Current Immunotherapy Approaches in Non-Hodgkin Lymphomas


Abstract
1. Introduction
2. Monoclonal Antibodies
2.1. Rituximab and Next-Generation Anti-CD20 Antibodies
2.2. Anti-CD19 Antibodies
2.3. Anti-CD52 Alemtuzumab
2.4. Anti-CCR4 Mogamulizumab
3. Bispecific Antibodies
3.1. T-cell Redirecting Bispecific Antibodies
3.2. First-Generation Bispecific Antibiodies: Blinatumomab
3.3. Second-Generation Bispecific Antibodies
3.3.1. Mosunetuzumab
3.3.2. Glofitamab
3.3.3. REGN1979
3.4. Natural Killer Cell-Activating Antibodies
4. Immune Checkpoint Inhibitors
4.1. CTLA-4 Inhibition and Dual Immune Checkpoint Inhibition in NHL
4.2. PD-1, PD-L1/2 Inhibitors in Specific Subtypes of NHL
4.2.1. Diffuse Large B-Cell Lymphoma
4.2.2. B-NHL with Recurrent Gains of 9p24.1
4.2.3. EBV-Positive NHL
4.2.4. Indolent Lymphomas
4.2.5. T-Cell Lymphomas
4.3. 4-1BB/CD137 Activators and Gain of Effector Functions
4.4. Innate Immune Checkpoint Blockade in NHL
5. Chimeric Antigen Receptor-Based Adoptive Immunotherapy
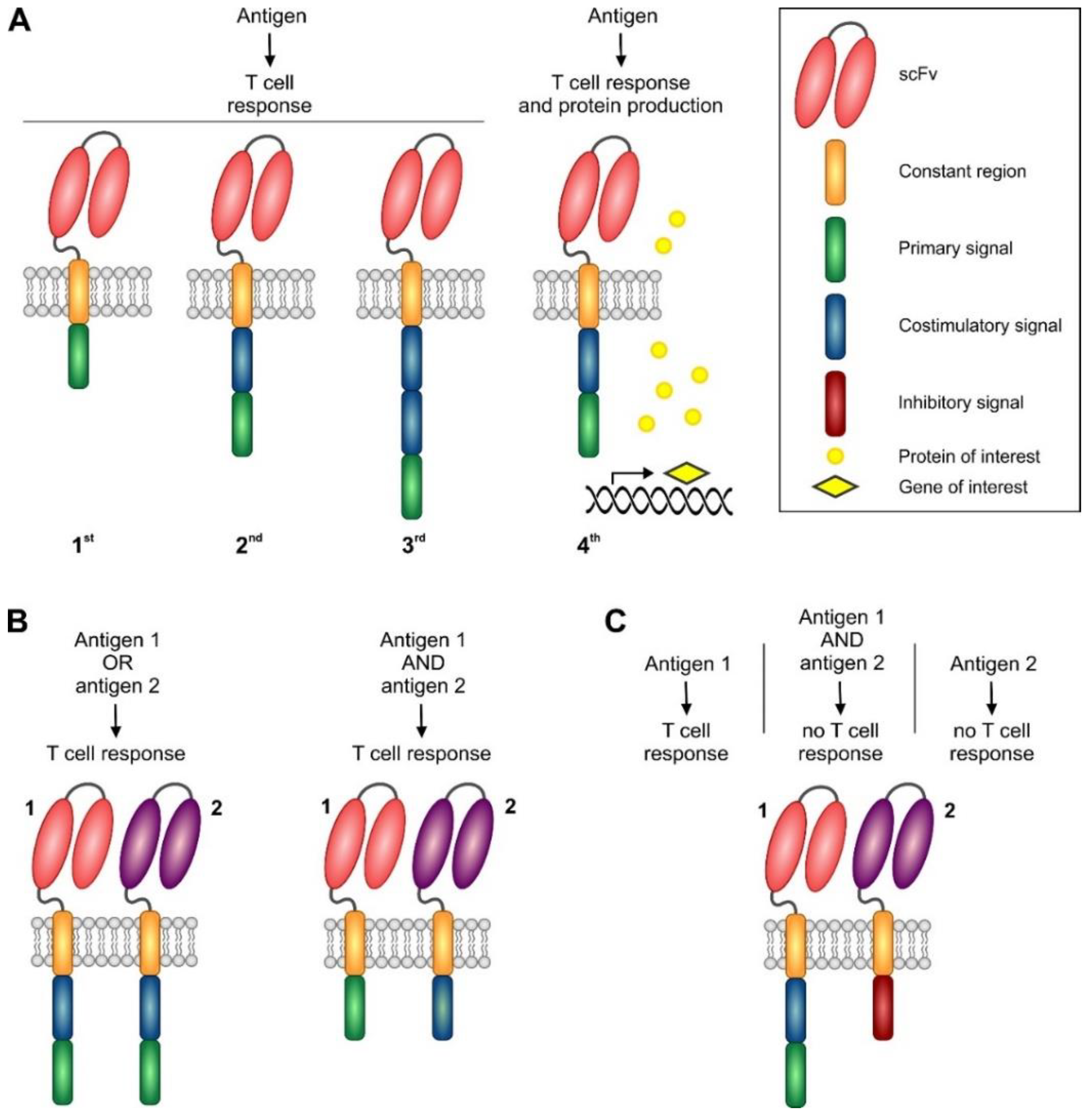
5.1. Basic Principles of CAR Design
5.1.1. CAR Spacer and Transmembrane Domains
5.1.2. First and Second Generation CARs
5.1.3. Third Generation CARs
5.1.4. Next Generation CARs and Future Approaches
5.2. Clinical Results with Registered CAR Products
5.3. Future Directions
5.3.1. CAR T-Cells in Low-Grade Lymphomas
5.3.2. CAR T-Cells in T-NHL
5.3.3. CAR T-Cells as a Consolidation Therapy
5.4. Mechanisms of Resistance to CAR T-Cells
5.5. Universal CAR T-Cells
5.6. CAR-Engineered NK Cells
6. Immunomodulatory Agents (IMiDs)
6.1. Lenalidomide
6.1.1. Lenalidomide and Anti-CD20 Antibodies: R2 and GALEN Regimen
6.1.2. Lenalidomide as a Maintenance Therapy
6.2. Avadomide
7. Toxicity Associated with Immunotherapy
7.1. Infusion Reactions
7.2. Immune-Mediated Adverse Events Associated with Checkpoint Inhibitors
7.3. Cytokine Release Syndrome
7.4. Neurotoxicity
8. Conclusions
Funding
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Bronstein, E.; Geller, W. Combined therapy in malignant lymphoma. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1966, 96, 167–170. [Google Scholar] [CrossRef]
- Griffin, M.M.; Morley, N. Rituximab in the treatment of non-Hodgkin’s lymphoma--a critical evaluation of randomized controlled trials. Expert Opin. Biol. Ther. 2013, 13, 803–811. [Google Scholar] [CrossRef]
- Motta, G.; Cea, M.; Moran, E.; Carbone, F.; Augusti, V.; Patrone, F.; Nencioni, A. Monoclonal antibodies for non-Hodgkin’s lymphoma: State of the art and perspectives. Clin. Dev. Immunol. 2010, 2010, 428253. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef]
- Rogers, L.M.; Veeramani, S.; Weiner, G.J. Complement in monoclonal antibody therapy of cancer. Immunol. Res. 2014, 59, 203–210. [Google Scholar] [CrossRef]
- Hilchey, S.P.; Hyrien, O.; Mosmann, T.R.; Livingstone, A.M.; Friedberg, J.W.; Young, F.; Fisher, R.I.; Kelleher, R.J., Jr.; Bankert, R.B.; Bernstein, S.H. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: Support for a “vaccinal effect” of rituximab. Blood 2009, 113, 3809–3812. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Jung, S.T. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Link, B.K.; Friedberg, J.W. Monoclonal antibodies in lymphoma: The first decade. Semin. Hematol. 2008, 45, 71–74. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, W.S.; Buske, C.; Ogura, M.; Jurczak, W.; Sancho, J.M.; Zhavrid, E.; Kim, J.S.; Hernández-Rivas, J.; Prokharau, A.; Vasilica, M.; et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: A randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017, 4, e362–e373. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.S.; Illmer, T.; et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Chua, N.; Mayer, J.; Dueck, G.; Trněný, M.; Bouabdallah, K.; Fowler, N.; Delwail, V.; Press, O.; Salles, G.; et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): A randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016, 17, 1081–1093. [Google Scholar] [CrossRef]
- Sehn, L.H.; Martelli, M.; Trněný, M.; Liu, W.; Bolen, C.R.; Knapp, A.; Sahin, D.; Sellam, G.; Vitolo, U. A randomized, open-label, Phase III study of obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-Cell lymphoma: Final analysis of GOYA. J. Hematol. Oncol. 2020, 13, 71. [Google Scholar] [CrossRef]
- Coiffier, B.; Lepretre, S.; Pedersen, L.M.; Gadeberg, O.; Fredriksen, H.; van Oers, M.H.; Wooldridge, J.; Kloczko, J.; Holowiecki, J.; Hellmann, A.; et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: A phase 1-2 study. Blood 2008, 111, 1094–1100. [Google Scholar] [CrossRef]
- Wierda, W.G.; Kipps, T.J.; Mayer, J.; Stilgenbauer, S.; Williams, C.D.; Hellmann, A.; Robak, T.; Furman, R.R.; Hillmen, P.; Trneny, M.; et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 1749–1755. [Google Scholar] [CrossRef]
- Hillmen, P.; Robak, T.; Janssens, A.; Babu, K.G.; Kloczko, J.; Grosicki, S.; Doubek, M.; Panagiotidis, P.; Kimby, E.; Schuh, A.; et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): A randomised, multicentre, open-label phase 3 trial. Lancet 2015, 385, 1873–1883. [Google Scholar] [CrossRef]
- Sawas, A.; Farber, C.M.; Schreeder, M.T.; Khalil, M.Y.; Mahadevan, D.; Deng, C.; Amengual, J.E.; Nikolinakos, P.G.; Kolesar, J.M.; Kuhn, J.G.; et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br. J. Haematol. 2017, 177, 243–253. [Google Scholar] [CrossRef]
