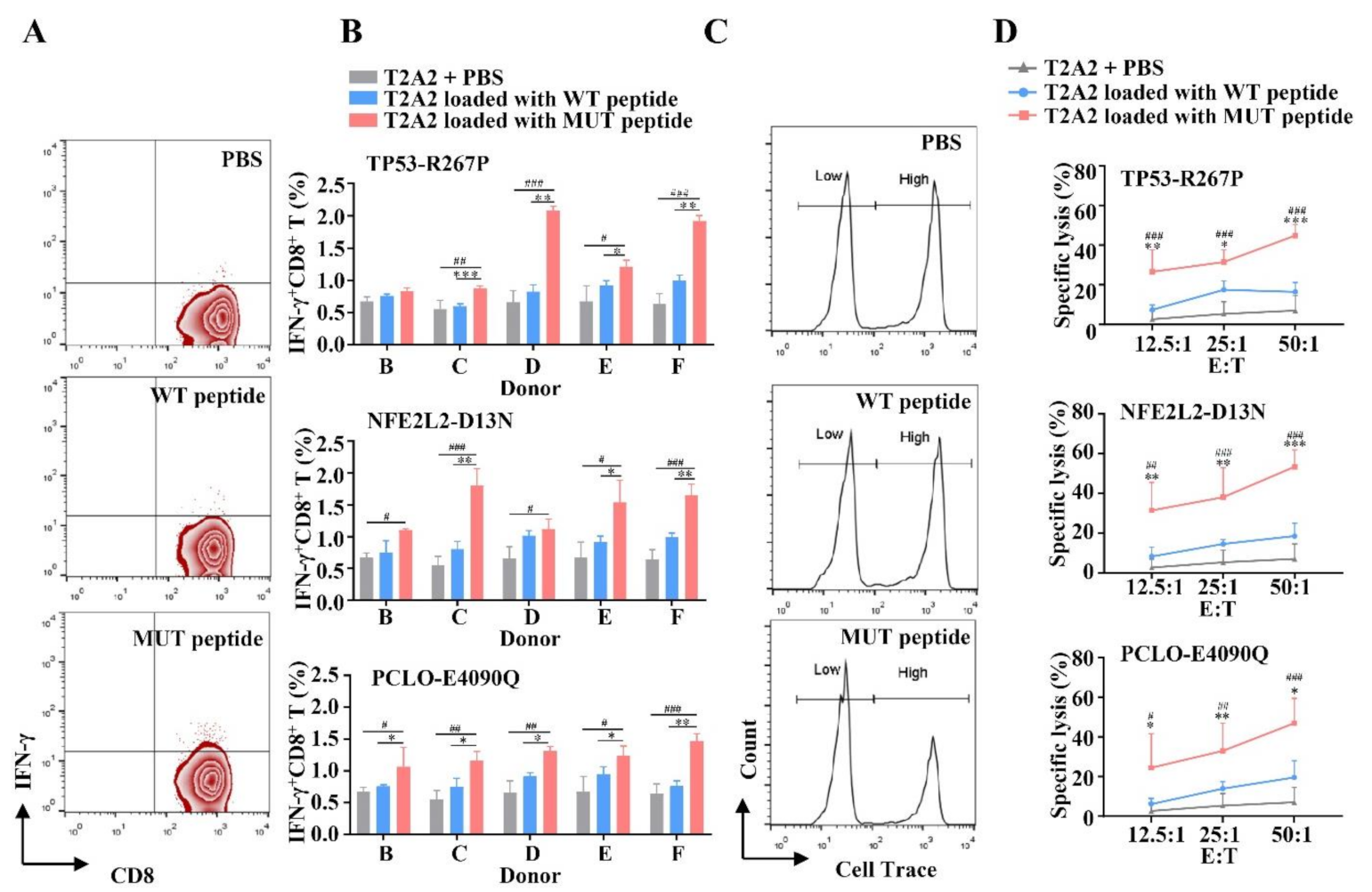
3.2. MUT Peptide-Specific T Cells Can Recognize MUT Peptide-Pulsing T2A2 Cells in MUT Peptide-Specific Manner In Vitro
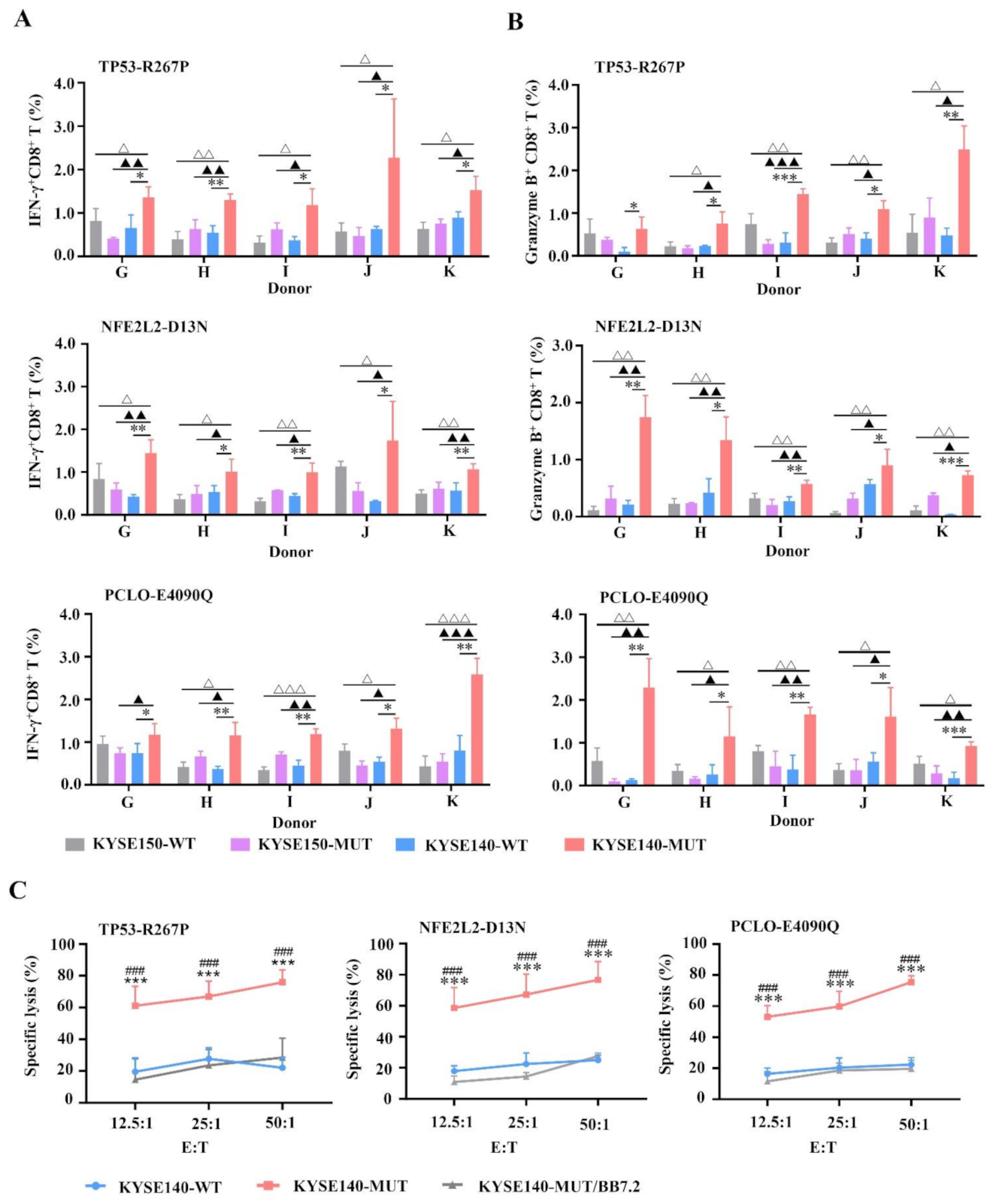
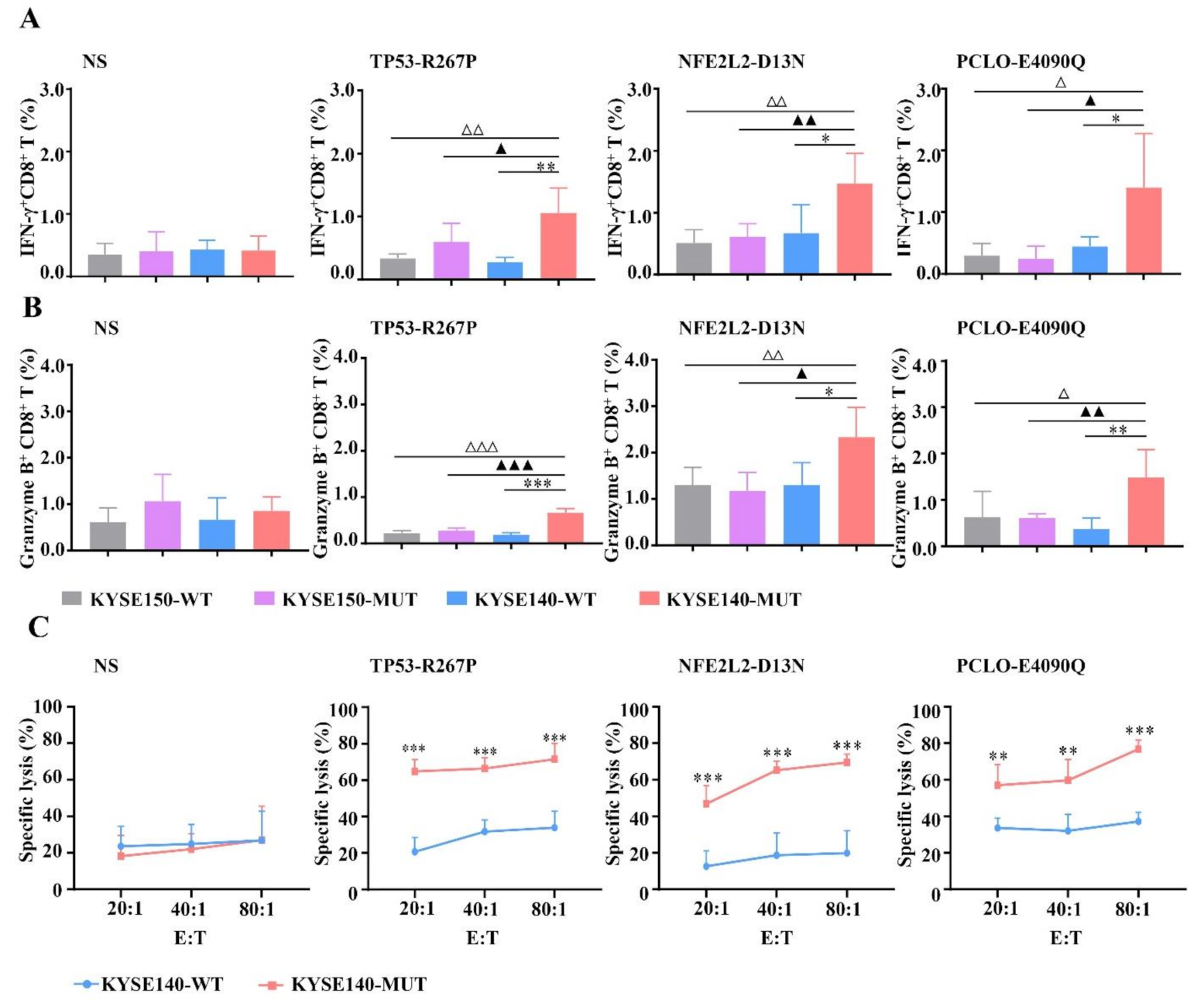
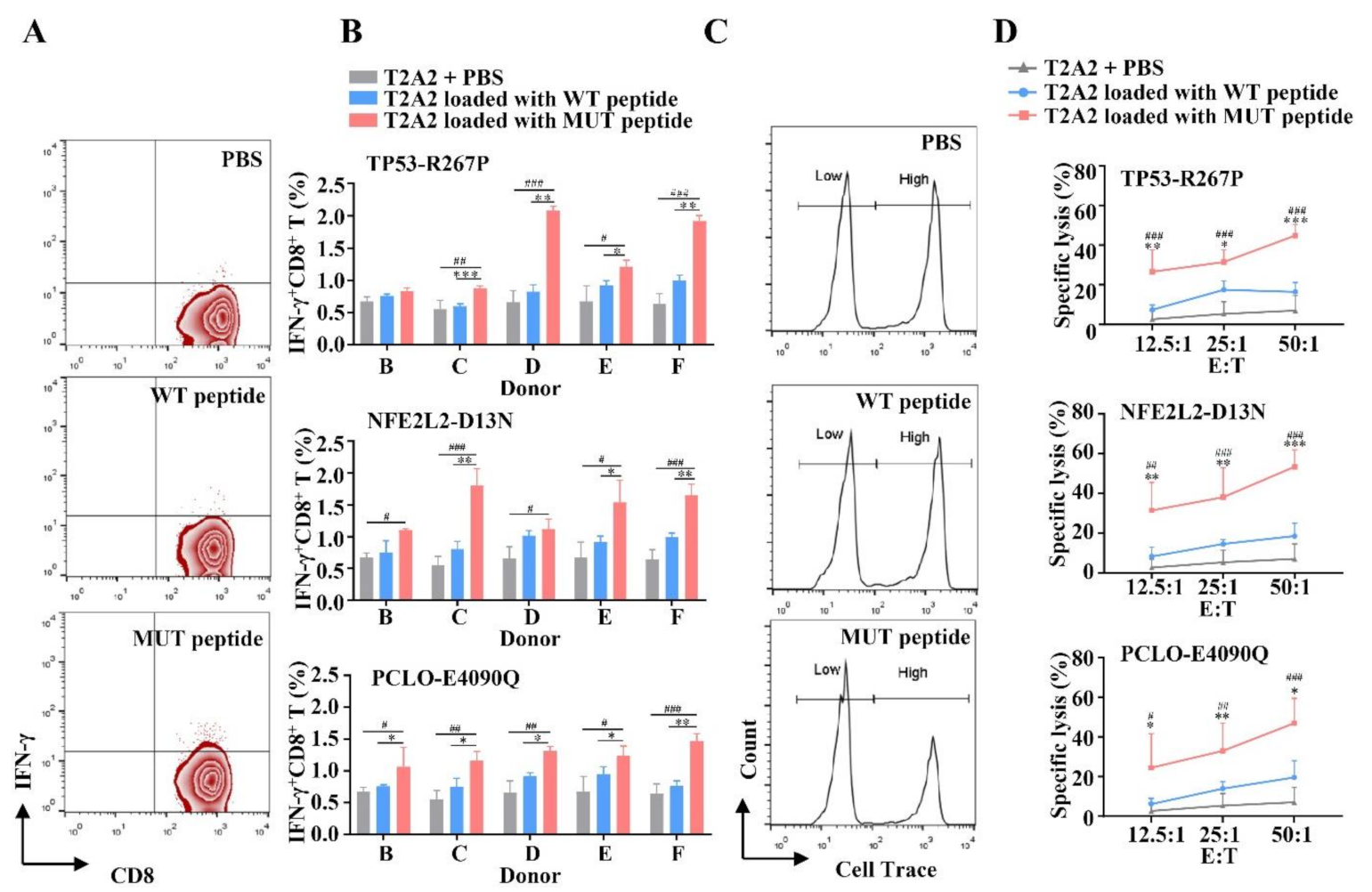
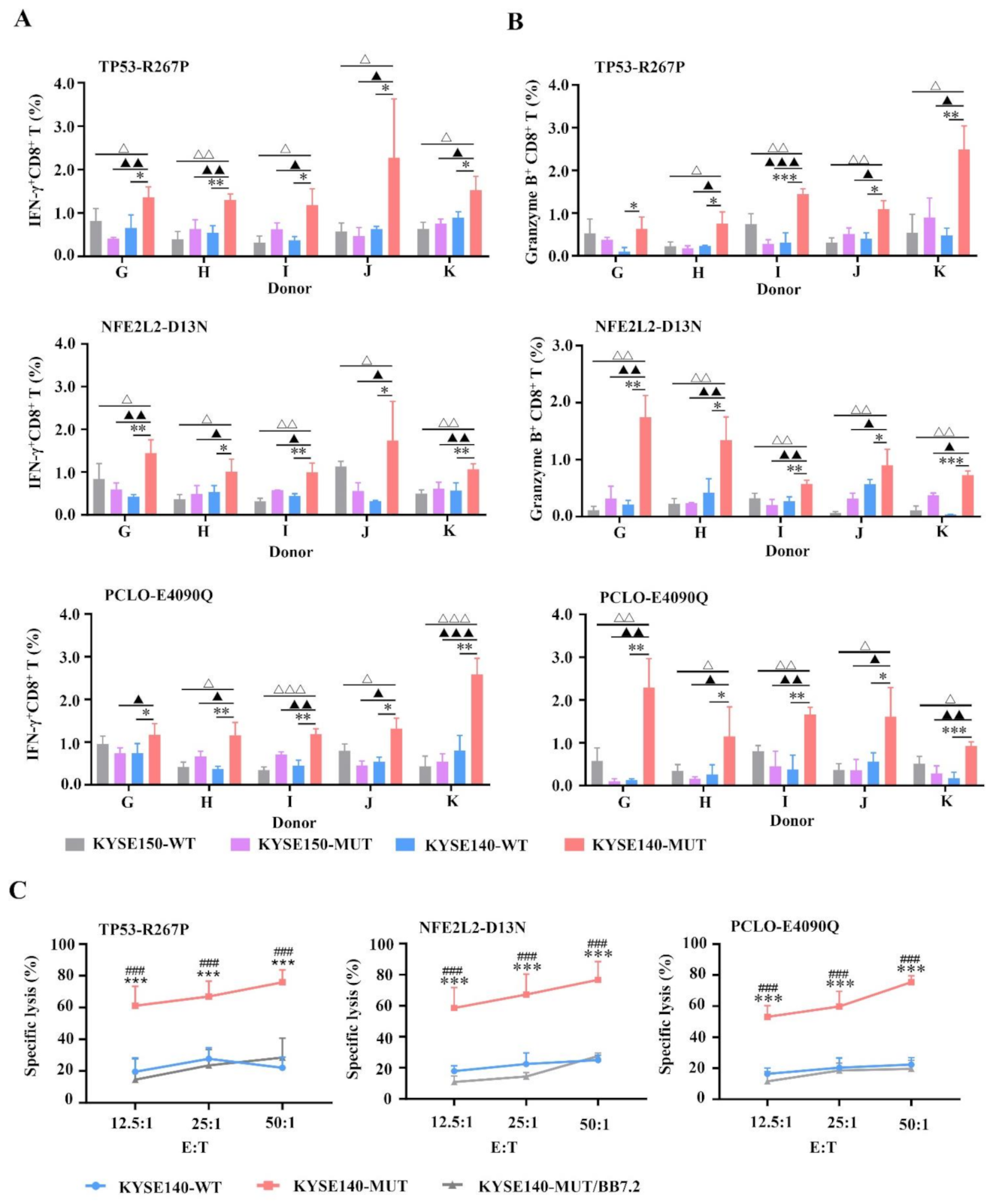
3.3. MUT Peptide-Specific T Cells Can Recognize MUT Peptides Presented in Tumor CELL Line in an HLA-A2-Restricted and MUT Peptide-Specific Manner In Vitro
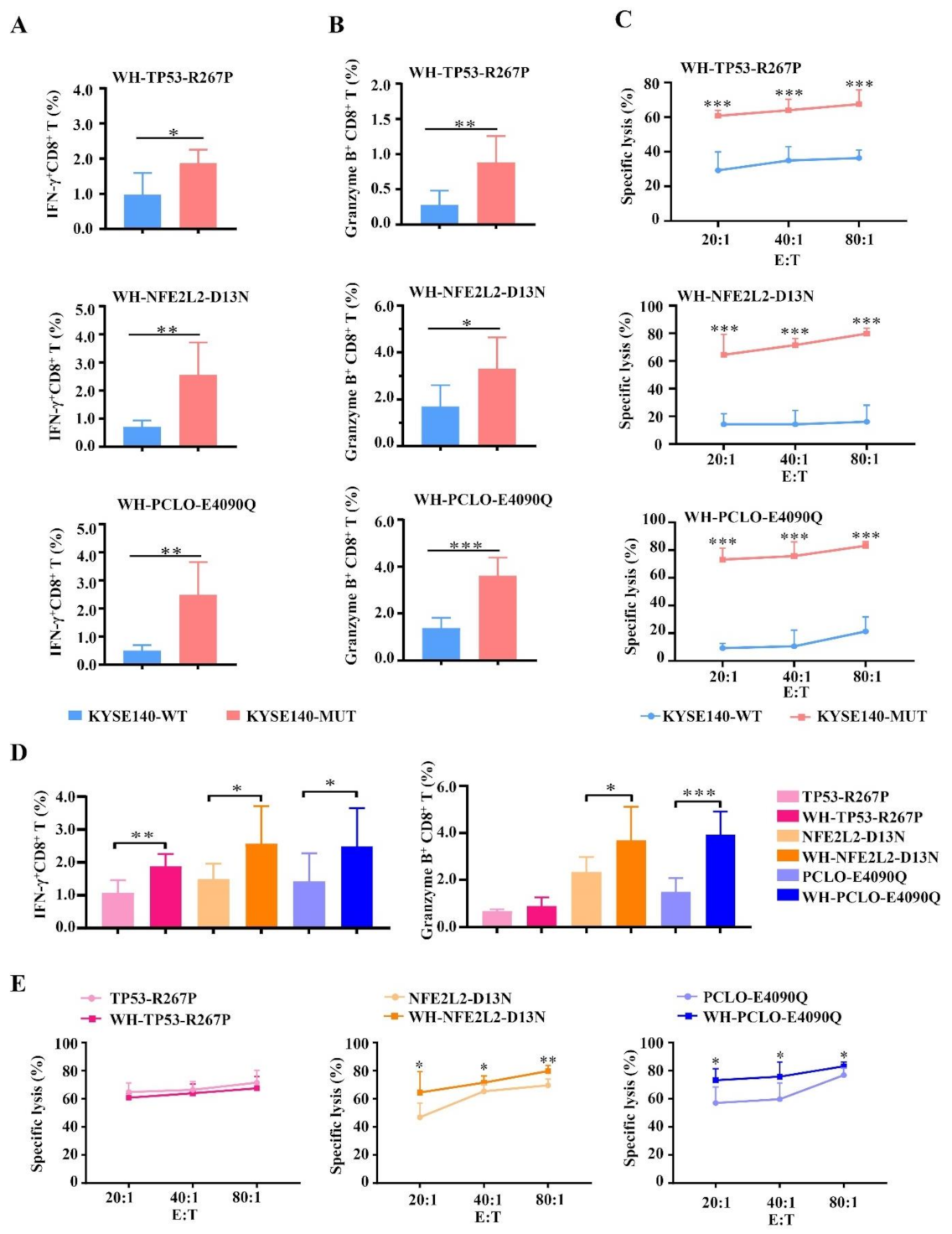
3.4. HLA-A2-Restricted and MUT Peptide-Specific CTL Responses Can Be Induced from HLA-A2.1/Kb Tg Mice In Vivo
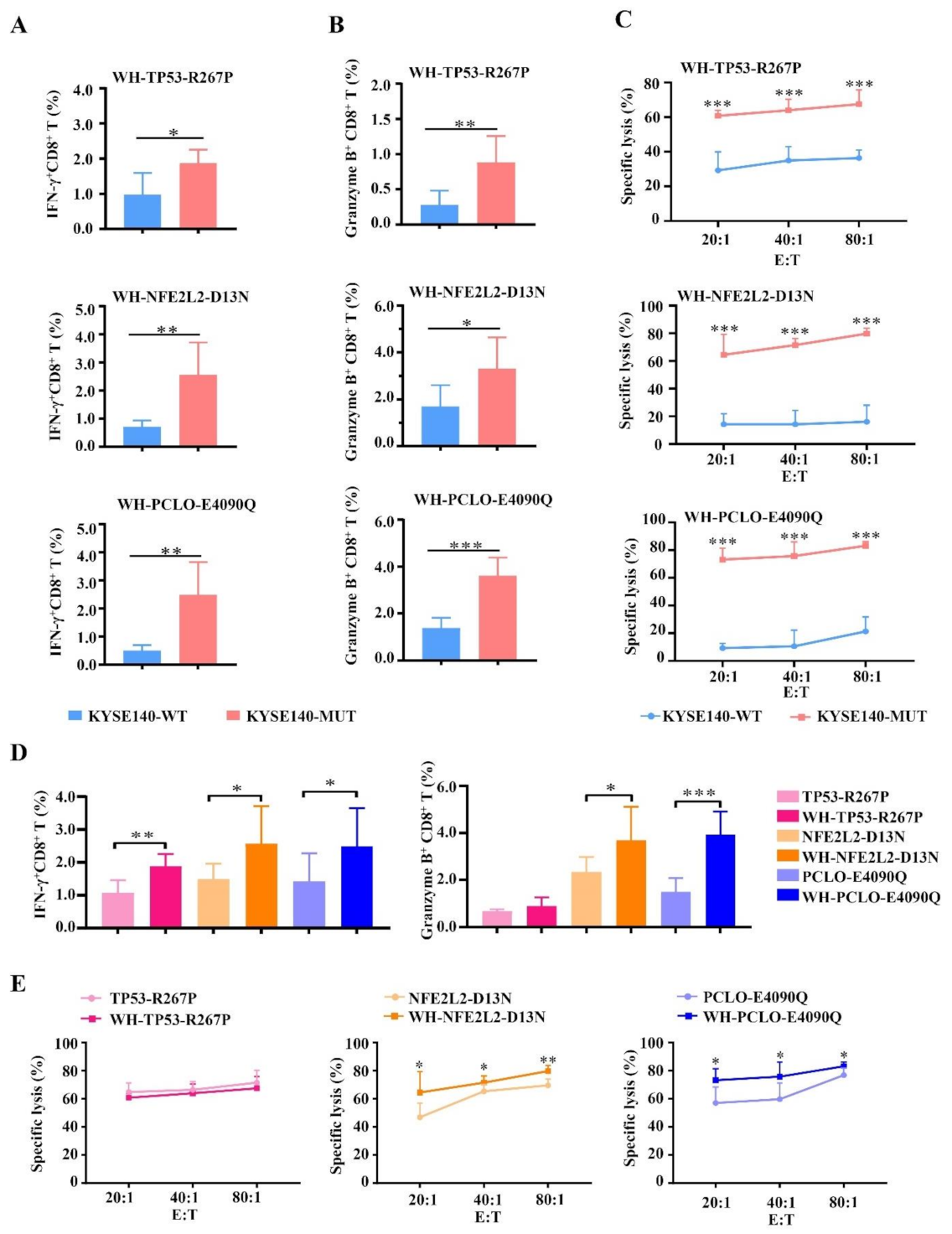
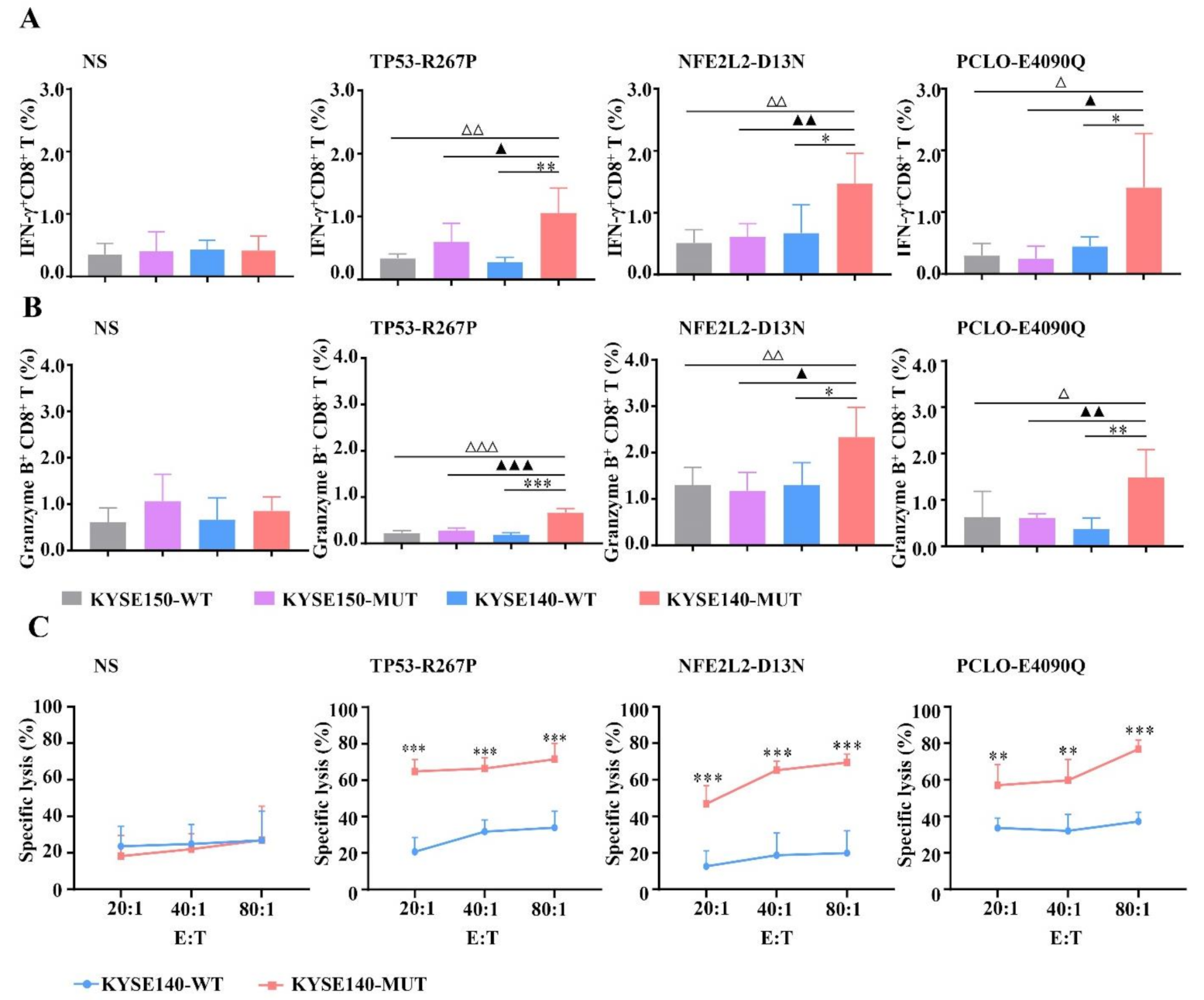
3.5. Enhanced CTL Responses by WH-MUT Peptides Induced from HLA-A2.1/Kb Tg Mice
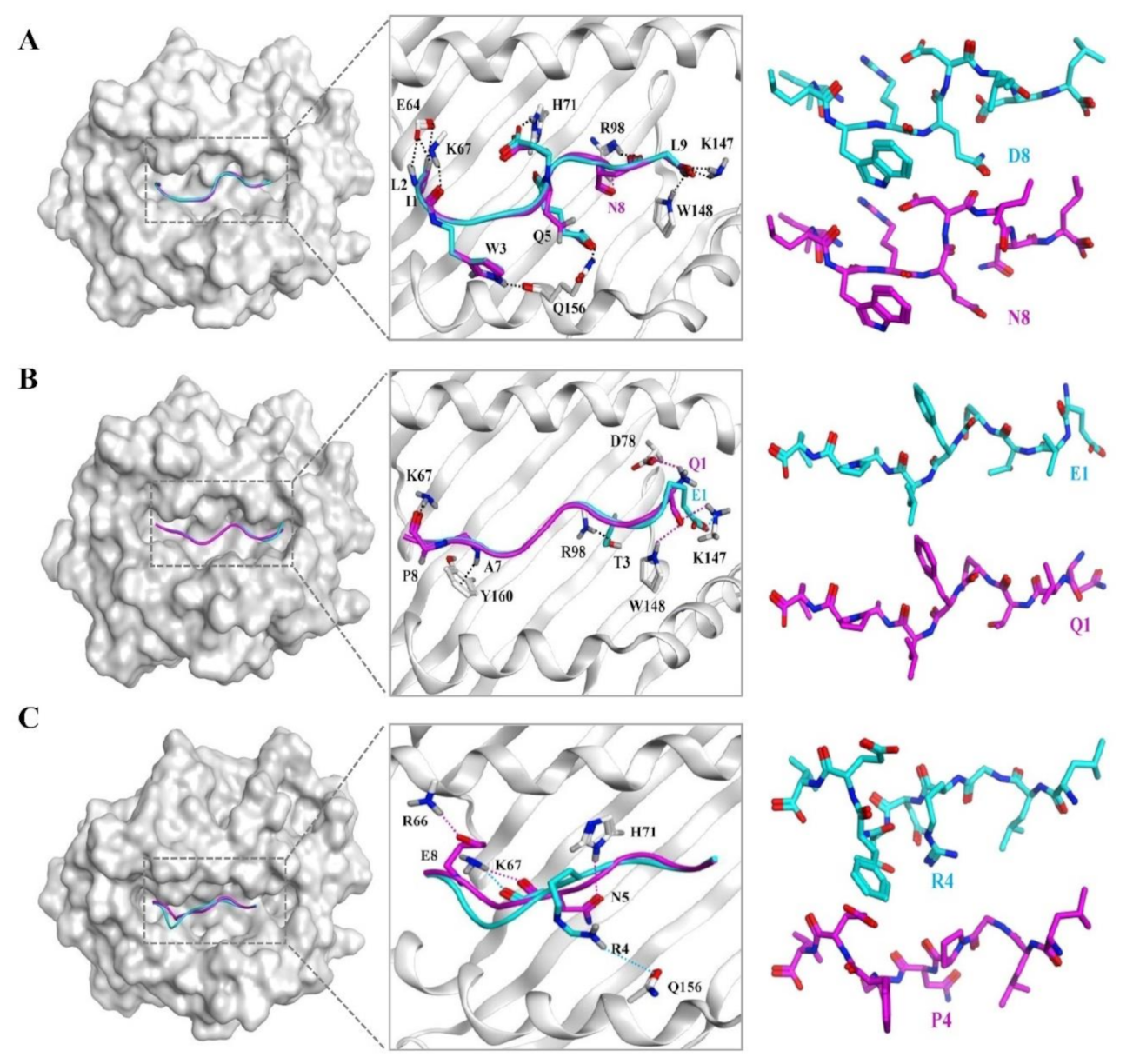
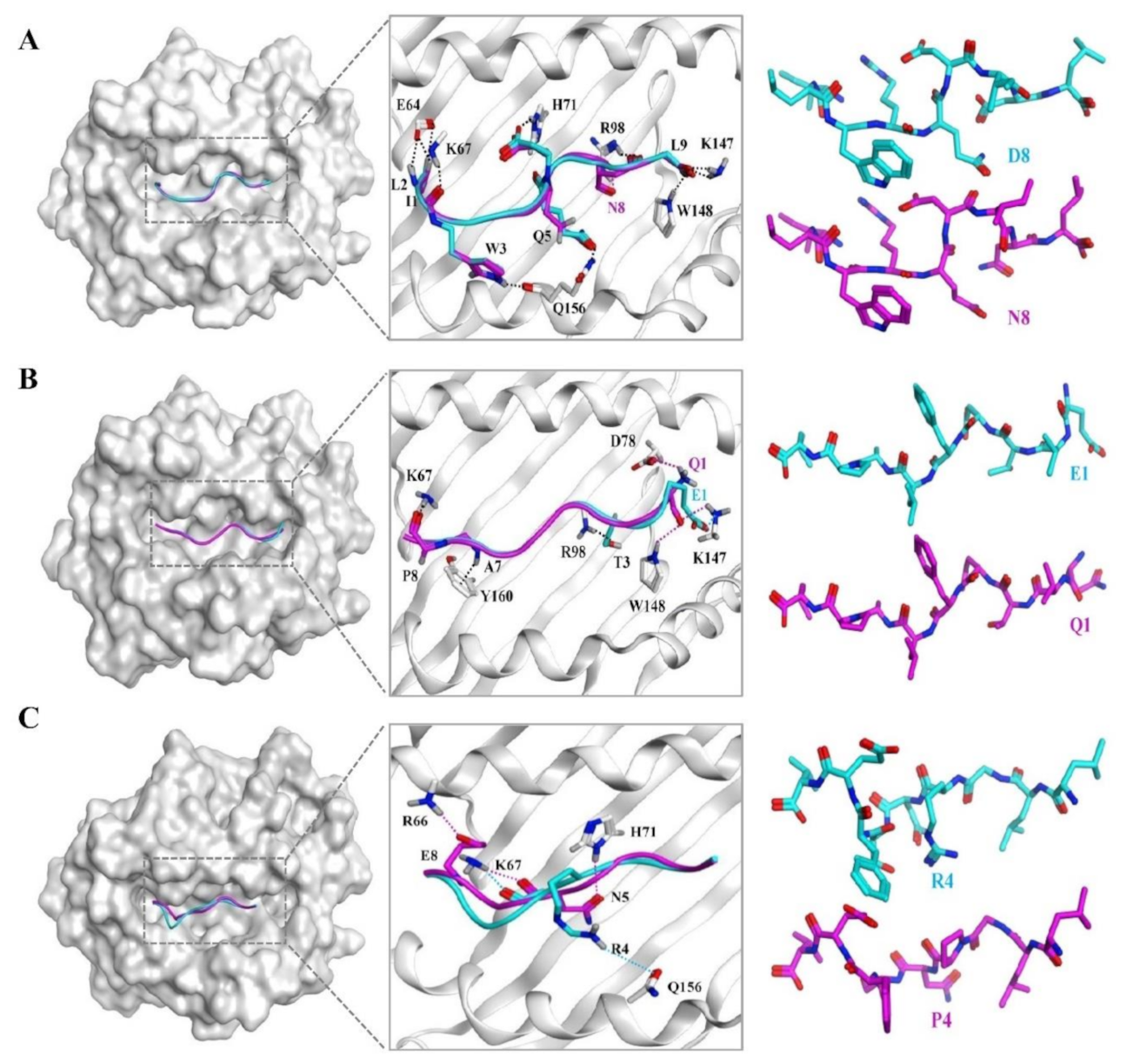
3.6. The Docking Results of the Candidate Peptide and HLA-A*0201 Molecule
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, W.; Xia, C.; Zheng, R.; Zhou, M.; Lin, C.; Zeng, H.; Zhang, S.; Wang, L.; Yang, Z.; Sun, K.; et al. Disparities by province, age, and sex in site-specific cancer burden attributable to 23 potentially modifiable risk factors in China: A comparative risk assessment. Lancet Glob. Health 2019, 7, e257–e269. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustgi, A.K.; El-Serag, H.B. Esophageal carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Pennathur, A.; Gibson, M.K.; Jobe, A.B.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, P.S.N.; Mohammad, N.H.; Vleggaar, F.P.; Van Hillegersberg, R. Treatment for unresectable or metastatic oesophageal cancer: Current evidence and trends. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A. Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.-C.; Hao, J.-J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.-W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-B.; Chen, Z.-L.; Li, J.-G.; Hu, X.-D.; Shi, X.-J.; Sun, Z.-M.; Zhang, F.; Zhao, Z.-R.; Li, Z.-T.; Liu, Z.-Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, Y.; Cheng, C.; Cui, H.; Cheng, L.; Kong, P.; Wang, J.; Li, Y.; Chen, W.; Song, B.; et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2015, 96, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Wu, Y.; Du, J.; Li, G.; Wang, S.; Cao, W.; Zhou, X.; Wu, C.; Zhang, D.; Jing, X.; et al. A novel peptide targeting Clec9a on dendritic cell for cancer immunotherapy. Oncotarget 2016, 7, 40437–40450. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhai, W.; Zhou, X.; Wang, Z.; Lin, Y.; Ran, L.; Qi, Y.; Gao, Y. HLA-A2-Restricted epitopes identified from MTA1 could elicit antigen-specific cytotoxic T lymphocyte response. J. Immunol. Res. 2018, 2018, 2942679. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Li, Y.; Ran, L.; Dong, Y.; Zhou, X.; Tang, J.; Han, L.; Wang, M.; Pang, L.; Qi, Y.; et al. Screening and identification of HLA-A2-restricted neoepitopes for immunotherapy of non-microsatellite instability-high colorectal cancer. Sci. China Life Sci. 2021, 1–16. [Google Scholar] [CrossRef]
- Plebanski, M.; Lee, E.A.; Hannan, C.M.; Flanagan, K.L.; Gilbert, S.C.; Gravenor, M.B.; Hill, A.V. Altered peptide ligands narrow the repertoire of cellular immune responses by interfering with T-cell priming. Nat. Med. 1999, 5, 565–571. [Google Scholar] [CrossRef]
- Zhu, B.; Chen, Z.; Cheng, X.; Lin, Z.; Guo, J.; Jia, Z.; Zou, L.; Wang, Z.; Hu, Y.; Wang, N.; et al. Identification of HLA-A*0201-restricted cytotoxic T lymphocyte epitope from TRAG-3 antigen. Clin. Cancer Res. 2003, 9, 1850–1857. [Google Scholar] [PubMed]
