
Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens

 ,
,  ,
,  and
and
Abstract
:1. Introduction
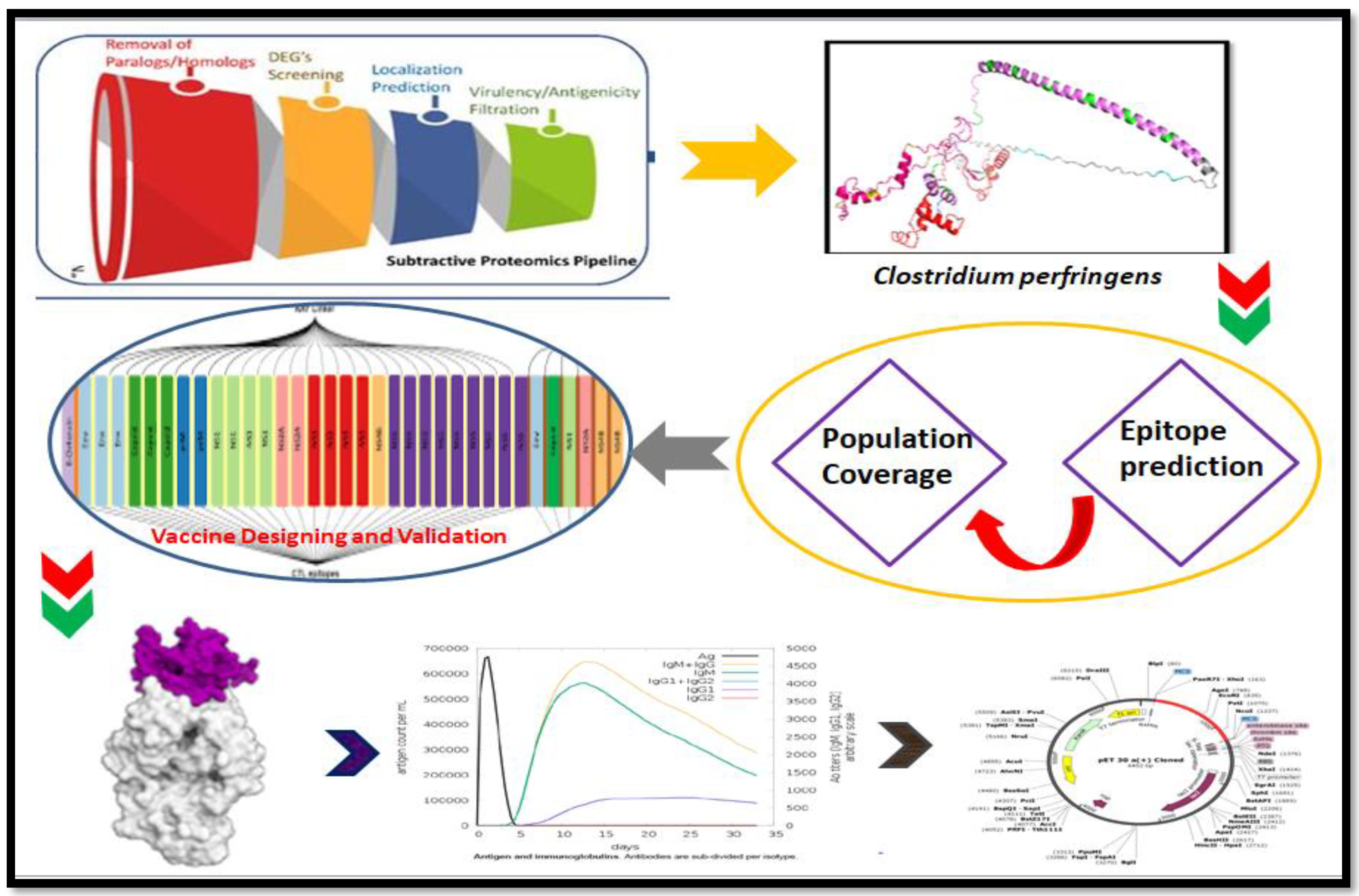
2. Methodology
2.1. Whole Proteome Retrieval
2.2. Essential Protein Retrieval
2.3. Virulent Factor Identification
2.4. Antigenicity Prediction
2.5. Subcellular Location
2.6. CTL Epitopes Prediction & Validation
2.7. HTL Epitope Prediction & Validation
2.8. B-Cell Epitope Prediction & Validation
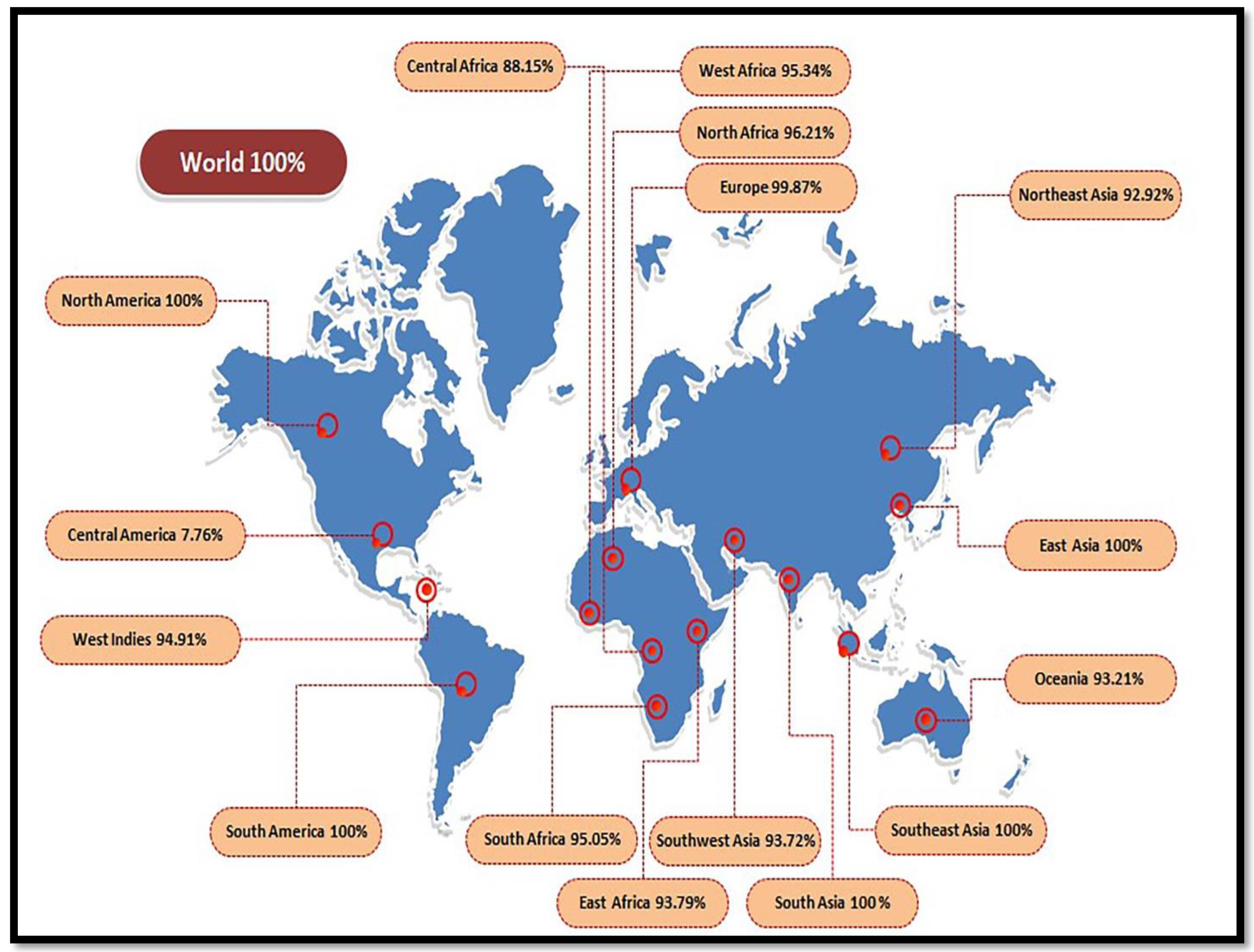
2.9. Population Coverage
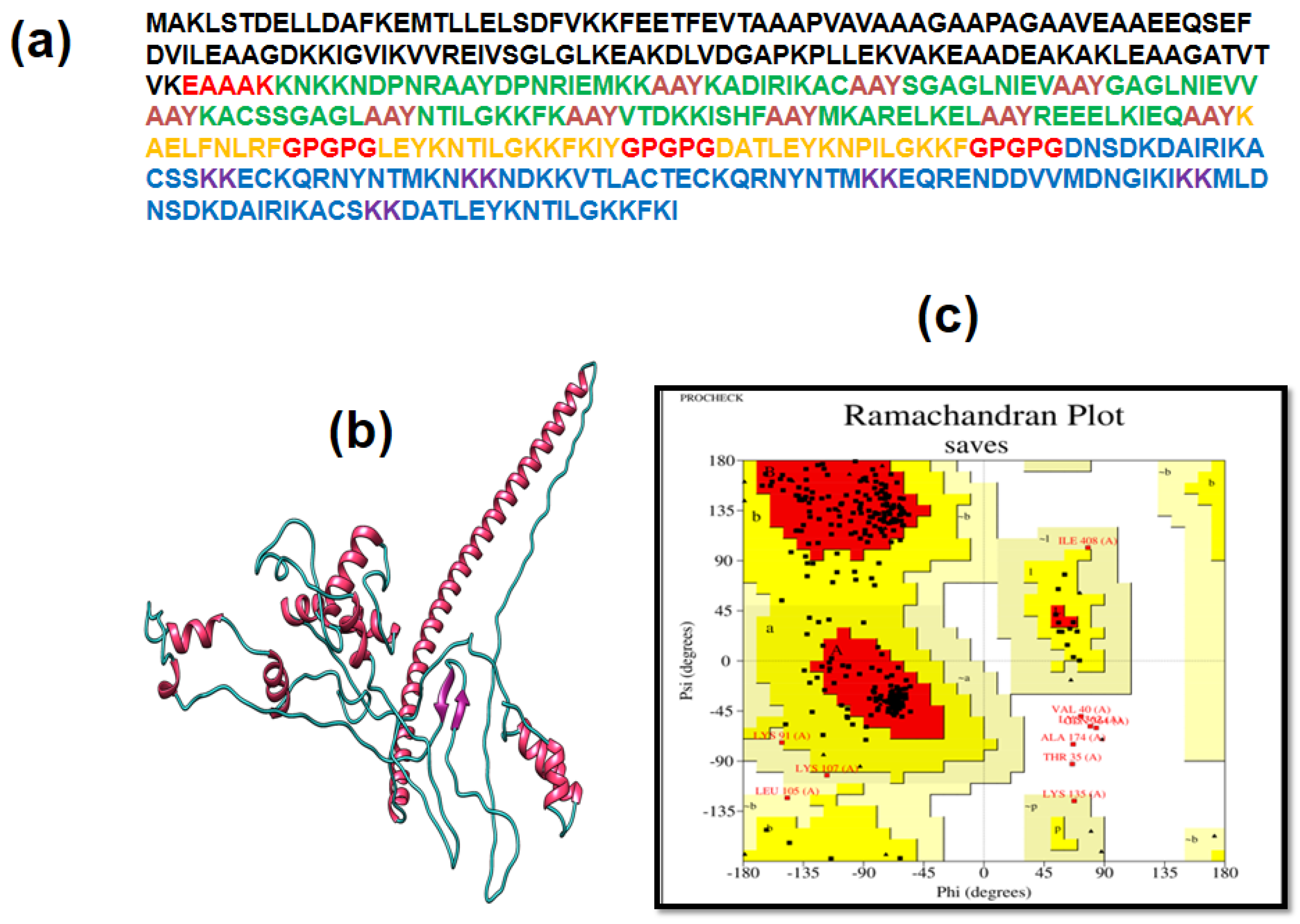
2.10. Construction of Vaccines
2.11. MEV Structure Analysis
2.12. 3D Structure Determination, Refinement, and Validation
2.13. B-Cell Epitope Screening
2.14. Disulfide Engineering
2.15. MEV Docking with TLR4 Receptors
2.16. MD Simulation
2.17. Immune-Simulation
2.18. In Silico Cloning & Codon Optimization
3. Results
3.1. Protein Selection
3.2. Evaluation and Identification of B- and T-Cell Epitopes
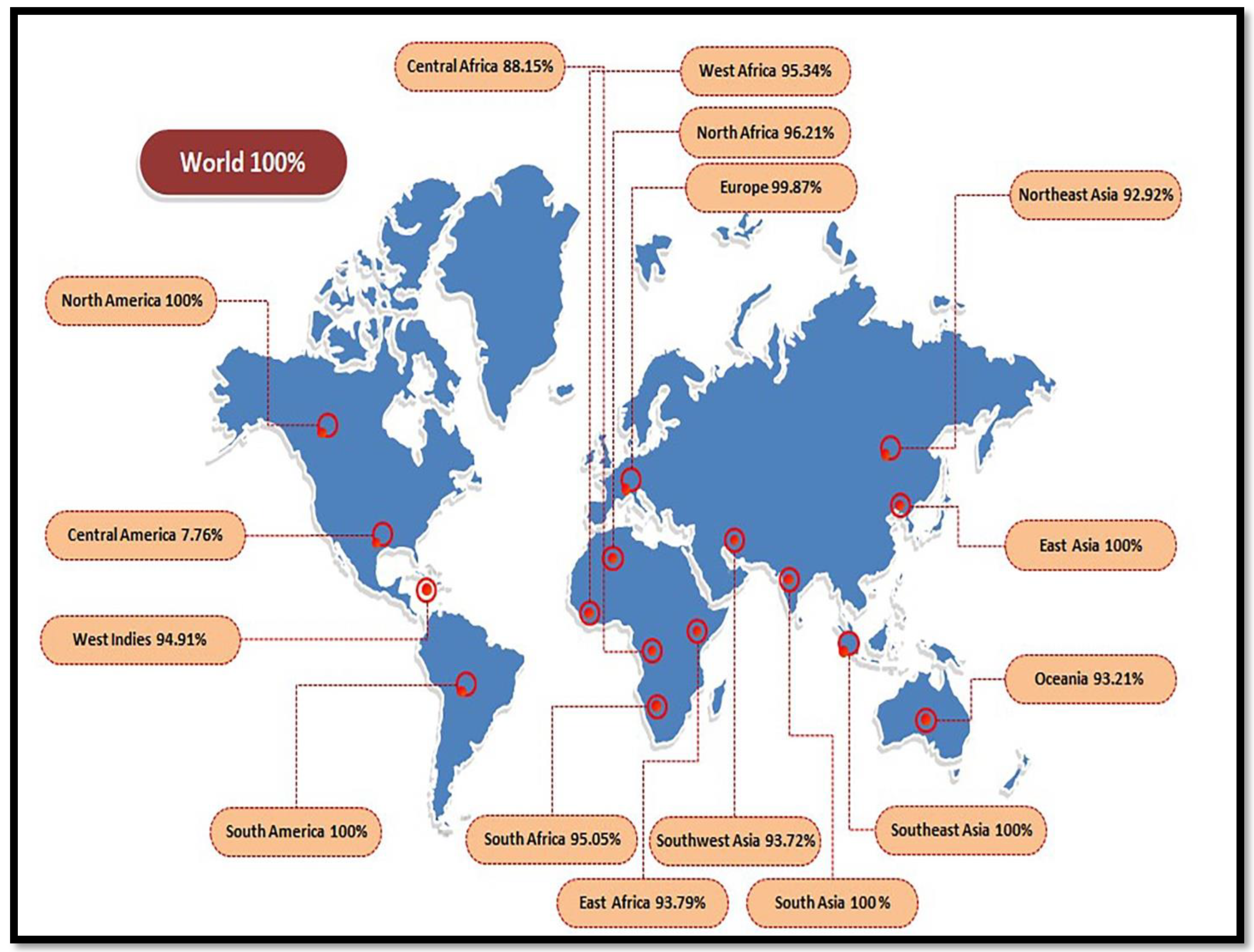
3.3. Population Coverage
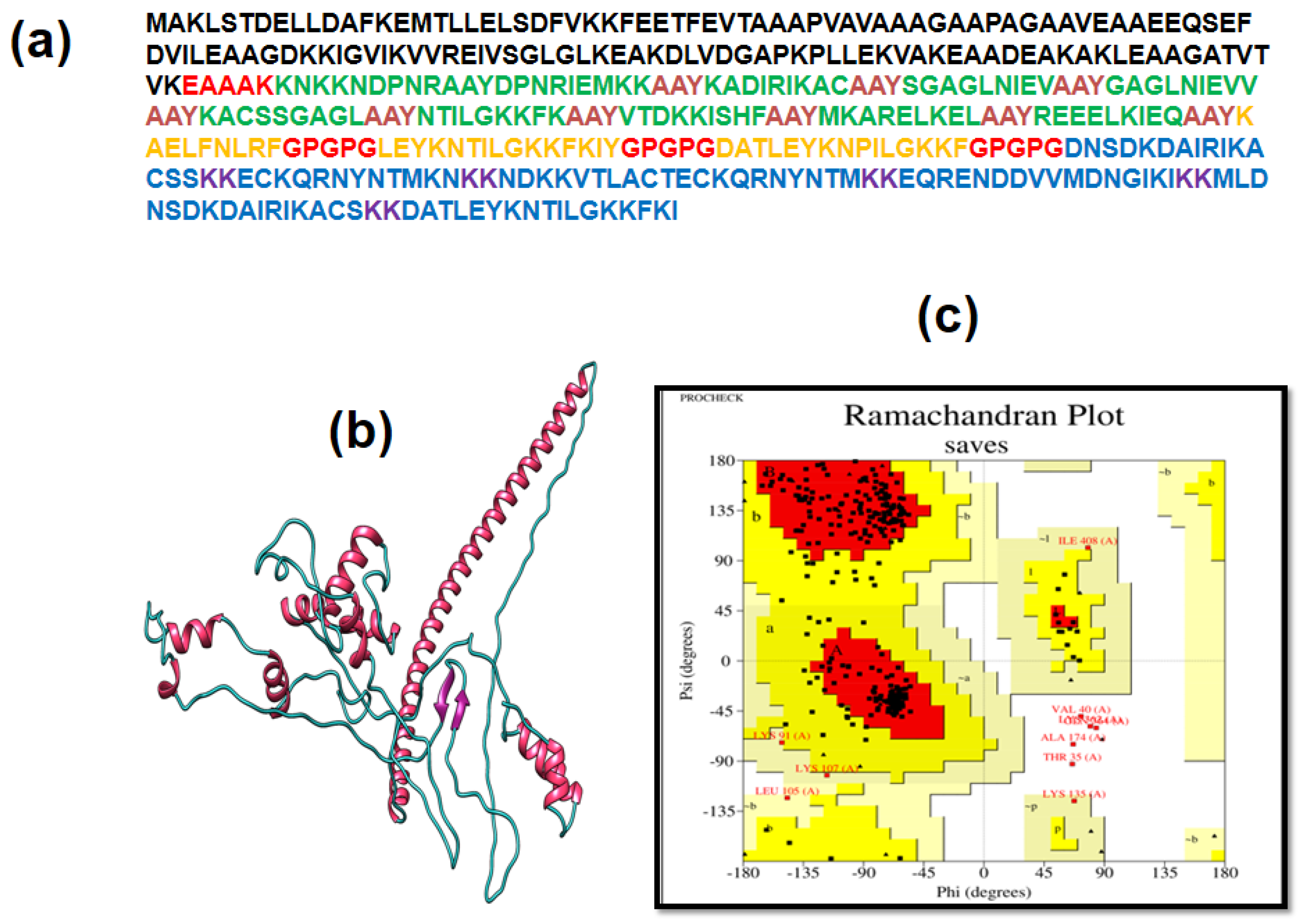
3.4. Multiple Epitope Vaccine Construction
3.5. Immunogenic and Physicochemical Analysis
3.6. Analysis of Structure
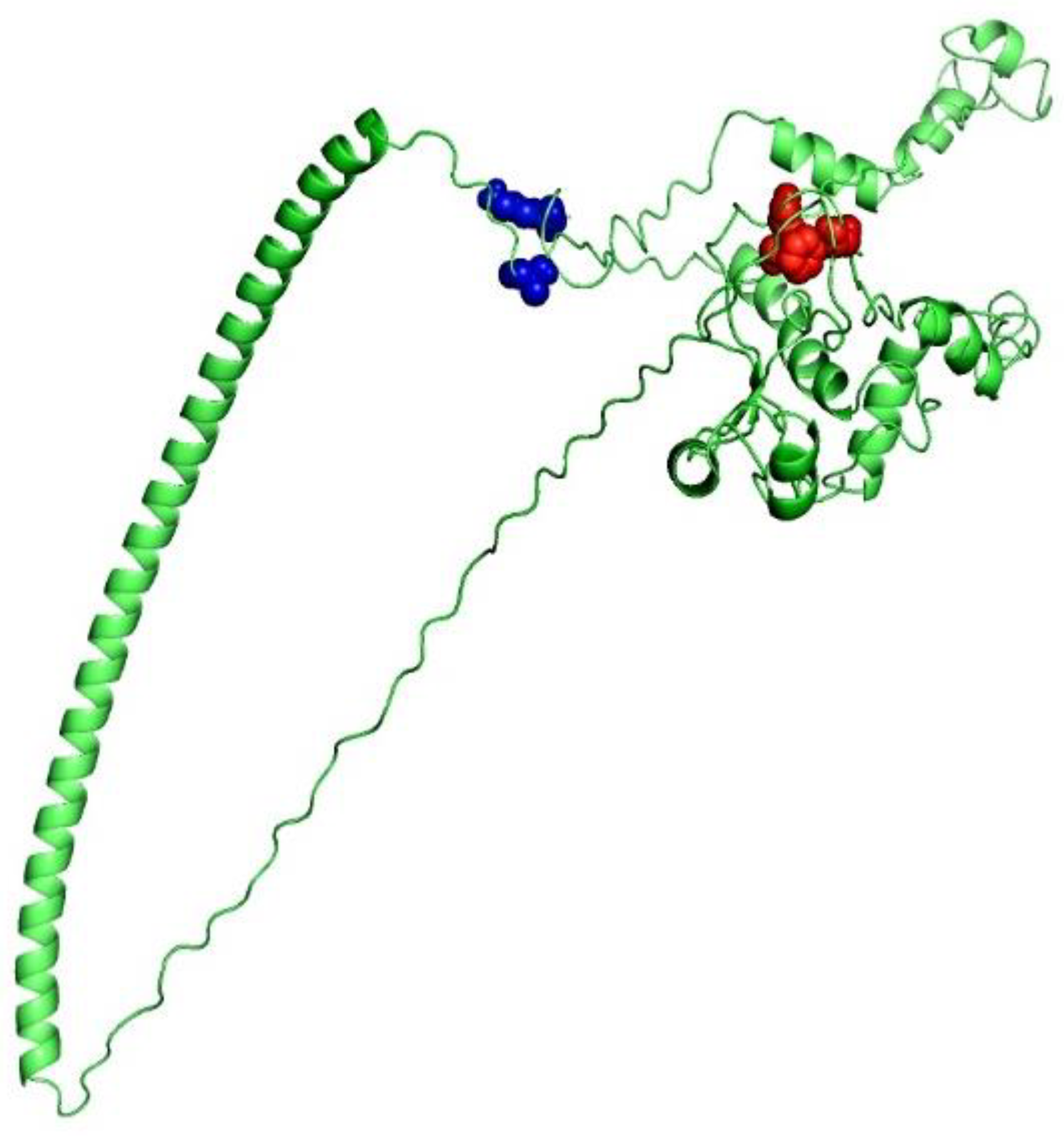
3.7. 3D Structure Determination, Refinement, and Verification
3.8. Screening of B-Cell Epitopes
3.9. Disulfide Engineering
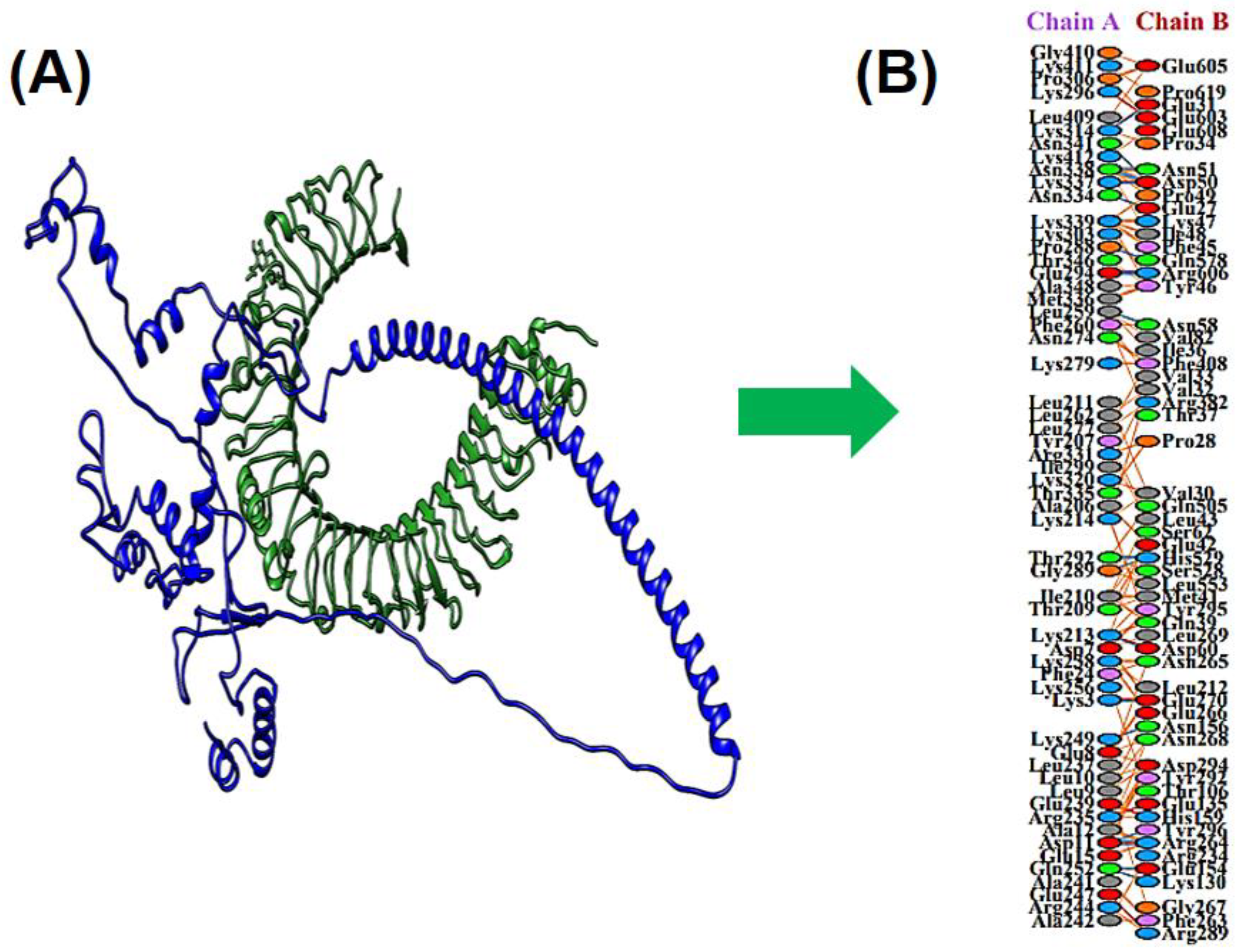
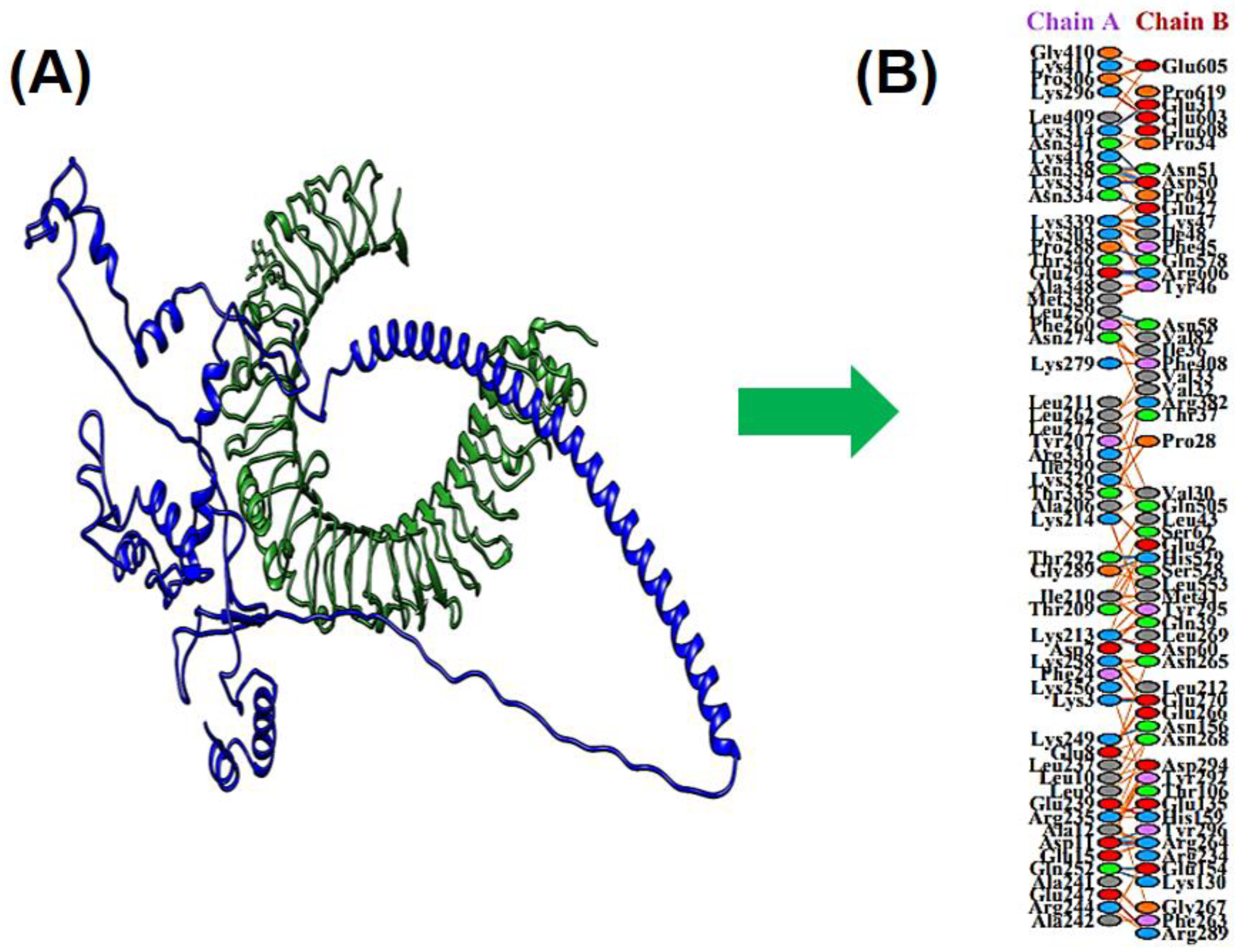
3.10. Docking between MEV & TLR4
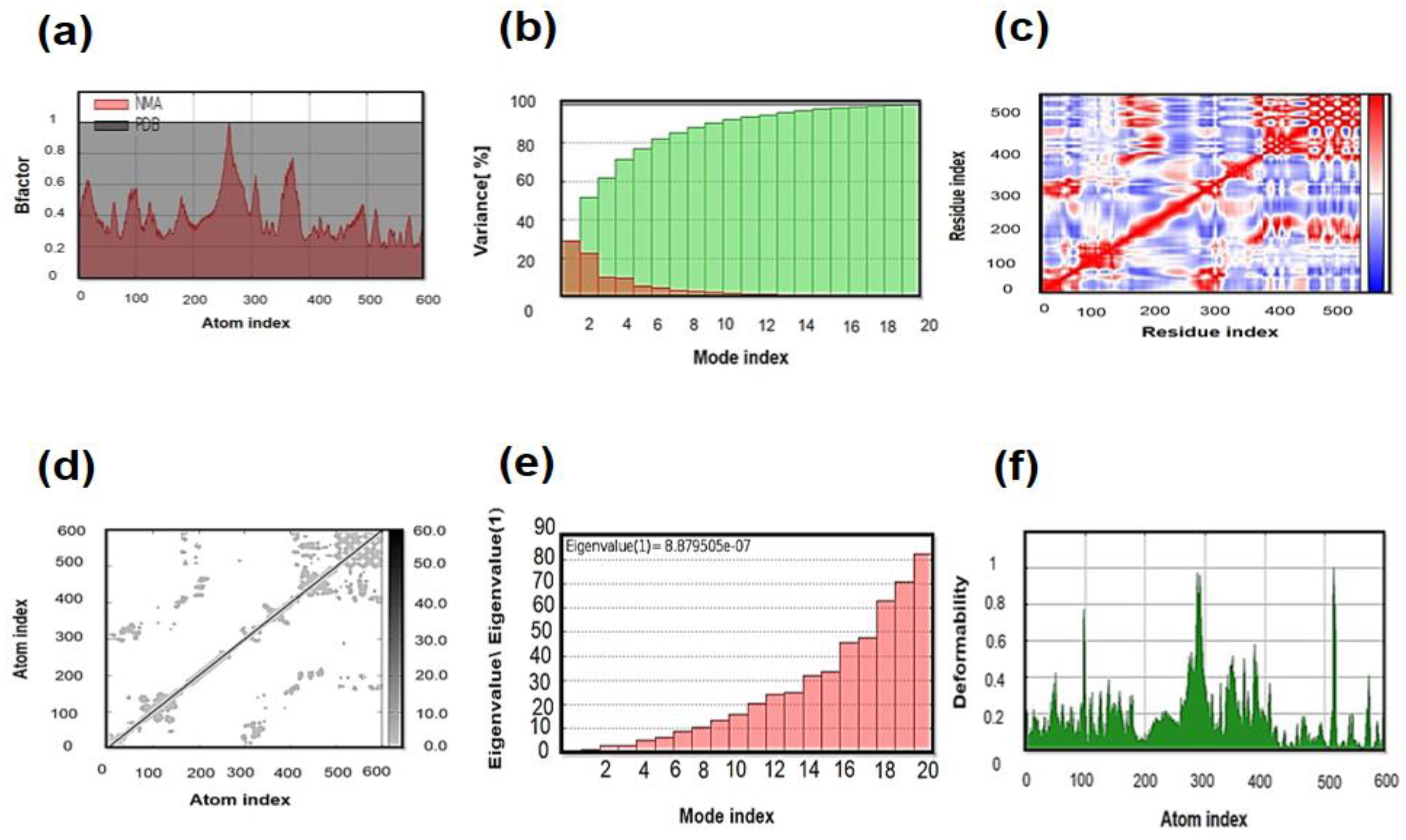
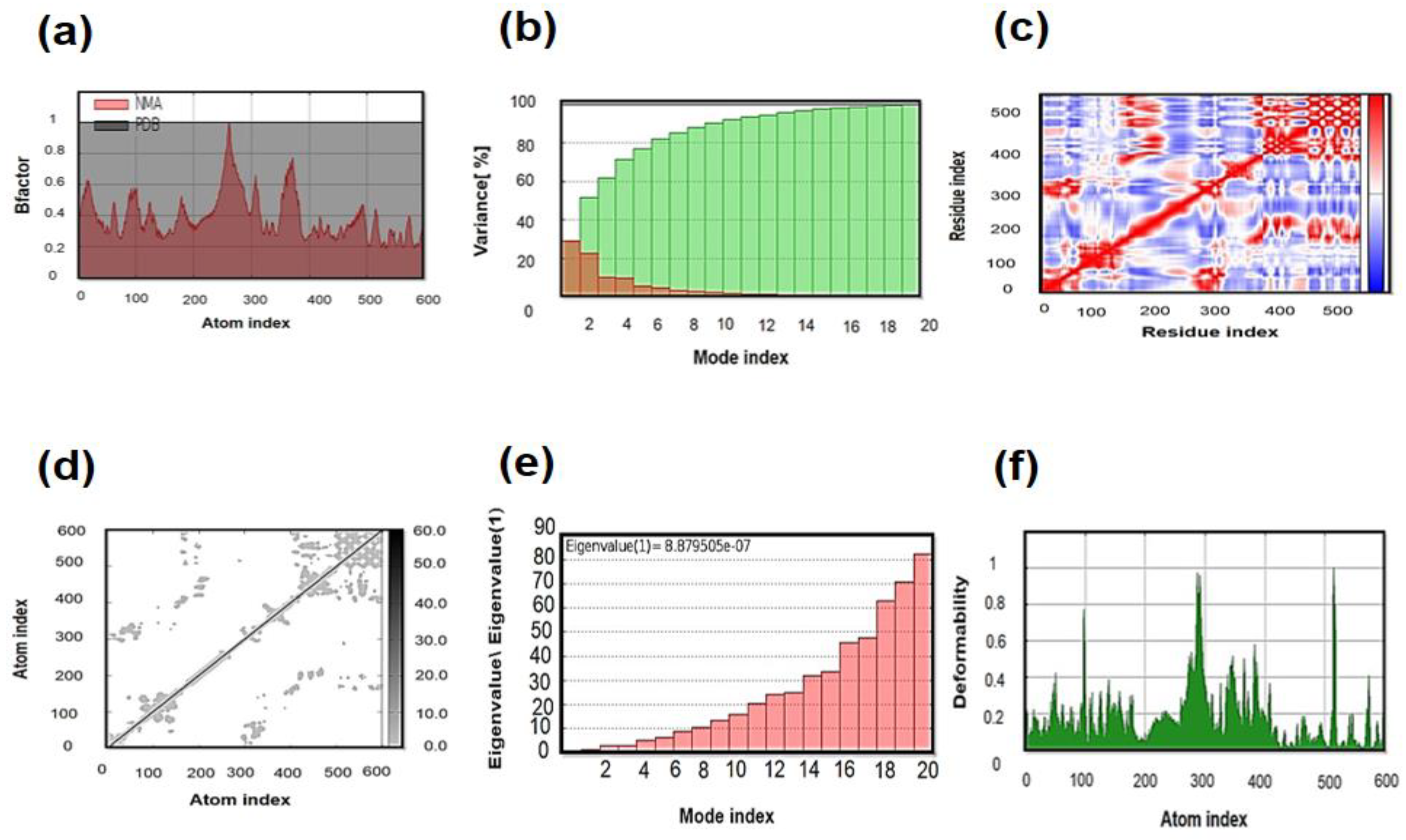
3.11. Molecular Dynamics (MD) Simulation
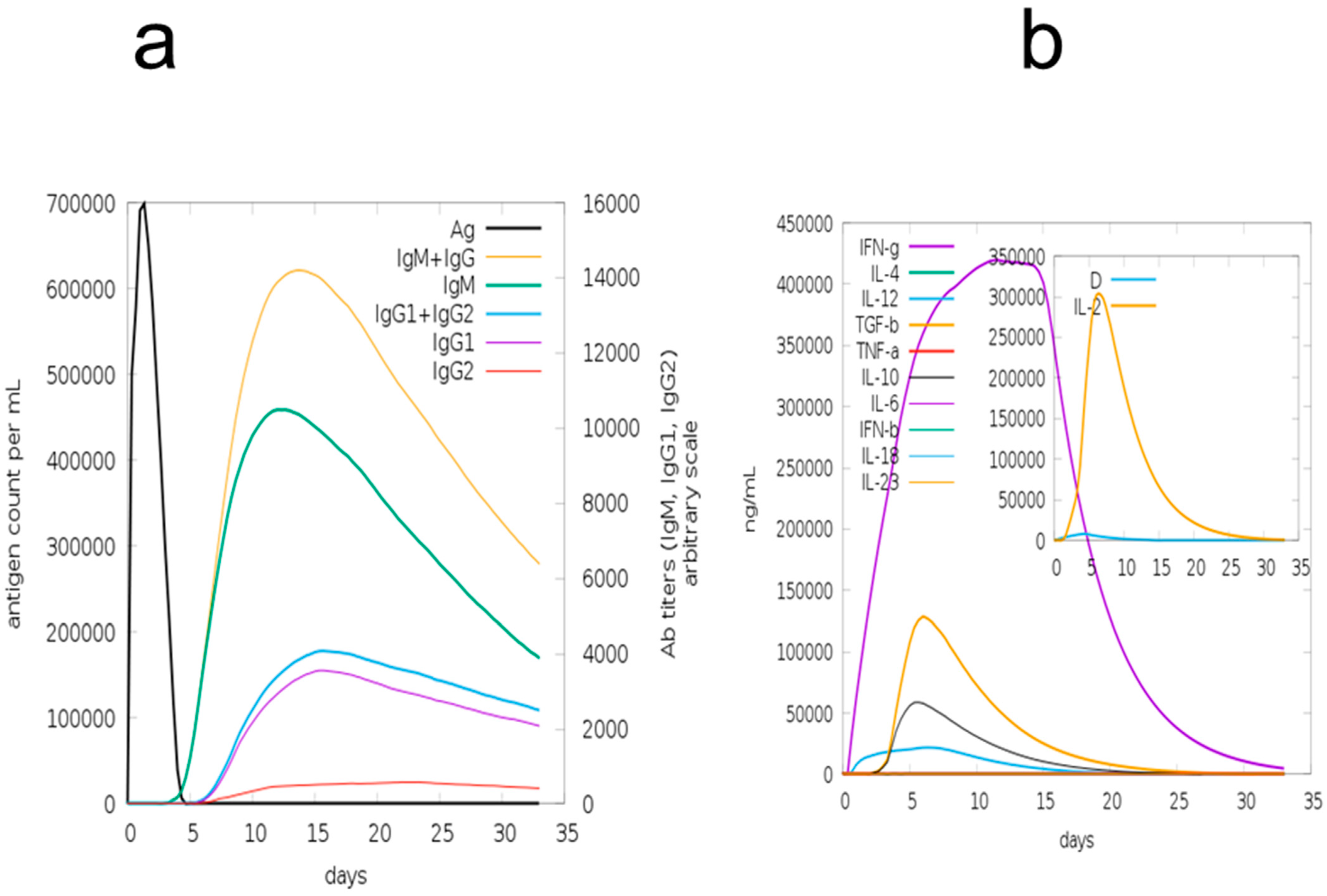
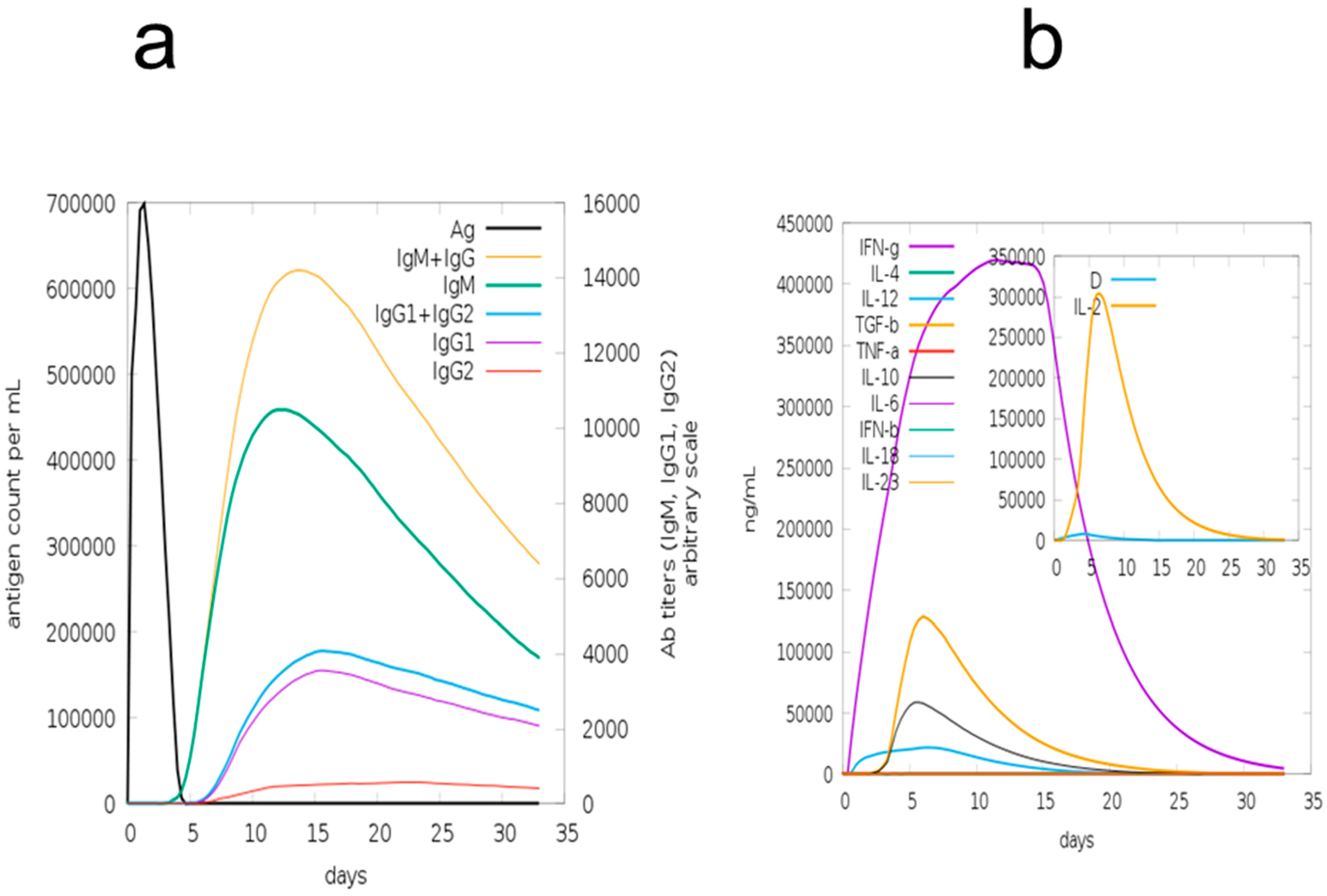
3.12. Immune Simulation
3.13. In Silico Cloning
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forti, K.; Ferroni, L.; Pellegrini, M.; Cruciani, D.; De Giuseppe, A.; Crotti, S.; Papa, P.; Maresca, C.; Severi, G.; Marenzoni, M.L.; et al. Molecular characterization of clostridium perfringens strains isolated in Italy. Toxins 2020, 12, 650. [Google Scholar] [CrossRef] [PubMed]
