Bacterial Immunogenicity Prediction by Machine Learning Methods



Abstract
1. Introduction
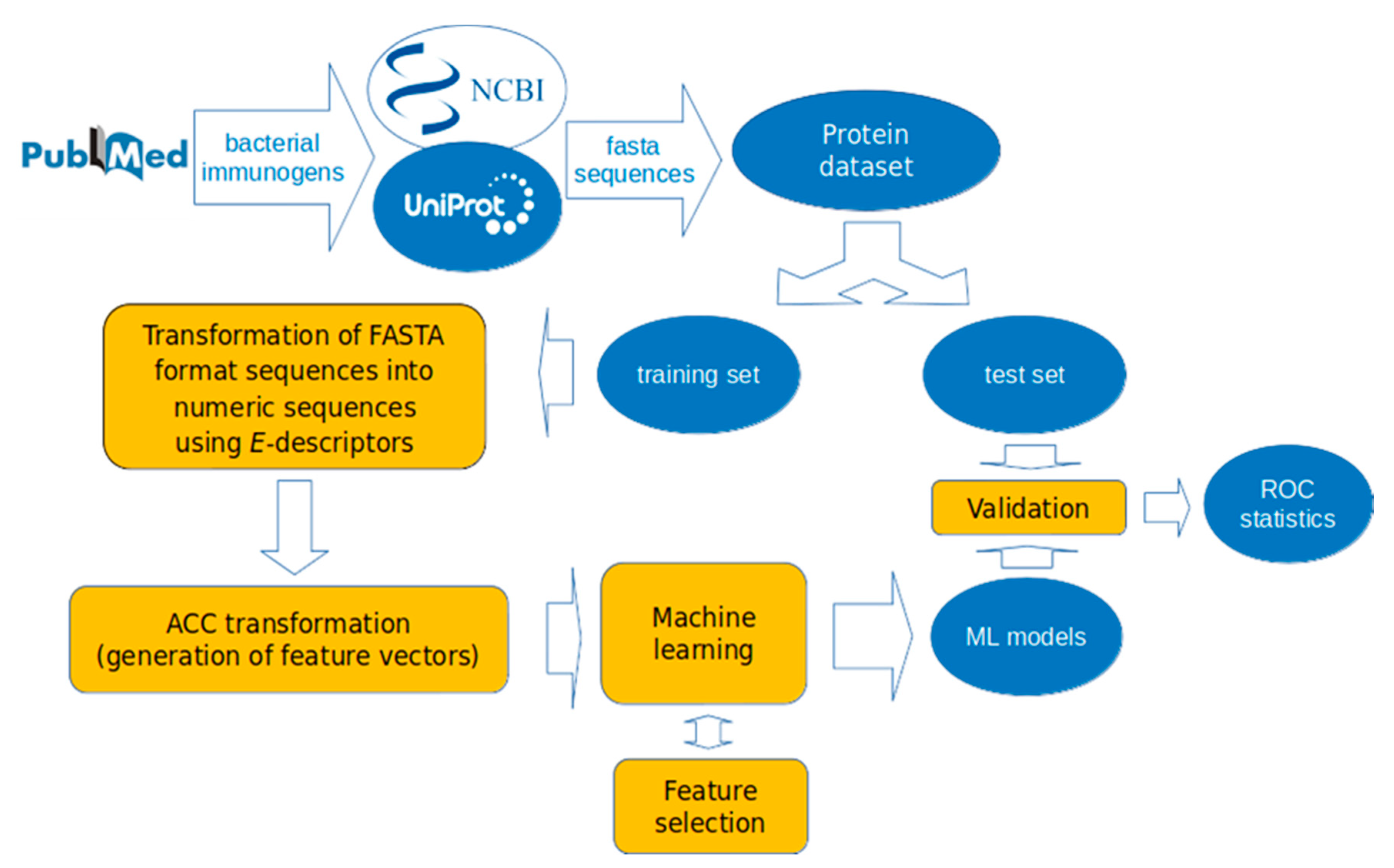
2. Materials and Methods
2.1. Datasets
2.1.1. Dataset of Immunogenic Proteins (Positive Training Set)
2.1.2. Dataset of Non-Immunogenic Proteins (Negative Training Set)
2.1.3. Training and Test Datasets
2.1.4. Dataset for External Evaluation of the Server VaxiJen v3.0
2.2. Descriptors
2.3. Auto-Cross Covariance (ACC) Transformation
2.4. Machine Learning Methods
2.4.1. Partial Least Squares-Based Discriminant Analysis (PLS-DA)
2.4.2. k Nearest Neighbor (kNN)
2.4.3. Support Vector Machine (SVM)
2.4.4. Random Forest (RF)
2.4.5. Random Subspace Method (RSM)
2.4.6. Extreme Gradient Boosting (Xgboost)
2.5. Feature Selection
2.6. Validation of the ML Models
2.7. Implementation of the Best ML Models on a Web Server
2.8. Evaluation of the Server VaxiJen v.3.0
- Fraction of proteome called potential vaccine candidates (PVC) (PVCs/proteome);
- Fraction of BPA identified within the set of PVCs (sensitivity).
- Fold-enrichment expressed as ratio between number of BPAs observed in the set of PVCs and the number expected drawing from the proteome a random sample of the same size of the set of PVCs (statistical significance of the fold-enrichment assessed through an hypergeometric test).
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arnon, R. Overview of vaccine strategies. In Vaccine Design. Innovative Approaches and Novel Strategies; Rappuoli, R., Ed.; Caister Academic Press: Norfolk, UK, 2011; pp. 1–20. [Google Scholar]
- Bagnoli, F.; Norais, N.; Ferlenghi, I.; Scarselli, M.; Donati, C.; Savino, S.; Barocchi, M.A.; Rappuoli, R. Designing vaccines in the era of genomics. In Vaccine Design. Innovative Approaches and Novel Strategies; Rappuoli, R., Ed.; Caister Academic Press: Norfolk, UK, 2011; pp. 21–54. [Google Scholar]
- Zaharieva, N.; Dimitrov, I.; Flower, D.R.; Doytchinova, I. Immunogenicity prediction by VaxiJen: A ten year overview. J. Proteom. Bioinform. 2017, 10, 11. [Google Scholar] [CrossRef]