- Xie, X.; Zhou, W.; Hu, Y.; Chen, Y.; Zhang, H.; Li, Y. A dual-function epidermal growth factor receptor pathway substrate 8 (Eps8)-derived peptide exhibits a potent cytotoxic T lymphocyte-activating effect and a specific inhibitory activity. Cell Death Dis. 2018, 9, 379. [Google Scholar] [CrossRef] [Green Version]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis, E.; Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Mennonna, D.; Maccalli, C.; Romano, M.C.; Garavaglia, C.; Capocefalo, F.; Bordoni, R.; Severgnini, M.; De Bellis, G.; Sidney, J.; Sette, A.; et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut 2015, 66, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [Green Version]
- Filby, A.; Begum, J.; Jalal, M.; Day, W. Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods 2015, 82, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Watari, E.; Shimizu, M.; Takahashi, H. One-step simple assay to determine antigen-specific cytotoxic activities by single-color flow cytometry. Biomed. Res. 2011, 32, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xue, S.-A.; Behboudi, S.; Mohammad, G.H.; Pereira, S.; Morris, E.C. Ex Vivo PD-L1/PD-1 Pathway blockade reverses dysfunction of circulating CEA-specific T cells in pancreatic cancer patients. Clin. Cancer Res. 2017, 23, 6178–6189. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Ramishetti, S.; Tseng, Y.-C.; Guo, S.; Wang, Y.; Huang, L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J. Control. Release 2013, 172, 259–265. [Google Scholar] [CrossRef]