- Heida, F.H.; Van Zoonen, A.G.J.F.; Hulscher, J.B.F.; Kiefte, B.J.C.T.; Wessels, R.; Kooi, E.M.W.; Bos, A.F.; Harmsen, H.J.M.; de Goffau, M. A Necrotizing enterocolitis-associated gut microbiota is present in the meconium: Results of a prospective study. Clin. Infect. Dis. 2016, 62, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Glil, M.Y.; Thomas, P.; Linde, J.; Busch, A.; Wieler, L.H.; Neubauer, H.; Seyboldt, C. Comparative in silico genome analysis of Clostridium perfringens unravels stable phylogroups with different genome characteristics and pathogenic potential. Sci. Rep. 2021, 11, 6756. [Google Scholar] [CrossRef]
- Broughan, J.; Anderson, R.; Anderson, A.S. Strategies for and advances in the development of Staphylococcus aureusprophylactic vaccines. Expert Rev. Vaccines 2011, 10, 695–708. [Google Scholar] [CrossRef]
- Proctor, R.A. Is there a future for a Staphylococcus aureus vaccine? Vaccine 2012, 30, 2921–2927. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Bruno, L.; Cortese, M.; Rappuoli, R.; Merola, M. Lessons from reverse vaccinology for viral vaccine design. Curr. Opin. Virol. 2015, 11, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [Green Version]
- Rappuoli, R. Reverse vaccinology, a genome-based approach to vaccine development. Vaccine 2001, 19, 2688–2691. [Google Scholar] [CrossRef]
- Bin Sayed, S.; Nain, Z.; Khan, S.A.; Abdulla, F.; Tasmin, R.; Adhikari, U.K. Exploring lassa virus proteome to design a multi-epitope vaccine through immunoinformatics and immune simulation analyses. Int. J. Pept. Res. Ther. 2020, 26, 2089–2107. [Google Scholar] [CrossRef]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel Immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, D.N.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [Green Version]
- Kazi, A.; Chuah, C.; Majeed, A.B.A.; Leow, C.H.; Lim, B.H. Current progress of immunoinformatics approach harnessed for cellular- and antibody-dependent vaccine design. Pathog. Glob. Health 2018, 112, 123–131. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: How to use the entry view. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef] [PubMed]