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and umbralisib in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2019, 134, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Leonard, J.P.; Fayad, L.; Coiffier, B.; Petillon, M.O.; Coleman, M.; Schuster, S.J.; Dyer, M.J.; Horne, H.; Teoh, N.; et al. Humanized anti-CD20 antibody, veltuzumab, in refractory/recurrent non-Hodgkin’s lymphoma: Phase I/II results. J. Clin. Oncol. 2009, 27, 3346–3353. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Marlton, P.; Vitolo, U.; Lindén, O.; Seymour, J.F.; Crump, M.; Coiffier, B.; Foà, R.; Wassner, E.; Burger, H.U.; et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann. Oncol. 2010, 21, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef]
- Jurczak, W.; Zinzani, P.L.; Gaidano, G.; Goy, A.; Provencio, M.; Nagy, Z.; Robak, T.; Maddocks, K.; Buske, C.; Ambarkhane, S.; et al. Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2018, 29, 1266–1272. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; González Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Hamadani, M.; Forero, A.; Kipps, T.J.; Fanale, M.A.; Cuneo, A.; Oteyza, J.P.d.; Gladstone, D.; Goswami, T.; Ibrahim, R.A.; Liang, M.; et al. MEDI-551, an anti-CD19 antibody active in chronic lymphocytic leukemia (CLL) patients previously treated with rituximab. J. Clin. Oncol. 2013, 31, 7045. [Google Scholar] [CrossRef]
- Ohmachi, K.; Ogura, M.; Suehiro, Y.; Ando, K.; Uchida, T.; Choi, I.; Ogawa, Y.; Kobayashi, M.; Fukino, K.; Yokoi, Y.; et al. A multicenter phase I study of inebilizumab, a humanized anti-CD19 monoclonal antibody, in Japanese patients with relapsed or refractory B-cell lymphoma and multiple myeloma. Int. J. Hematol. 2019, 109, 657–664. [Google Scholar] [CrossRef]
- Lepretre, S.; Aurran, T.; Mahé, B.; Cazin, B.; Tournilhac, O.; Maisonneuve, H.; Casasnovas, O.; Delmer, A.; Leblond, V.; Royer, B.; et al. Excess mortality after treatment with fludarabine and cyclophosphamide in combination with alemtuzumab in previously untreated patients with chronic lymphocytic leukemia in a randomized phase 3 trial. Blood 2012, 119, 5104–5110. [Google Scholar] [CrossRef]
- Binder, C.; Ziepert, M.; Pfreundschuh, M.; Dührsen, U.; Eimermacher, H.; Aldaoud, A.; Rosenwald, A.; Loeffler, M.; Schmitz, N.; Truemper, L. CHO(E)P-14 followed by alemtuzumab consolidation in untreated peripheral T cell lymphomas: Final analysis of a prospective phase II trial. Ann. Hematol. 2013, 92, 1521–1528. [Google Scholar] [CrossRef]
- Roswarski, J.; Roschewski, M.; Melani, C.; Pittaluga, S.; Lucas, A.; Steinberg, S.M.; Jaffe, E.S.; Waldmann, T.A.; Wilson, W.H. Phase 1/2 study of alemtuzumab with dose-adjusted EPOCH in untreated aggressive T and NK cell lymphomas. Leuk Lymphoma 2019, 60, 2062–2066. [Google Scholar] [CrossRef] [PubMed]
- Lundin, J.; Hagberg, H.; Repp, R.; Cavallin-Ståhl, E.; Fredén, S.; Juliusson, G.; Rosenblad, E.; Tjønnfjord, G.; Wiklund, T.; Osterborg, A. Phase 2 study of alemtuzumab (anti-CD52 monoclonal antibody) in patients with advanced mycosis fungoides/Sezary syndrome. Blood 2003, 101, 4267–4272. [Google Scholar] [CrossRef] [PubMed]
- Dearden, C.E.; Matutes, E.; Cazin, B.; Tjønnfjord, G.E.; Parreira, A.; Nomdedeu, B.; Leoni, P.; Clark, F.J.; Radia, D.; Rassam, S.M.; et al. High remission rate in T-cell prolymphocytic leukemia with CAMPATH-1H. Blood 2001, 98, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Aribi, A.; O’Brien, S.; Faderl, S.; Jones, D.; Ferrajoli, A.; Huang, X.; York, S.; Pierce, S.; Wierda, W.; et al. Phase II study of alemtuzumab in combination with pentostatin in patients with T-cell neoplasms. J. Clin. Oncol. 2009, 27, 5425–5430. [Google Scholar] [CrossRef]
- Ishida, T.; Utsunomiya, A.; Iida, S.; Inagaki, H.; Takatsuka, Y.; Kusumoto, S.; Takeuchi, G.; Shimizu, S.; Ito, M.; Komatsu, H.; et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: Its close association with skin involvement and unfavorable outcome. Clin. Cancer Res. 2003, 9, 3625–3634. [Google Scholar]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: A multicenter phase II study. J. Clin. Oncol. 2012, 30, 837–842. [Google Scholar] [CrossRef]
- Ishida, T.; Jo, T.; Takemoto, S.; Suzushima, H.; Uozumi, K.; Yamamoto, K.; Uike, N.; Saburi, Y.; Nosaka, K.; Utsunomiya, A.; et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: A randomized phase II study. Br. J. Haematol. 2015, 169, 672–682. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Duell, J.; Lammers, P.E.; Djuretic, I.; Chunyk, A.G.; Alekar, S.; Jacobs, I.; Gill, S. Bispecific Antibodies in the Treatment of Hematologic Malignancies. Clin. Pharmacol. Ther. 2019, 106, 781–791. [Google Scholar] [CrossRef]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umaña, P.; Mössner, E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Tita-Nwa, F.; Moldenhauer, G.; Herbst, M.; Kleist, C.; Ho, A.D.; Kornacker, M. Cytokine-induced killer cells targeted by the novel bispecific antibody CD19xCD5 (HD37xT5.16) efficiently lyse B-lymphoma cells. Cancer Immunol. Immunother. 2007, 56, 1911–1920. [Google Scholar] [CrossRef]
- Sun, L.L.; Ellerman, D.; Mathieu, M.; Hristopoulos, M.; Chen, X.; Li, Y.; Yan, X.; Clark, R.; Reyes, A.; Stefanich, E.; et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci. Transl. Med. 2015, 7, 287ra270. [Google Scholar] [CrossRef]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Coyle, L.; Morley, N.J.; Rambaldi, A.; Mason, K.D.; Verhoef, G.; Furness, C.L.; Zhang, A.; Jung, A.S.; Cohan, D.; Franklin, J.L. Open-Label, phase 2 study of blinatumomab as second salvage therapy in adults with relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Leuk Lymphoma 2020, 61, 2103–2112. [Google Scholar] [CrossRef]