- Ito, H.; Ando, T.; Arioka, Y.; Saito, K.; Seishima, M. Inhibition of indoleamine 2,3-dioxygenase activity enhances the anti-tumour effects of a Toll-like receptor 7 agonist in an established cancer model. Immunology 2015, 144, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Fasterde novostructure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [Green Version]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Rustgi, A.K. Squamous cell cancers: A unified perspective on biology and genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prickett, T.D.; Crystal, J.S.; Cohen, C.J.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Yao, X.; Wang, R.; Gros, A.; Li, Y.F.; et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol. Res. 2016, 4, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Middelberg, A.P.; Gemiarto, A.; Macdonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T Cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Mints, M.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2018, 565, 234–239. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position | Sequence | Scores | ||
|---|---|---|---|---|---|
| IEDB | NetCTL | SYFPEITHI | |||
| ABCA13 | D1303H | NLHSINDFL | 2 | 1.0131 | 20 |
| E1359Q | ILQDGFLYV | 0.2 | 1.3979 | 26 | |
| DNAH5 | S3587Y | GLPNDDLYI | 3.3 | 0.9645 | 20 |
| D4110N | FMNELMD I | 0.6 | 1.2466 | 21 | |
| L4406H | RMQRVLSHV | 1.6 | 0.9677 | 22 | |
| M4495T | FLTATRQEI | 0.8 | 0.9167 | 20 | |
| KMT2D | R4198Q | QLQAQLQGV | 0.9 | 0.9111 | 25 |
| F4722L | ILGEEAPRL | 2.5 | 0.8820 | 25 | |
| LRP1B | C2479Y | YLLTPNGRV | 1.2 | 1.0572 | 25 |
| R3362L | GLFQCGTGL | 2.4 | 1.0649 | 23 | |
| P3707L | ALDMCVKFL | 1.6 | 0.9640 | 25 | |
| LRP2 | D1744Y | CLRDYQPFL | 1.2 | 1.2251 | 23 |
| MUC16 | A4579D | SMGDALDSI | 2 | 1.1824 | 25 |
| Q5024H | LMSRIPHDV | 1.8 | 0.8792 | 21 | |
| S5361F | SIPSFPLPV | 1.8 | 1.1484 | 22 | |
| A9832D | VLDDSETTI | 2.1 | 1.1934 | 22 | |
| MUC17 | A2414V | TMPVVSSEV | 2.3 | 0.9571 | 20 |
| T3809M | TMSERSTLL | 2.4 | 1.1026 | 20 | |
| NEB | D3282V | VISDYKYKV | 1.3 | 1.1003 | 24 |
| NFE2L2 | D13N | ILWRQDINL | 1.6 | 1.0632 | 23 |
| I28T | ILWRQDTDL | 3.6 | 0.7755 | 23 | |
| NOTCH1 | G1995V | RMHDVTTPL | 0.8 | 1.4064 | 21 |
| S2202F | GMLSPVDFL | 1.8 | 1.1812 | 26 | |
| PCDH15 | S628L | TLTATVNIV | 1.7 | 0.8437 | 24 |