- Vivona, S.; Bernante, F.; Filippini, F. NERVE: New Enhanced Reverse Vaccinology Environment. BMC Biotechnol. 2006, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiang, Z.; Mobley, H.L.T. Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed Biotechnol. 2010, 297505. [Google Scholar] [CrossRef] [PubMed]
- Goodswen, S.J.; Kennedy, P.J.; Ellis, J.T. Vacceed: A high-throughput in silico vaccine candidate discovery pipeline for eukaryotic pathogens based on reverse vaccinology. Bioinformatics 2014, 30, 2381–2383. [Google Scholar] [CrossRef]
- Jaiswal, V.; Chanumolu, S.K.; Gupta, A.; Chauhan, R.S.; Rout, C. Jenner-predict server: Prediction of protein vaccine candidates (PVCs) in bacteria based on host-pathogen interactions. BMC Bioinform. 2013, 14, 211. [Google Scholar] [CrossRef]
- Moise, L.; Gutierrez, A.; Kibria, F.; Martin, R.; Tassone, R.; Liu, R.; Terry, F.; Martin, B.; De Groot, A.S. iVAX: An integrated toolkit for the selection and optimization of antigens and the design of epitope-driven vaccines. Hum. Vaccines Immunother. 2015, 11, 2312–2321. [Google Scholar] [CrossRef]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef]
- Altindis, E.; Cozzi, R.; Di Palo, B.; Necchi, F.; Mishra, R.P.; Fontana, M.R.; Soriani, M.; Bagnoli, F.; Maione, D.; Grandi, G.; et al. Protectome analysis: A new selective bioinformatics tool for bacterial vaccine candidate discovery. Mol. Cell. Proteom. 2015, 14, 418–429. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Hellberg, S.; Sjöström, M.; Skagerberg, B.; Wold, S. Peptide quantitative structure-activity relationships, a multivariate approach. J. Med. Chem. 1987, 30, 1126–1135. [Google Scholar] [CrossRef]
- Wold, S.; Jonsson, J.; Sjöström, M.; Sandberg, M.; Rännar, S. DNA and peptide sequences and chemical processes multivariately modelled by principal component analysis and partial least squares projections to latent structures. Anal. Chim. Acta 1993, 277, 239–253. [Google Scholar] [CrossRef]