- Ferl, G.Z.; Reyes, A.; Sun, L.L.; Cheu, M.; Oldendorp, A.; Ramanujan, S.; Stefanich, E.G. A Preclinical Population Pharmacokinetic Model for Anti-CD20/CD3 T-Cell-Dependent Bispecific Antibodies. Clin. Transl. Sci. 2018, 11, 296–304. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bartlett, N.L.; Assouline, S.; Yoon, S.-S.; Bosch, F.; Sehn, L.H.; Cheah, C.Y.; Shadman, M.; Gregory, G.P.; Ku, M.; et al. Mosunetuzumab Induces Complete Remissions in Poor Prognosis Non-Hodgkin Lymphoma Patients, Including Those Who Are Resistant to or Relapsing After Chimeric Antigen Receptor T-Cell (CAR-T) Therapies, and Is Active in Treatment through Multiple Lines. Blood 2019, 134, 6. [Google Scholar] [CrossRef]
- Bartlett, N.L.; Sehn, L.H.; Assouline, S.E.; Bosch, F.; Diefenbach, C.S.M.; Flinn, I.; Hong, J.; Kim, W.S.; Matasar, M.J.; Nastoupil, L.J.; et al. Managing cytokine release syndrome (CRS) and neurotoxicity with step-fractionated dosing of mosunetuzumab in relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (NHL). J. Clin. Oncol. 2019, 37, 7518. [Google Scholar] [CrossRef]
- Bacac, M.; Colombetti, S.; Herter, S.; Sam, J.; Perro, M.; Chen, S.; Bianchi, R.; Richard, M.; Schoenle, A.; Nicolini, V.; et al. CD20-TCB with Obinutuzumab Pretreatment as Next-Generation Treatment of Hematologic Malignancies. Clin. Cancer Res. 2018, 24, 4785–4797. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Klein, C.; Umana, P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology 2016, 5, e1203498. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.J.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martinez, J.; Crump, M.; Thomas, D.N.; et al. Cd20-Tcb (Rg6026), A Novel “2:1” Format T-Cell-Engaging Bispecific Antibody, Induces Complete Remissions in Relapsed/Refractory B-Cell Non-Hodgkin’s Lymphoma. Hematol. Oncol. 2019, 37, 92–93. [Google Scholar] [CrossRef]
- Hutchings, M.; Iacoboni, G.; Morschhauser, F.; Offner, F.; Sureda, A.; Salles, G.A.; Carlo-Stella, C.; Martinez Lopez, J.; Thomas, D.; Morcos, P.N.; et al. CD20-Tcb (RG6026), a Novel “2:1” Format T-Cell-Engaging Bispecific Antibody, Induces Complete Remissions in Relapsed/Refractory B-Cell Non-Hodgkin’s Lymphoma: Preliminary Results from a Phase I First in Human Trial. Blood 2018, 132, 226. [Google Scholar] [CrossRef]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.H.; Barnes, J.A.; Ansell, S.M.; O’Brien, S.M.; Chavez, J.; Duell, J.; et al. Clinical Activity of REGN1979, a Bispecific Human, Anti-CD20 x Anti-CD3 Antibody, in Patients with Relapsed/Refractory (R/R) B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood 2019, 134, 762. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Y.; Liu, L.L.; Lundqvist, A. Strategies to Augment Natural Killer (NK) Cell Activity against Solid Tumors. Cancers 2019, 11, 1040. [Google Scholar] [CrossRef]
- Grzywacz, B.; Kataria, N.; Verneris, M.R. CD56dimCD16+ NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia 2007, 21, 356–359. [Google Scholar] [CrossRef]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef] [PubMed]
- Tun, A.M.; Ansell, S.M. Immunotherapy in Hodgkin and non-Hodgkin lymphoma: Innate, adaptive and targeted immunological strategies. Cancer Treat. Rev. 2020, 88, 102042. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Kuzume, A.; Chi, S.; Yamauchi, N.; Minami, Y. Immune-Checkpoint Blockade Therapy in Lymphoma. Int. J. Mol. Sci. 2020, 21, 5456. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Okazaki, I.M.; Sugiura, D.; Takahashi, S.; Maeda, T.K.; Shimizu, K.; Okazaki, T. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 2018, 19, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Song, Y.; Zhu, J. Immune checkpoint inhibitors in malignant lymphoma: Advances and perspectives. Chin. J. Cancer Res. 2020, 32, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef]
- Ansell, S.M.; Hurvitz, S.A.; Koenig, P.A.; LaPlant, B.R.; Kabat, B.F.; Fernando, D.; Habermann, T.M.; Inwards, D.J.; Verma, M.; Yamada, R.; et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2009, 15, 6446–6453. [Google Scholar] [CrossRef]
- Ansell, S.; Gutierrez, M.E.; Shipp, M.A.; Gladstone, D.; Moskowitz, A.; Borello, I.; Popa-Mckiver, M.; Farsaci, B.; Zhu, L.; Lesokhin, A.M.; et al. A Phase 1 Study of Nivolumab in Combination with Ipilimumab for Relapsed or Refractory Hematologic Malignancies (CheckMate 039). Blood 2016, 128, 183. [Google Scholar] [CrossRef]
- Jelinek, T.; Mihalyova, J.; Kascak, M.; Duras, J.; Hajek, R. PD-1/PD-L1 inhibitors in haematological malignancies: Update 2017. Immunology 2017, 152, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Minnema, M.C.; Johnson, P.; Timmerman, J.M.; Armand, P.; Shipp, M.A.; Rodig, S.J.; Ligon, A.H.; Roemer, M.G.M.; Reddy, N.; et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Nagler, A.; Weller, E.A.; Devine, S.M.; Avigan, D.E.; Chen, Y.B.; Kaminski, M.S.; Holland, H.K.; Winter, J.N.; Mason, J.R.; et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: Results of an international phase II trial. J. Clin. Oncol. 2013, 31, 4199–4206. [Google Scholar] [CrossRef]
- Chapuy, B.; Roemer, M.G.; Stewart, C.; Tan, Y.; Abo, R.P.; Zhang, L.; Dunford, A.J.; Meredith, D.M.; Thorner, A.R.; Jordanova, E.S.; et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 2016, 127, 869–881. [Google Scholar] [CrossRef]
- Nayak, L.; Iwamoto, F.M.; LaCasce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073. [Google Scholar] [CrossRef]
- Shi, M.; Roemer, M.G.; Chapuy, B.; Liao, X.; Sun, H.; Pinkus, G.S.; Shipp, M.A.; Freeman, G.J.; Rodig, S.J. Expression of programmed cell death 1 ligand 2 (PD-L2) is a distinguishing feature of primary mediastinal (thymic) large B-cell lymphoma and associated with PDCD1LG2 copy gain. Am. J. Surg. Pathol. 2014, 38, 1715–1723. [Google Scholar] [CrossRef]