| PCLO | E4090Q | QVTDFLAPL | 3.8 | 1.0399 | 21 |
| SYNE1 | A65S | KLLSLLEVL | 0.8 | 1.2351 | 28 |
| V7402I | FLIQTEQKL | 0.5 | 1.0359 | 25 | |
| S7628Y | YLPDHHEEL | 0.8 | 1.3406 | 25 | |
| N4342I | TILEELNVV | 3 | 0.9190 | 27 | |
| TP53 | C135F | ALNKMFFQL | 1.2 | 1.2333 | 23 |
| P190L | ALPQHLIRV | 1 | 1.2385 | 26 | |
| G244V | YMCNSSCMV | 0.5 | 1.0763 | 20 | |
| G266A | LLARNSFEV | 0.4 | 1.2765 | 26 | |
| R267P | LLGPNSFEV | 0.4 | 1.2604 | 26 | |
| V272L | LLGRNSFEL | 1.6 | 0.9715 | 24 | |
| Gene | Position | ESI-MS [M+H]+ | FI a | DC 50 b | |
|---|---|---|---|---|---|
| Calculated | Observed | ||||
| ABCA13 | E1359Q | 1067.25 | 1067.87 | 1.56 | >6 h |
| KMT2D | R4198Q | 984.12 | 985.29 | 1.12 | <2 h |
| MUC16 | A4579D | 908 | 909.05 | 1.35 | <2 h |
| A9832D | 992.05 | 993.04 | 3.56 | >6 h | |
| S5361F | 956.15 | 957.03 | 2.25 | <2 h | |
| MUC17 | A2414V | 948.11 | 948.80 | 1.54 | <4 h |
| NFE2L2 | D13N | 1170.38 | 1171.03 | 1.67 | >6 h |
| PCLO | E4090Q | 1003.16 | 1003.87 | 1.85 | >4 h |
| SYNE1 | S7628Y | 1152.23 | 1153.55 | 1.67 | <2 h |
| N4342I | 1029.2 | 1030.16 | 1.81 | <4 h | |
| V7402I | 1119.33 | 1120.6 | 1.27 | >6 h | |
| TP53 | P190L | 1046.28 | 1047.51 | 1.26 | >4 h |
| R267P | 975.11 | 976.26 | 1.81 | >6 h | |