- Solanki, V.; Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 9044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.-F.; Liu, S.; Dong, C.; Guo, H.-X.; Gao, Y.-Z.; Guo, F.-B. Geptop 2.0: An updated, more precise, and faster geptop server for identification of prokaryotic essential genes. Front. Microbiol. 2019, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Azhagesan, K.; Ravindran, B.; Raman, K. Network-based features enable prediction of essential genes across diverse organisms. PLoS ONE 2018, 13, e0208722. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardy, J.L.; Laird, M.; Brinkman, F.S.L.; Chen, F.; Rey, S.; Walsh, C.J.; Ester, M. PSORTb v2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 2004, 21, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-S.; Cheng, C.-W.; Su, W.-C.; Chang, S.-C.; Huang, S.-W.; Hwang, J.-K.; Lu, C.-H. CELLO2GO: A web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.; Sidney, J.; Bui, H.-H.; Grey, H.M.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wildt, K.F.; Zhu, J.; Zhang, X.; Feigenbaum, L.; Tessarollo, L.; Paul, W.E.; Fowlkes, B.J.; Bosselut, R. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4+ T cells. Nat. Immunol. 2008, 9, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, J.J.; Wang, W.Y.; Mei, Z.G.; Shang, S.L.; Chen, L.-Q.; Liu, Z.-K. A mixed-space approach to first-principles calculations of phonon frequencies for polar materials. J. Phys. Condens. Matter 2010, 22, 202201. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.D. The early history of B cells. Nat. Rev. Immunol. 2015, 15, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Gupta, N.; Kumar, A. Identification of potent vaccine candidates against campylobacter jejuni using immunoinformatics approach. Int. J. Pept. Res. Ther. 2019, 26, 1303–1312. [Google Scholar] [CrossRef]
- Dicker, D.; Nguyen, G.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; et al. Global, regional, and national age-sex-specific mortality and life expectancy, 1950–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1684–1735. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Nezafat, N.; Ghasemi, Y.; Javadi, G.; Khoshnoud, M.J.; Omidinia, E. A novel multi-epitope peptide vaccine against cancer: An in silico approach. J. Theor. Biol. 2014, 349, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Mahram, A.; Herbordt, M.C. Fast and accurate NCBI BLASTP: Acceleration with multiphase FPGA-based prefiltering. In Proceedings of the 24th ACM International Conference on Supercomputing, Tsukuba, Japan, 2–4 June 2010; pp. 73–82. [Google Scholar]
- Bjellqvist, B.; Hughes, G.J.; Pasquali, C.; Paquet, N.; Ravier, F.; Sanchez, J.-C.; Frutiger, S.; Hochstrasser, D. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 1993, 14, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Zheng, W.; Li, Y.; Zhang, C.; Pearce, R.; Mortuza, S.M.; Zhang, Y. Deep-learning contact-map guided protein structure prediction in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1149–1164. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Ponomarenko, J.V.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fu, W.; Zheng, L.; Wang, Y.; Liang, G. Recent progress in the discovery of myeloid differentiation 2 (MD2) modulators for inflammatory diseases. Drug Discov. Today 2018, 23, 1187–1202. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the toll like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A.M. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, N.; Woetzel, N.; Meiler, J. Bcl:Cluster: A method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. In Proceedings of the 2011 IEEE 1st International Conference on Computational Advances in Bio and Medical Sciences (ICCABS), Orlando, FL, USA, 3–5 February 2011; pp. 13–18. [Google Scholar]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef] [PubMed]
- van Aalten, D.; De Groot, B.L.; Findlay, J.B.C.; Berendsen, H.J.C.; Amadei, A. A comparison of techniques for calculating protein essential dynamics. J. Comput. Chem. 1997, 18, 169–181. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Orti, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arumugam, S.; Varamballi, P. In-silico design of envelope based multi-epitope vaccine candidate against Kyasanur forest disease virus. Sci. Rep. 2021, 11, 17118. [Google Scholar] [CrossRef]