- Melani, C.; Major, A.; Schowinsky, J.; Roschewski, M.; Pittaluga, S.; Jaffe, E.S.; Pack, S.D.; Abdullaev, Z.; Ahlman, M.A.; Kwak, J.J.; et al. PD-1 Blockade in Mediastinal Gray-Zone Lymphoma. N. Engl. J. Med. 2017, 377, 89–91. [Google Scholar] [CrossRef]
- Armand, P.; Rodig, S.; Melnichenko, V.; Thieblemont, C.; Bouabdallah, K.; Tumyan, G.; Ozcan, M.; Portino, S.; Fogliatto, L.; Caballero, M.D.; et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 3291–3299. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Ribrag, V.; Moskowitz, C.H.; Michot, J.M.; Kuruvilla, J.; Balakumaran, A.; Zhang, Y.; Chlosta, S.; Shipp, M.A.; Armand, P. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017, 130, 267–270. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Santoro, A.; Gritti, G.; Brice, P.; Barr, P.M.; Kuruvilla, J.; Cunningham, D.; Kline, J.; Johnson, N.A.; Mehta-Shah, N.; et al. Nivolumab Combined With Brentuximab Vedotin for Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma: Efficacy and Safety From the Phase II CheckMate 436 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.; Raffeld, M.; Jaffe, E.S. EBV-positive large B-cell lymphomas in young patients: A nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of Efficacy of Pembrolizumab between Epstein-Barr VirusPositive and Negative Relapsed or Refractory Non-Hodgkin Lymphomas. Cancer Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef]
- Richendollar, B.G.; Pohlman, B.; Elson, P.; Hsi, E.D. Follicular programmed death 1-positive lymphocytes in the tumor microenvironment are an independent prognostic factor in follicular lymphoma. Hum. Pathol. 2011, 42, 552–557. [Google Scholar] [CrossRef]
- Armand, P.; Janssens, A.M.; Gritti, G.; Radford, J.; Timmerman, J.M.; Pinto, A.; Mercadal Vilchez, S.; Johnson, P.W.M.; Cunningham, D.; Leonard, J.P.; et al. Efficacy and safety results from CheckMate 140, a phase 2 study of nivolumab for relapsed/refractory follicular lymphoma. Blood 2020. [Google Scholar] [CrossRef]
- Ding, W.; Laplant, B.; Witzig, T.E.; Johnston, P.B.; Colgan, J.P.; Rech, K.L.; Leis, J.F.; Feldman, A.L.; He, R.; Nowakowski, G.S.; et al. PD-1 Blockade with Pembrolizumab in Relapsed Low Grade Non-Hodgkin Lymphoma. Blood 2017, 130, 4055. [Google Scholar] [CrossRef]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Westin, J.R.; Fowler, N.H.; Fanale, M.A.; Samaniego, F.; Oki, Y.; Obi, C.; Cao, J.; Cheng, X.; Ma, M.C.J.; et al. Response rates with pembrolizumab in combination with rituximab in patients with relapsed follicular lymphoma: Interim results of an on open-label, phase II study. J. Clin. Oncol. 2017, 35, 7519. [Google Scholar] [CrossRef]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, A.; Al-Juhaishi, T.; Davila, E.; Haverkos, B. Enhancing antitumor immunity through checkpoint blockade as a therapeutic strategy in T-cell lymphomas. Blood Adv. 2020, 4, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sézary Syndrome: A Multicenter Phase II Study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.P.; Neelapu, S.S.; Burns, E.; Nair, R.; Hosing, C.; Nieto, Y.; Westin, J.R.; Parmar, S.; Fowler, N.H.; Nastoupil, L.J.; et al. A Phase I/II Study to Examine the Safety and Efficacy of Pembrolizumab 200 Mg Fixed Dose Administered Every 3 Weeks (Q3W) in Combination with Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma (PTCL). Blood 2019, 134, 1546. [Google Scholar] [CrossRef]
- Fisher, T.S.; Kamperschroer, C.; Oliphant, T.; Love, V.A.; Lira, P.D.; Doyonnas, R.; Bergqvist, S.; Baxi, S.M.; Rohner, A.; Shen, A.C. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol. Immunother. 2012, 61, 1721–1733. [Google Scholar] [CrossRef]
- Gopal, A.K.; Levy, R.; Houot, R.; Patel, S.P.; Popplewell, L.; Jacobson, C.; Mu, X.J.; Deng, S.; Ching, K.A.; Chen, Y.; et al. First-in-Human Study of Utomilumab, a 4-1BB/CD137 Agonist, in Combination with Rituximab in Patients with Follicular and Other CD20+ Non-Hodgkin Lymphomas. Clin. Cancer Res. 2020, 26, 2524–2534. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Hüsser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Compte, M.; Harwood, S.L.; Muñoz, I.G.; Navarro, R.; Zonca, M.; Perez-Chacon, G.; Erce-Llamazares, A.; Merino, N.; Tapia-Galisteo, A.; Cuesta, A.M.; et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat. Commun. 2018, 9, 4809. [Google Scholar] [CrossRef]
- Seiffert, M.; Cant, C.; Chen, Z.; Rappold, I.; Brugger, W.; Kanz, L.; Brown, E.J.; Ullrich, A.; Bühring, H.-J. Human Signal-Regulatory Protein Is Expressed on Normal, But Not on Subsets of Leukemic Myeloid Cells and Mediates Cellular Adhesion Involving Its Counterreceptor CD47. Blood 1999, 94, 3633–3643. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.N.; Van den Berg, T.K. The Interaction Between Signal Regulatory Protein Alpha (SIRPα) and CD47: Structure, Function, and Therapeutic Target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Flinn, I.W.; Maris, M.B.; O’Connor, O.A.; Lesokhin, A.; Advani, A.S.; Minden, M.D.; Percival, M.B.M.; Johnson, L.D.; Catalano, T.; et al. TTI-621 (SIRPαFc), an Immune Checkpoint Inhibitor Blocking the CD47 “Do Not Eat” Signal, Induces Objective Responses in Patients with Advanced, Relapsed/Refractory Diffuse Large B-Cell Lymphoma (DLBCL). Blood 2017, 130, 4116. [Google Scholar] [CrossRef]
- Querfeld, C.; Thompson, J.A.; Taylor, M.; PILLAI, R.K.; DS, L.; JOHNSON, T.C.; Petrova, P.S.; UGER, R.A.; Irwin, M.; Thompson, T. Intralesional Injection of the CD47-blocking immune checkpoint inhibitor TTI-621 (SIRPαFc) induces antitumor activity in patients with relapsed/refractory mycosis fungoides and Sezary syndrome: Interim results of a multicenter Phase 1 trial. Eur. J. Cancer 2018, 101, S34. [Google Scholar] [CrossRef]