- Leardi, R.; Boggia, R.; Terrile, M. Genetic algorithms as a strategy for feature selection. J. Chemom. 1992, 6, 267–281. [Google Scholar] [CrossRef]
- Dalsass, M.; Brozzi, A.; Medini, D.; Rappuoli, R. Comparison of open-source reverse vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.N.; McAdam, P.R.; Vivona, S.; Zhang, J.X.; Luong, T.; Belew, R.K.; Sahota, H.; Guiney, D.; Valafar, F.; Fierer, J.; et al. Improving reverse vaccinology with a machine learning approach. Vaccine 2011, 29, 8156–8164. [Google Scholar] [CrossRef]
- Heinson, A.I.; Gunawardana, Y.; Moesker, B.; Hume, C.C.D.; Vataga, E.; Hall, Y.; Stylianou, E.; McShane, H.; Williams, A.; Niranjan, M.; et al. Enhancing the biological relevance of machine learning classifiers for reverse vaccinology. Int. J. Mol. Sci. 2017, 18, 312. [Google Scholar] [CrossRef]
- Zaharieva, N.; Dimitrov, I.; Flower, D.R.; Doytchinova, I. VaxiJen dataset of bacterial immunogens: An update. Curr. Comp. Aided Drug Des. 2019, 15, 398–400. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Venkatarajan, M.S.; Braun, W. New quantitative descriptors of amino acids based on multidimensional scaling of a large number of physical-chemical properties. J. Mol. Modeling 2001, 7, 445–453. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. In Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”, 4th ed.; Morgan Kaufmann: Burlington, MA, USA, 2016. [Google Scholar]
- Umetrics, A.B. PLS. In Multi- and Megavariate Data Analysis; Part I; Umetrics Academy: Umea, Sweden, 2006; pp. 63–102. [Google Scholar]
- Song, Y.; Liang, J.; Lu, J.; Zhao, X. An efficient instance selection algorithm for k nearest neighbor regression. Neurocomputing 2017, 251, 26–34. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- El-Manzalawy, Y. WLSVM. Available online: http://www.cs.iastate.edu/~yasser/ (accessed on 25 September 2005).