- Mahmood, M.; Javaid, A.; Shahid, F.; Ashfaq, U.A. Rational design of multimeric based subunit vaccine against Mycoplasma pneumonia: Subtractive proteomics with immunoinformatics framework. Infect. Genet. Evol. 2021, 91, 104795. [Google Scholar] [CrossRef]
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. Des. Sel. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Lund, F.E. Cytokine-producing B lymphocytes—Key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, J.A.; Chacón, P.; Abagyan, R. Predictions of protein flexibility: First-order measures. Proteins Struct. Funct. Bioinform. 2004, 56, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, M.A.; Li, J.; Beingesser, J.; McClane, B.A.; Uzal, F.A. The Agr-like quorum-sensing system is important for clostridium perfringens type A strain ATCC 3624 to cause gas gangrene in a mouse model. mSphere 2020, 5, e00500–e00520. [Google Scholar] [CrossRef] [PubMed]
- Depla, E.; Van der Aa, A.; Livingston, B.D.; Crimi, C.; Allosery, K.; De Brabandere, V.; Krakover, J.; Murthy, S.; Huang, M.; Power, S.; et al. Rational design of a multiepitope vaccine encoding T-lymphocyte epitopes for treatment of chronic hepatitis B virus infections. J. Virol. 2008, 82, 435–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- María, R.R.; Arturo, C.J.; Alicia, J.A.; Paulina, M.G.; Gerardo, A.O. The impact of bioinformatics on vaccine design and development. Vaccines 2017, 2, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.L.; Zhao, X.; Rappuoli, R.; Seib, K.L.; Zhao, X.; Rappuoli, R.; Seib, K.L.; Zhao, X.; Rappuoli, R.; Seib, K.L.; et al. Developing vaccines in the era of genomics: A decade of reverse vaccinology. Clin. Microbiol. Infect. 2012, 18, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zom, G.G.; Khan, S.; Filippov, D.V.; Ossendorp, F. TLR ligand–peptide conjugate vaccines: Toward clinical application. Adv. Immunol. 2012, 114, 177–201. [Google Scholar] [CrossRef]
- Chew, M.-F.; Poh, K.-S.; Poh, C.-L. Peptides as therapeutic agents for dengue virus. Int. J. Med Sci. 2017, 14, 1342–1359. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.U.; Rafique, S.; Ali, A.; Munir, M.; Ikram, N.; Manan, A.; Salo-Ahen, O.M.H.; Idrees, M. Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016, 6, 37313. [Google Scholar] [CrossRef]