| COX-2 | 321–329 | 999.6 | 1000.3 | 2.12 | >6 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Ran, L.; Chen, C.; Shi, R.; Dong, Y.; Li, Y.; Zhou, X.; Qi, Y.; Zhu, P.; Gao, Y.; et al. Identification of HLA-A2-Restricted Mutant Epitopes from Neoantigens of Esophageal Squamous Cell Carcinoma. Vaccines 2021, 9, 1118. https://doi.org/10.3390/vaccines9101118
Wang Z, Ran L, Chen C, Shi R, Dong Y, Li Y, Zhou X, Qi Y, Zhu P, Gao Y, et al. Identification of HLA-A2-Restricted Mutant Epitopes from Neoantigens of Esophageal Squamous Cell Carcinoma. Vaccines. 2021; 9(10):1118. https://doi.org/10.3390/vaccines9101118
Chicago/Turabian StyleWang, Zhiwei, Ling Ran, Chunxia Chen, Ranran Shi, Yu Dong, Yubing Li, Xiuman Zhou, Yuanming Qi, Pingping Zhu, Yanfeng Gao, and et al. 2021. "Identification of HLA-A2-Restricted Mutant Epitopes from Neoantigens of Esophageal Squamous Cell Carcinoma" Vaccines 9, no. 10: 1118. https://doi.org/10.3390/vaccines9101118
APA StyleWang, Z., Ran, L., Chen, C., Shi, R., Dong, Y., Li, Y., Zhou, X., Qi, Y., Zhu, P., Gao, Y., & Wu, Y. (2021). Identification of HLA-A2-Restricted Mutant Epitopes from Neoantigens of Esophageal Squamous Cell Carcinoma. Vaccines, 9(10), 1118. https://doi.org/10.3390/vaccines9101118