- Schneider, C.K.; Salmikangas, P.; Jilma, B.; Flamion, B.; Todorova, L.R.; Paphitou, A.; Haunerova, I.; Maimets, T.; Trouvin, J.H.; Flory, E.; et al. Challenges with advanced therapy medicinal products and how to meet them. Nat. Rev. Drug Discov. 2010, 9, 195–201. [Google Scholar] [CrossRef]
- Lock, D.; Mockel-Tenbrinck, N.; Drechsel, K.; Barth, C.; Mauer, D.; Schaser, T.; Kolbe, C.; Al Rawashdeh, W.; Brauner, J.; Hardt, O.; et al. Automated Manufacturing of Potent CD20-Directed Chimeric Antigen Receptor T Cells for Clinical Use. Hum. Gene Ther. 2017, 28, 914–925. [Google Scholar] [CrossRef]
- Zhu, F.; Shah, N.; Xu, H.; Schneider, D.; Orentas, R.; Dropulic, B.; Hari, P.; Keever-Taylor, C.A. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy 2018, 20, 394–406. [Google Scholar] [CrossRef]
- Jackson, Z.; Roe, A.; Sharma, A.A.; Lopes, F.B.T.P.; Talla, A.; Kleinsorge-Block, S.; Zamborsky, K.; Schiavone, J.; Manjappa, S.; Schauner, R.; et al. Automated Manufacture of Autologous CD19 CAR-T Cells for Treatment of Non-hodgkin Lymphoma. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Romeo, C.; Seed, B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell 1991, 64, 1037–1046. [Google Scholar] [CrossRef]
- Letourneur, F.; Klausner, R.D. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Abken, H. CAR T Cells: A Snapshot on the Growing Options to Design a CAR. Hemasphere 2019, 3, e172. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Faitschuk, E.; Nagy, V.; Hombach, A.A.; Abken, H. A dual chain chimeric antigen receptor (CAR) in the native antibody format for targeting immune cells towards cancer cells without the need of an scFv. Gene Ther. 2016, 23, 718–726. [Google Scholar] [CrossRef]
- Yang, E.Y.; Shah, K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front. Oncol. 2020, 10, 1182. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar]
- Demoulin, B.; Cook, W.J.; Murad, J.; Graber, D.J.; Sentman, M.L.; Lonez, C.; Gilham, D.E.; Sentman, C.L.; Agaugue, S. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017, 13, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 2017, 7, e1368604. [Google Scholar] [CrossRef]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [PubMed]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, J.S.; Ladell, K.; Sheard, V.E.; Miners, K.; Hawkins, R.E.; Price, D.A.; Gilham, D.E. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: Implications for optimization of receptor structure for adoptive cell therapy. Clin. Exp. Immunol. 2014, 175, 258–267. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal 2018, 11. [Google Scholar] [CrossRef]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D., Jr.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Cheng, Z.; Wei, R.; Ma, Q.; Shi, L.; He, F.; Shi, Z.; Jin, T.; Xie, R.; Wei, B.; Chen, J.; et al. In Vivo Expansion and Antitumor Activity of Coinfused CD28- and 4-1BB-Engineered CAR-T Cells in Patients with B Cell Leukemia. Mol. Ther. 2018, 26, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Strömberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Hombach, A.A.; Rappl, G.; Abken, H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol. Ther. 2013, 21, 2268–2277. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Sesques, P.; Ferrant, E.; Safar, V.; Wallet, F.; Tordo, J.; Dhomps, A.; Karlin, L.; Brisou, G.; Vercasson, M.; Hospital-Gustem, C.; et al. Commercial anti-CD19 CAR T cell therapy for patients with relapsed/refractory aggressive B cell lymphoma in a European center. Am. J. Hematol. 2020. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Jacobson, C.A.; Oluwole, O.O.; Munoz, J.; Deol, A.; Miklos, D.B.; Bartlett, N.L.; Braunschweig, I.; Jiang, Y.; Kim, J.J.; et al. Outcomes of older patients in ZUMA-1, a pivotal study of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood 2020, 135, 2106–2109. [Google Scholar] [CrossRef]
- Frigault, M.J.; Dietrich, J.; Martinez-Lage, M.; Leick, M.; Choi, B.D.; DeFilipp, Z.; Chen, Y.B.; Abramson, J.; Crombie, J.; Armand, P.; et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood 2019, 134, 860–866. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- CAR T-Cell Therapy Shows Durable Responses in Indolent NHL. Oncologist 2020, 25 (Suppl. 1), S6–S7. [CrossRef]
- Lemal, R.; Tournilhac, O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J. Immunother. Cancer 2019, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Sardar, M.; Malik, S.U.; Khan, A.; Idrees, M.; Ahmad, Q.; Sohail, C.; Naseer, R.; Amin, S.; McBride, A.; Abuzar, M.; et al. Efficacy of Ibrutinib-Based Regimen in Chronic Lymphocytic Leukemia: A Systematic Review. J. Hematol. 2019, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Klichinsky, M.; Melenhorst, J.J.; Wasik, M.A.; Lacey, S.F.; June, C.H.; Gill, S. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia 2017, 31, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C.; et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Legut, M.; Cole, D.K.; Sewell, A.K.; Gritti, G.; et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017, 23, 1416–1423. [Google Scholar] [CrossRef]
- Pinz, K.; Liu, H.; Golightly, M.; Jares, A.; Lan, F.; Zieve, G.W.; Hagag, N.; Schuster, M.; Firor, A.E.; Jiang, X.; et al. Preclinical targeting of human T-cell malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia 2016, 30, 701–707. [Google Scholar] [CrossRef]