- Unni, P.A.; Ali, A.M.M.T.; Rout, M.; Thabitha, A.; Vino, S.; Lulu, S.S. Designing of an epitope-based peptide vaccine against walking pneumonia: An immunoinformatics approach. Mol. Biol. Rep. 2019, 46, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Cooper, N.R.; Nemerow, G.R. The role of antibody and complement in the control of viral infections. J. Investig. Dermatol. 1984, 83, 121s–127s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.; Fikes, J.; Hoffman, S.; Franke, E.; Sacci, J.; Appella, E.; Chisari, F.; Guidotti, L.G.; Chesnut, R.W.; Livingston, B.; et al. The optimization of helper T lymphocyte (HTL) function in vaccine development. Immunol. Res. 1998, 18, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An overview of novel adjuvants designed for improving vaccine efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef]
- Lee, S.; Nguyen, M.T. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015, 15, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-based subunit vaccine design and evaluation against respiratory syncytial virus using reverse vaccinology approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immuno-informatics characterization SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. bioRxiv 2020. [Google Scholar] [CrossRef]
- Durdagi, S.; Qamar, M.T.U.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Gori, A.; Longhi, R.; Peri, C.; Colombo, G. Peptides for immunological purposes: Design, strategies and applications. Amino Acids 2013, 45, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol. Adv. 2012, 30, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
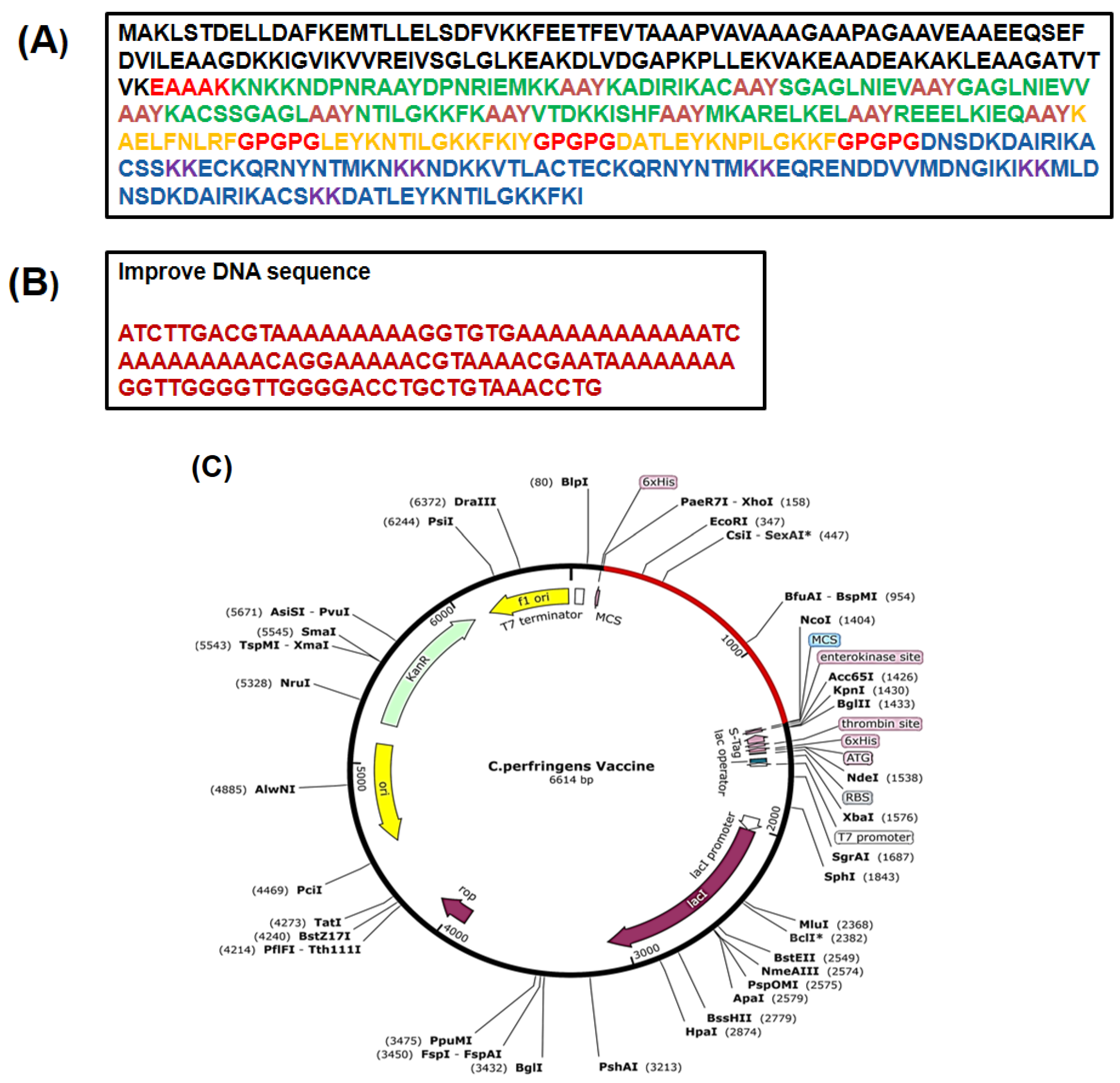

{kind=link}
{kind=link}
{kind=link}
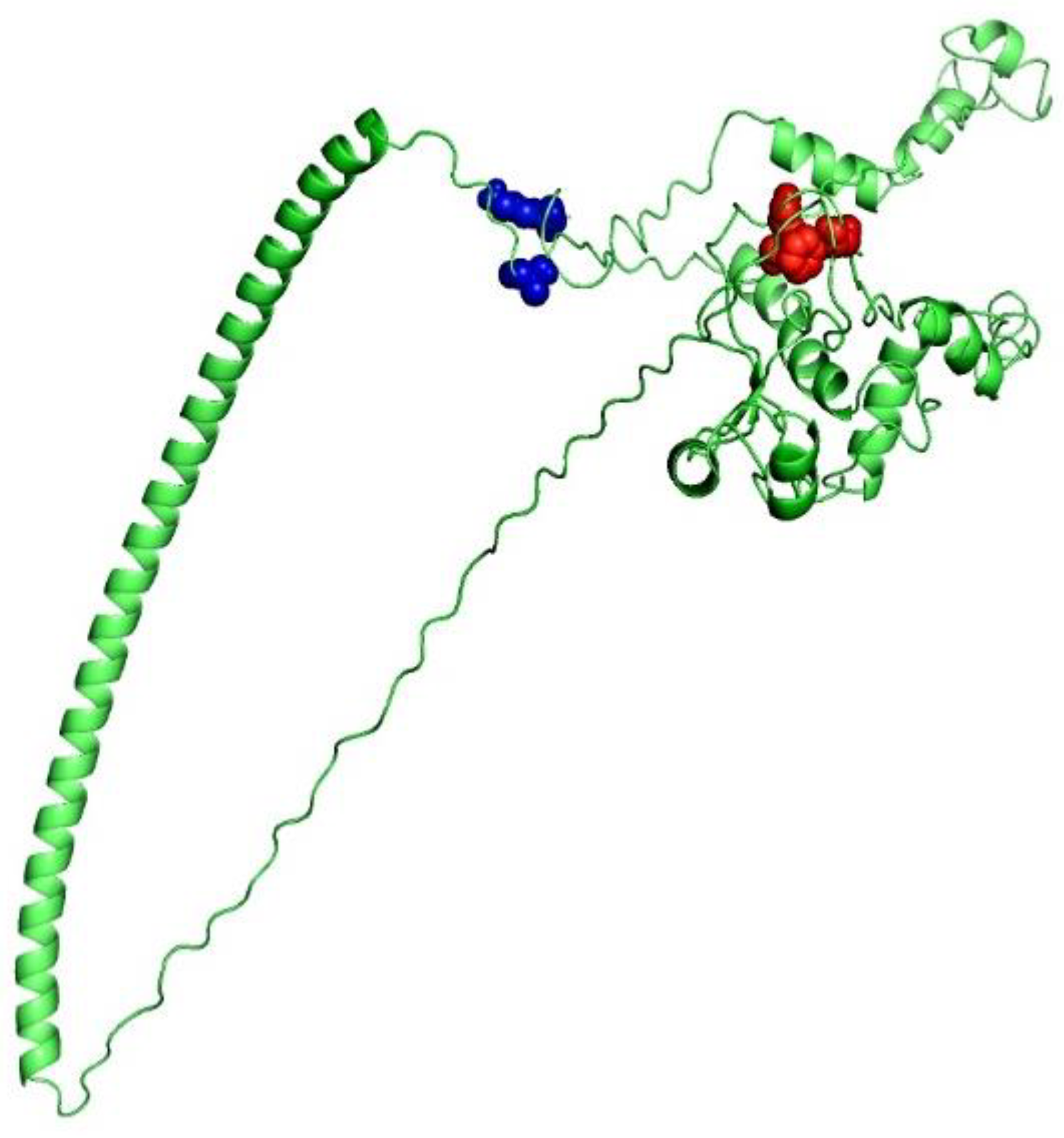{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTL | Antigenicity | Allergenicity | Toxicity | Acc No. | Alleles |
|---|---|---|---|---|---|
| KNKKNDPNR | 0.9965 | NP | Non-toxic | Q0TMN1 | HLA-A*31:01 HLA-E*01:03 HLA-A*30:01 HLA-C*12:03 HLA-B*58:02 |