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5. [Google Scholar] [CrossRef]
- Ho, T.K. The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832–844. [Google Scholar] [CrossRef]
- Li, S.; Harner, E.J.; Adjeroh, D.A. Random KNN. In Proceedings of the IEEE International Conference on Data Mining Workshop, Shenzhen, China, 14 December 2014; pp. 629–636. [Google Scholar] [CrossRef]
- Breiman, L. Arcing the Edge Technical Report 486; Statistics Department, University of California: Berkeley, CA, USA, 1997. [Google Scholar]
- Friedman, J. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 779–785. [Google Scholar] [CrossRef]
- Singh, D.A.G.; Balamurugan, S.A.; Leavline, E.J. Literature review on feature selection methods for high-dimensional data. Int. J. Comput. Appl. 2016, 136, 9–17. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta—Prot. Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]


{kind=link}
| Model | TP | TN | FP | FN | Sensitivity (Recall) | Specificity | Accuracy | Precision | AROC | APR | MCC | F1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLS-DA | ||||||||||||
| Training set | 160 | 168 | 82 | 90 | 0.64 | 0.67 | 0.65 | 0.66 | 0.70 | 0.66 | 0.31 | 0.65 |
| Test set | 41 | 53 | 14 | 26 | 0.61 | 0.79 | 0.70 | 0.74 | 0.74 | 0.76 | 0.41 | 0.67 |
| RF | ||||||||||||
| without FS | ||||||||||||
| Training set | 177 | 191 | 59 | 73 | 0.71 | 0.76 | 0.74 | 0.75 | 0.82 | 0.82 | 0.47 | 0.73 |
| Test set | 47 | 53 | 14 | 20 | 0.70 | 0.79 | 0.75 | 0.77 | 0.83 | 0.84 | 0.50 | 0.73 |
| with FS | ||||||||||||
| Training set | 185 | 190 | 60 | 65 | 0.74 | 0.76 | 0.75 | 0.76 | 0.82 | 0.82 | 0.50 | 0.75 |
| Test set | 48 | 55 | 12 | 19 | 0.72 | 0.82 | 0.77 | 0.80 | 0.85 | 0.83 | 0.54 | 0.76 |
| kNN | ||||||||||||
| Training set | 191 | 181 | 69 | 59 | 0.76 | 0.72 | 0.74 | 0.74 | 0.81 | 0.81 | 0.49 | 0.75 |
| Test set | 50 | 56 | 11 | 17 | 0.75 | 0.84 | 0.79 | 0.82 | 0.83 | 0.84 | 0.58 | 0.78 |
| SVM | ||||||||||||
| Training set | 174 | 199 | 51 | 76 | 0.70 | 0.80 | 0.75 | 0.77 | 0.75 | 0.69 | 0.49 | 0.73 |
| Test set | 49 | 56 | 11 | 18 | 0.73 | 0.84 | 0.78 | 0.82 | 0.78 | 0.73 | 0.57 | 0.77 |
| RSM-1NN | ||||||||||||
| Training set | 190 | 198 | 52 | 60 | 0.76 | 0.79 | 0.78 | 0.78 | 0.85 | 0.87 | 0.55 | 0.77 |
| Test set | 48 | 62 | 5 | 19 | 0.72 | 0.92 | 0.82 | 0.91 | 0.88 | 0.89 | 0.66 | 0.80 |
| xgboost | ||||||||||||
| Training set | 178 | 179 | 71 | 72 | 0.71 | 0.72 | 0.71 | 0.72 | 0.79 | 0.80 | 0.43 | 0.71 |
| Test set | 56 | 50 | 17 | 11 | 0.84 | 0.75 | 0.79 | 0.77 | 0.86 | 0.88 | 0.58 | 0.80 |
| Performances’ Measure | VaxiJen v2.0 | VaxiJen v3.0 |
|---|---|---|
| Number of proteins | 27,055 | 27,055 |
| PVCs | 17,256 | 4825 |
| Fraction of PVC, % | 63.78 | 17.83 |
| Sensitivity, % | 76 | 80 |
| Fold-enrichment | 1.2 | 4.5 |
| p-value | 0.00611 | 8.33x10-42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitrov, I.; Zaharieva, N.; Doytchinova, I. Bacterial Immunogenicity Prediction by Machine Learning Methods. Vaccines 2020, 8, 709. https://doi.org/10.3390/vaccines8040709
Dimitrov I, Zaharieva N, Doytchinova I. Bacterial Immunogenicity Prediction by Machine Learning Methods. Vaccines. 2020; 8(4):709. https://doi.org/10.3390/vaccines8040709
Chicago/Turabian StyleDimitrov, Ivan, Nevena Zaharieva, and Irini Doytchinova. 2020. "Bacterial Immunogenicity Prediction by Machine Learning Methods" Vaccines 8, no. 4: 709. https://doi.org/10.3390/vaccines8040709
APA StyleDimitrov, I., Zaharieva, N., & Doytchinova, I. (2020). Bacterial Immunogenicity Prediction by Machine Learning Methods. Vaccines, 8(4), 709. https://doi.org/10.3390/vaccines8040709