- Sauter, C.S.; Senechal, B.; Rivière, I.; Ni, A.; Bernal, Y.; Wang, X.; Purdon, T.; Hall, M.; Singh, A.N.; Szenes, V.Z.; et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2019, 134, 626–635. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, X.; Tian, Y.; Li, F.; Zhao, X.; Liu, J.; Yao, C.; Zhang, Y. Point mutation in CD19 facilitates immune escape of B cell lymphoma from CAR-T cell therapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Chow, V.A.; Gopal, A.K.; Gauthier, J.; Tseng, Y.D.; Turtle, C.J.; Maloney, D.G.; Shadman, M. Axicabtagene ciloleucel for relapsed or refractory lymphoma after prior treatment with a different CD19-directed CAR T-cell therapy. Blood Adv. 2020, 4, 4869–4872. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.; Shi, M.; Yang, J.; Cao, J.; Xu, L.; Yan, D.; Yao, M.; Liu, H.; Li, W.; Zhang, B.; et al. Phase II trial of co-administration of CD19- and CD20-targeted chimeric antigen receptor T cells for relapsed and refractory diffuse large B cell lymphoma. Cancer Med. 2020, 9, 5827–5838. [Google Scholar] [CrossRef] [PubMed]
- Rataj, F.; Kraus, F.B.T.; Chaloupka, M.; Grassmann, S.; Heise, C.; Cadilha, B.L.; Duewell, P.; Endres, S.; Kobold, S. PD1-CD28 Fusion Protein Enables CD4+ T Cell Help for Adoptive T Cell Therapy in Models of Pancreatic Cancer and Non-hodgkin Lymphoma. Front. Immunol. 2018, 9, 1955. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Mailliard, R.; Kashii, Y.; Reichert, T.E.; Herberman, R.B.; Robbins, P.; Whiteside, T.L. Stable transduction of the interleukin-2 gene into human natural killer cell lines and their phenotypic and functional characterization in vitro and in vivo. Blood 1998, 91, 3850–3861. [Google Scholar] [CrossRef] [PubMed]
- Sutlu, T.; Nyström, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: Implications for gene therapy. Hum. Gene Ther. 2012, 23, 1090–1100. [Google Scholar] [CrossRef]
- Li, L.; Liu, L.N.; Feller, S.; Allen, C.; Shivakumar, R.; Fratantoni, J.; Wolfraim, L.A.; Fujisaki, H.; Campana, D.; Chopas, N.; et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010, 17, 147–154. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef] [PubMed]
- Yamshon, S.; Ruan, J. IMiDs New and Old. Curr. Hematol. Malig. Rep. 2019, 14, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Gribben, J.G.; Fowler, N.; Morschhauser, F. Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2803–2811. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Hagner, P.R.; Chiu, H.; Ortiz, M.; Apollonio, B.; Wang, M.; Couto, S.; Waldman, M.F.; Flynt, E.; Ramsay, A.G.; Trotter, M.; et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell-mediated cytotoxicity. Br. J. Haematol. 2017, 179, 399–409. [Google Scholar] [CrossRef]
- Trneny, M.; Lamy, T.; Walewski, J.; Belada, D.; Mayer, J.; Radford, J.; Jurczak, W.; Morschhauser, F.; Alexeeva, J.; Rule, S.; et al. Lenalidomide versus investigator’s choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): A phase 2, randomised, multicentre trial. Lancet Oncol. 2016, 17, 319–331. [Google Scholar] [CrossRef]
- Goy, A.; Sinha, R.; Williams, M.E.; Kalayoglu Besisik, S.; Drach, J.; Ramchandren, R.; Zhang, L.; Cicero, S.; Fu, T.; Witzig, T.E. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: Phase II MCL-001 (EMERGE) study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3688–3695. [Google Scholar] [CrossRef]
- Leonard, J.P.; Trneny, M.; Izutsu, K.; Fowler, N.H.; Hong, X.; Zhu, J.; Zhang, H.; Offner, F.; Scheliga, A.; Nowakowski, G.S.; et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1188–1199. [Google Scholar] [CrossRef]
- Morschhauser, F.; Fowler, N.H.; Feugier, P.; Bouabdallah, R.; Tilly, H.; Palomba, M.L.; Fruchart, C.; Libby, E.N.; Casasnovas, R.O.; Flinn, I.W.; et al. Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N. Engl. J. Med. 2018, 379, 934–947. [Google Scholar] [CrossRef]
- Ruan, J.; Martin, P.; Shah, B.; Schuster, S.J.; Smith, S.M.; Furman, R.R.; Christos, P.; Rodriguez, A.; Svoboda, J.; Lewis, J.; et al. Lenalidomide plus Rituximab as Initial Treatment for Mantle-Cell Lymphoma. N. Engl. J. Med. 2015, 373, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Martin, P.; Christos, P.; Cerchietti, L.; Tam, W.; Shah, B.; Schuster, S.J.; Rodriguez, A.; Hyman, D.; Calvo-Vidal, M.N.; et al. Five-year follow-up of lenalidomide plus rituximab as initial treatment of mantle cell lymphoma. Blood 2018, 132, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Ghesquieres, H.; Chevrier, M.; Laadhari, M.; Chinot, O.; Choquet, S.; Moluçon-Chabrot, C.; Beauchesne, P.; Gressin, R.; Morschhauser, F.; Schmitt, A.; et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: A multicenter prospective ‘proof of concept’ phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA). Ann. Oncol. 2019, 30, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Pellegrini, C.; Argnani, L.; Broccoli, A. Prolonged disease-free survival in elderly relapsed diffuse large B-cell lymphoma patients treated with lenalidomide plus rituximab. Haematologica 2016, 101, e385–e386. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, M.; Fowler, N.; Wagner-Bartak, N.; Feng, L.; Romaguera, J.; Neelapu, S.S.; Hagemeister, F.; Fanale, M.; Oki, Y.; Pro, B.; et al. Oral lenalidomide with rituximab in relapsed or refractory diffuse large cell, follicular and transformed lymphoma: A phase II clinical trial. Leukemia 2013, 27, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fayad, L.; Wagner-Bartak, N.; Zhang, L.; Hagemeister, F.; Neelapu, S.S.; Samaniego, F.; McLaughlin, P.; Fanale, M.; Younes, A.; et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: A phase 1/2 clinical trial. Lancet Oncol. 2012, 13, 716–723. [Google Scholar] [CrossRef]