| CTECKQRNY | 1.1365 | NP | Non-toxic | Q0TMN1 | HLA-A*01:01 HLA-A*30:02 HLA-A*29:02 HLA-A*25:01 HLA-A*26:01 |
| DPNRIEMKK | 1.0997 | NP | Non-toxic | Q0TMN1 | HLA-B*35:03 HLA-A*68:01 HLA-B*53:01 HLA-B*18:01 HLA-A*11:01 |
| ENDDVVMDN | 0.7866 | NP | Non-toxic | A0A0H2YRY0 | HLA-C*05:01 HLA-C*08:02 HLA-C*07:01 HLA-C*04:01 HLA-C*12:03 |
| KDAIRIKAC | 1.2592 | NP | Non-toxic | A0A0H2YRY0 | HLA-B*14:02 HLA-B*40:02 HLA-B*44:02 HLA-B*58: HLA-C*14:02 |
| VVMDNGIKI | 1.1108 | NP | Non-toxic | A0A0H2YRY0 | HLA-B*51:01 HLA-A*02:06 HLA-C*06:02 HLA-A*02:01 HLA-A*23:01 HLA-A*32:01 |
| ILGKKFKIY | 0.9003 | NP | Non-toxic | A0A0H2YRY0 | HLA-A*30:02 HLA-B*08:01 HLA-A*29:02 HLA-E*01:03 HLA-E*01:01 |
| NSDKDAIRI | 1.1108 | NP | Non-toxic | A0A0H2YRY0 | HLA-C*08:02 HLA-C*15:02 HLA-A*01:01 HLA-C*12:03 HLA-B*38:01 HLA-B*39:01 |
| SGAGLNIEV | 2.9786 | NP | Non-toxic | A0A0H2YRY0 | HLA-A*68:02 HLA-A*02:06 HLA-A*02:01 HLA-B*39:01 HLA-B*46:01 |
| GAGLNIEVV | 2.6123 | NP | Non-toxic | A0A0H2YRY0 | HLA-C*12:03 HLA-B*46:01 HLA-C*15:02 HLA-C*07:01 HLA-B*14:02 |
| DVVMDNGIK | 0.7657 | NP | Non-toxic | A0A0H2YRY0 | HLA-A*68:01 HLA-A*25:01 HLA-A*11:01 HLA-A*03:01 HLA-B*18:01 |
| KACSSGAGL | 2.0380 | NP | Non-toxic | A0A0H2YRY0 | HLA-B*48:01 HLA-A*30:01 HLA-B*58:01 HLA-C*15:02 HLA-B*57:01 |
| NTILGKKFK | 0.7983 | NP | Non-toxic | A0A0H2YRY0 | HLA-A*68:01 HLA-A*11:01 HLA-A*30:01 HLA-A*03:01 HLA-A*31:01 HLA-A*25:01 |
| VTDKKISHF | 0.686 | NP | Non-toxic | A0A0H2YRY0 | HLA-B*58:02 HLA-A*01:01 HLA-C*05:01 HLA-A*23:01 HLA-B*58:01 HLA-C*04:01 |
| MKARELKEL | 0.5162 | NP | Non-toxic | Q0TMQ4 | HLA-A*23:01 HLA-B*58:01 HLA-C*04:01 HLA-A*32:01 HLA-B*15:01 HLA-B*46:01 |
| REEELKIEQ | 1.5844 | NP | Non-toxic | Q0TMQ4 | HLA-B*40:01 HLA-B*18:01 HLA-B*44:03 HLA-B*48:01 HLA-B*44:02 HLA-C*05:01 |
| KAELFNLRF | 2.1029 | NP | Non-toxic | Q0TMQ4 | HLA-C*05:01 HLA-A*32:01 HLA-B*58:01 HLA-B*57:01 HLA-A*01:01 HLA-A*24:02 |
| HTL | ALLELES | Antigenicity | P-ID | Toxicity |
|---|---|---|---|---|
| LEYKNTILGKKFKIY | HLA-DRB5*01:05, HLA-DPA1*02:01/DPB1*05:01, HLA-DRB1*11:01 HLA-DRB1*13:07 HLA-DPA1*03:01/DPB1*04:02 | 0.9003 | A0A0H2YRY0 | Non-toxic |