- Fowler, N.H.; Davis, R.E.; Rawal, S.; Nastoupil, L.; Hagemeister, F.B.; McLaughlin, P.; Kwak, L.W.; Romaguera, J.E.; Fanale, M.A.; Fayad, L.E.; et al. Safety and activity of lenalidomide and rituximab in untreated indolent lymphoma: An open-label, phase 2 trial. Lancet Oncol. 2014, 15, 1311–1318. [Google Scholar] [CrossRef]
- Morschhauser, F.; Le Gouill, S.; Feugier, P.; Bailly, S.; Nicolas-Virelizier, E.; Bijou, F.; Salles, G.A.; Tilly, H.; Fruchart, C.; Van Eygen, K.; et al. Obinutuzumab combined with lenalidomide for relapsed or refractory follicular B-cell lymphoma (GALEN): A multicentre, single-arm, phase 2 study. Lancet Haematol. 2019, 6, e429–e437. [Google Scholar] [CrossRef]
- Houot, R.; Cartron, G.; Bijou, F.; de Guibert, S.; Salles, G.A.; Fruchart, C.; Bouabdallah, K.; Maerevoet, M.; Feugier, P.; Le Gouill, S.; et al. Obinutuzumab plus Lenalidomide (GALEN) for the treatment of relapse/refractory aggressive lymphoma: A phase II LYSA study. Leukemia 2019, 33, 776–780. [Google Scholar] [CrossRef]
- Thieblemont, C.; Tilly, H.; Gomes da Silva, M.; Casasnovas, R.O.; Fruchart, C.; Morschhauser, F.; Haioun, C.; Lazarovici, J.; Grosicka, A.; Perrot, A.; et al. Lenalidomide Maintenance Compared With Placebo in Responding Elderly Patients With Diffuse Large B-Cell Lymphoma Treated With First-Line Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone. J. Clin. Oncol. 2017, 35, 2473–2481. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Geng, H.; Fraser, E.J.; Formaker, P.; Chen, L.; Sharma, J.; Killea, P.; Choi, K.; Ventura, J.; Kurhanewicz, J.; et al. Phase 1 investigation of lenalidomide/rituximab plus outcomes of lenalidomide maintenance in relapsed CNS lymphoma. Blood Adv. 2018, 2, 1595–1607. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Geng, H.; Vu, K.; Mannis, G.; Formaker, P.; Hwang, J.; Munster, P.N.; Damato, B. Maintenance lenalidomide in primary CNS lymphoma. Ann. Oncol. 2019, 30, 1397–1398. [Google Scholar] [CrossRef] [PubMed]
- Carpio, C.; Bouabdallah, R.; Ysebaert, L.; Sancho, J.M.; Salles, G.; Cordoba, R.; Pinto, A.; Gharibo, M.; Rasco, D.; Panizo, C.; et al. Avadomide monotherapy in relapsed/refractory DLBCL: Safety, efficacy, and a predictive gene classifier. Blood 2020, 135, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bouabdallah, R.; Vitolo, U.; Doorduijn, J.K.; Salles, G.; Chiappella, A.; Zinzani, P.L.; Bijou, F.; Kersten, M.J.; Sarmiento, R.; et al. Avadomide plus obinutuzumab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma (CC-122-NHL-001): A multicentre, dose escalation and expansion phase 1 study. Lancet Haematol. 2020, 7, e649–e659. [Google Scholar] [CrossRef]
- Dillman, R.O. Infusion reactions associated with the therapeutic use of monoclonal antibodies in the treatment of malignancy. Cancer Metastasis Rev. 1999, 18, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist 2008, 13, 725–732. [Google Scholar] [CrossRef]
- Patel, D.A.; Johanns, T.M.; Trinkaus, K.; Bartlett, N.L.; Wagner-Johnston, N.; Cashen, A.F. Implication of Rituximab Infusion Reactions on Clinical Outcomes in Patients With Diffuse Large B-cell Lymphoma: A Single Institution Experience. Clin. Lymphoma Myeloma Leuk 2019, 19, 806–811. [Google Scholar] [CrossRef]
- Jeyarajah, D.R.; Thistlethwaite, J.R., Jr. General aspects of cytokine-release syndrome: Timing and incidence of symptoms. Transplant. Proc. 1993, 25, 16–20. [Google Scholar]
- Fouda, G.E.; Bavbek, S. Rituximab Hypersensitivity: From Clinical Presentation to Management. Front. Pharmacol. 2020, 11, 572863. [Google Scholar] [CrossRef]
- Rombouts, M.D.; Swart, E.L.; AJM, V.D.E.; Crul, M. Systematic Review on Infusion Reactions to and Infusion Rate of Monoclonal Antibodies Used in Cancer Treatment. Anticancer Res. 2020, 40, 1201–1218. [Google Scholar] [CrossRef]
- Osterborg, A.; Karlsson, C.; Lundin, J.; Kimby, E.; Mellstedt, H. Strategies in the management of alemtuzumab-related side effects. Semin. Oncol. 2006, 33, S29–S35. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, V.; Giavina-Bianchi, P.; Picard, M.; Caiado, J.; Castells, M. Drug desensitization in the management of hypersensitivity reactions to monoclonal antibodies and chemotherapy. BioDrugs 2014, 28, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Vani, V.; Regge, D.; Cappello, G.; Gabelloni, M.; Neri, E. Imaging of Adverse Events Related to Checkpoint Inhibitor Therapy. Diagnostics 2020, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Wagner, D.H., Jr.; Stout, R.D.; Suttles, J. Role of the CD40-CD40 ligand interaction in CD4+ T cell contact-dependent activation of monocyte interleukin-1 synthesis. Eur. J. Immunol. 1994, 24, 3148–3154. [Google Scholar] [CrossRef]
- Wei, J.; Liu, Y.; Wang, C.; Zhang, Y.; Tong, C.; Dai, G.; Wang, W.; Rasko, J.E.J.; Melenhorst, J.J.; Qian, W.; et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal. Transduct Target. Ther. 2020, 5, 134. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, F.; Zhang, P.; Zhang, Y.; Chen, Y.; Fan, X.; Cao, X.; Liu, J.; Yang, Y.; Wang, B.; et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front. Med. 2019, 13, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Strati, P.; Ahmed, S.; Kebriaei, P.; Nastoupil, L.J.; Claussen, C.M.; Watson, G.; Horowitz, S.B.; Brown, A.R.T.; Do, B.; Rodriguez, M.A.; et al. Clinical efficacy of anakinra to mitigate CAR T-cell therapy-associated toxicity in large B-cell lymphoma. Blood Adv. 2020, 4, 3123–3127. [Google Scholar] [CrossRef] [PubMed]
- Huarte, E.; O’Connor, R.S.; Peel, M.T.; Nunez-Cruz, S.; Leferovich, J.; Juvekar, A.; Yang, Y.O.; Truong, L.; Huang, T.; Naim, A.; et al. Itacitinib (INCB039110), a JAK1 Inhibitor, Reduces Cytokines Associated with Cytokine Release Syndrome Induced by CAR T-cell Therapy. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J. Natl. Cancer Inst. 2019, 111, 646–654. [Google Scholar] [CrossRef]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef]