| DATLEYKNTILGKKF | HLA-DRB5*01:05 HLA-DRB1*08:17 HLA-DRB1*08:06 HLA-DRB5*01:01 HLA-DRB5*01:01 | 0.6102 | A0A0H2YRY0 | Non-toxic |
| DNSDKDAIRIKACSS | HLA-DQA1*01:02/DQB1*06:02 HLA-DRB1*11:01 HLA-DRB1*12:01 HLA-DQA1*05:01/DQB1*03:01 HLA-DQA1*03:01/DQB1*03:02 | 1.2464 | A0A0H2YRY0 | Non-toxic |
| B-Cell Epitopes | Protein Acc. No | Antigenicity | Allergenicity | Toxicity | Score |
|---|---|---|---|---|---|
| ECKQRNYNTMKNKKND | Q0TMN1 | 1.2041 | Non-allergen | Not toxic | 0.94 |
| VTLACTECKQRNYNTM | Q0TMN1 | 1.0367 | Non-allergen | Not toxic | 0.81 |
| EQRENDDVVMDNGIKI | A0A0H2YRY0 | 1.0655 | Non-allergen | Not toxic | 0.89 |
| MLDNSDKDAIRIKACS | A0A0H2YRY0 | 0.8408 | Non-allergen | Not toxic | 0.75 |
| DATLEYKNTILGKKFKI | A0A0H2YRY0 | 0.6620 | Non-allergen | Not toxic | 0.66 |
| Parameters | MEV-TLR4 |
|---|---|
| HADDOCK score | 84.2 ± 23.3 |
| Cluster size | 15 |
| RMSD from the overall lowest-energy structure | 0.7 ± 0.4 |
| Van der Waals energy | −150.8 ± 8.6 |
| Electrostatic energy | −538.7 ± 93.2 |
| Desolation energy | −5.3 ± 2.2 |
| Restraints violation energy | 3480.5 ± 117.1 |
| Buried surface area | 5373.2 ± 389.3 |
| Z-score | −2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldakheel, F.M.; Abrar, A.; Munir, S.; Aslam, S.; Allemailem, K.S.; Khurshid, M.; Ashfaq, U.A. Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens. Vaccines 2021, 9, 1079. https://doi.org/10.3390/vaccines9101079
Aldakheel FM, Abrar A, Munir S, Aslam S, Allemailem KS, Khurshid M, Ashfaq UA. Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens. Vaccines. 2021; 9(10):1079. https://doi.org/10.3390/vaccines9101079
Chicago/Turabian StyleAldakheel, Fahad M., Amna Abrar, Samman Munir, Sehar Aslam, Khaled S. Allemailem, Mohsin Khurshid, and Usman Ali Ashfaq. 2021. "Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens" Vaccines 9, no. 10: 1079. https://doi.org/10.3390/vaccines9101079
APA StyleAldakheel, F. M., Abrar, A., Munir, S., Aslam, S., Allemailem, K. S., Khurshid, M., & Ashfaq, U. A. (2021). Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens. Vaccines, 9(10), 1079. https://doi.org/10.3390/vaccines9101079