- Torre, M.; Solomon, I.H.; Sutherland, C.L.; Nikiforow, S.; DeAngelo, D.J.; Stone, R.M.; Vaitkevicius, H.; Galinsky, I.A.; Padera, R.F.; Trede, N.; et al. Neuropathology of a Case With Fatal CAR T-Cell-Associated Cerebral Edema. J. Neuropathol. Exp. Neurol. 2018, 77, 877–882. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Combination | Target Antigens | Mode of Action of the Combination Agent(s) Other Than Bispecific Antibody | Study Phase | Disease Status | Estimated Study Completion Date | ClinicalTrials.gov Identifier (Other Identifier) |
|---|---|---|---|---|---|---|
| blinatumomab | CD19/CD3 | 2 | R/R indolent B-NHL | December 2023 | NCT02811679 | |
| blinatumomab | CD19/CD3 | 1 | R/R indolent B-NHL | May 2022 | NCT02961881 | |
| blinatumomab | CD19/CD3 | 1 | DLBCL after ASCT | December 2023 | NCT03072771 | |
| blinatumomab + lenalidomide | CD19/CD3 | immunomodulatory agent lenalidomide | 1 | R/R B-NHL | December 2020 | NCT02568553 |
| blinatumomab + pembrolizumab | CD19/CD3 | immune check-point PD-1 inhibitor pembrolizumab | 1 | R/R DLBCL | January 2026 | NCT03340766 (KEYNOTE-348) |
| Drug Combination | Target Antigens | Mode of Action of the Combination Agent(s) Other Than Bispecific Antibody | Study Phase | Disease Status | Estimated Study Completion Date | ClinicalTrials.gov Identifier (Other Identifier) |
|---|---|---|---|---|---|---|
| Mosunetuzumab ± atezolizumab | CD20/CD3 | immune check-point PD-L1 inhibitor atezolizumab | 1 | R/R B-NHL and CLL | October 2021 | NCT02500407 |
| Mosunetuzumab + polatuzumab vedotin compared to bendamustine + rituxumab + polatuzumab vedotin | CD20/CD3 | anti-CD79B antibody-drug conjugate polatuzumab-vedotin, anti-CD20 rituximab, new cytostatic agent bendamustine | 1B/2 | R/R DLBCL and R/R FL | June 2022 | NCT03671018 |
| Mosunetuzumab + lenalidomide, glofitamab + lenalidomide or glofitamab + lenalidomide + obinutuzumab | CD20/CD3 | immunomodulatory agent lenalidomide, glycoengeneered anti-CD20 mAb obinutuzumab | 1 | newly dg DLBCL, R/R DLBCL and R/R FL | August 2022 | NCT04246086 |
| Mosunetuzumab + CHOP or mosunetuzumab + polatuzumab vedotin + CHP | CD20/CD3 | chemotherapy CHOP, anti-CD79B antibody drug conjugate polatuzumab-vedotin | 1B/2 | newly dg DLBCL, R/R B-NHL | June 2022 | NCT03677141 |
| Mosunetuzumab | CD20/CD3 | 1B/2 | newly dg DLBCL | April 2023 | NCT03677154 | |
| Mosunetuzumab or Glofitamab + GemOx | CD20/CD3 | chemotherapy gemcitabine and oxaliplatin (GemOx) | 1 | R/R DLBCL | March 2021 | NCT04313608 |
| Drug Combination | Target Antigens | Mode of Action of the Combination Agent(s) Other Than Bispecific Antibody | Study Phase | Disease Status | Estimated Study Completion Date | ClinicalTrials.gov Identifier (Other Identifier) |
|---|---|---|---|---|---|---|
| Glofitamab + GemOx compared to rituximab + GemOx | CD20/CD3 | cytostatics gemcitabine and oxaliplatin (GemOx), anti-CD20 rituximab | 3 | R/R DLBCL | March 2022 | NCT04408638 |
| Glofitamab + obintuzumab with obinutuzumab pretreatment | CD20/CD3 | glycoengeneered anti-CD20 obinutuzumab | 1 | R/R B-NHL | June 2022 | NCT03075696 |
| Glofitamab + obintuzumab or rituximab + CHOP with obinutuzumab pretreatment | CD20/CD3 | chemotherapy CHOP, anti-CD20 obintuzumab, anti-CD20 rituximab | 1 | newly dg and R/R B-NHL | December 2023 | NCT03467373 |
| Glofitamab + atezolizumab or polatuzumab-vedotin with obinutuzumab pretreatment | CD20/CD3 | PD-L1 inhibitor atezolizumab, anti-CD79B antibody-drug conjugate polatuzumab-vedotin | 1 | R/R B-NHL | August 2021 | NCT03533283 |
| Glofitamab + RO7227166 with obinutuzumab pretreatment | CD20/CD3 | CD19 Targeted 4-1BB Ligand RO7227166 | 1 | R/R B-NHL | January 2023 | NCT04077723 |
| Glofitamab + lenalidomide +/− obinutuzumab | CD20/CD3 | Immunomodulatory agent lenalidomide | 1 | R/R FL | August 2022 | NCT04246086 |
| Drug Combination | Target Antigens | Mode of Action of the Combination Agent(s) Other Than Bispecific Antibody | Study Phase | Disease Status | Estimated Study Completion Date | ClinicalTrials.gov Identifier (Other Identifier) |
|---|---|---|---|---|---|---|
| REGN1979 (odronextamab) | CD20/CD3 | 1 | R/R B-NHL | August 2026 | NCT03888105 | |
| REGN1979 (odronextamab) | CD20/CD3 | 1 | R/R B-NHL and CLL | April 2025 | NCT02290951 | |
| AFM13 + NK cells | CD30/CD16A | modified umbilical cord blood immune cells (natural killer [NK] cells) | 1 | R/R Hodgkin and CD30+ B-NHL | April 2023 | NCT04074746 |
| AFM13 | CD30/CD16A | 2 | R/R T-NHL and tMF | February 2023 | NCT04101331 | |
| JNJ-75348780 | CD22/CD3 | 1 | R/R MCL | May 2023 | NCT04540796 | |
| TG-1801 + ublituximab | CD19/CD47 | chimeric anti-CD20 mAb | 1 | R/R B-NHL | August 2021 | NCT03804996 |
| RO7227166 + obinutuzumab + glofitamab | CD19/4-1BB | glycoengineered anti-CD20 antibody obinutuzumab, anti-CD3/CD20 bispecific antibody glofitamab | 1 | R/R B-NHL | January 2023 | NCT04077723 |
| Name | Trade Name | Developed by | Structure | Target | First Approval by US FDA for the Treatment of Cancer | Number of Studies in Patients with NHL Registered at ClinicalTrials.gov |
|---|---|---|---|---|---|---|
| Ipilimumab | Yervoy | Bristol-Myers-Squibb | human IgG1 | CTLA-4 | 2011 | 13 |
| Tremelimumab | N/A | AstraZeneca | human IgG2 | CTLA-4 | N/A | 3 |
| Pembrolizumab | Keytruda | Merck | humanized IgG4 | PD-1 | 2014 | 60 |
| Nivolumab | Opdivo | Bristol-Myers-Squibb | human IgG4 | PD-1 | 2014 | 41 |
| Pidilizumab | N/A | Medivation | human IgG1 | PD-1 | N/A | 3 |
| Durvalumab | Imfinzi | AstraZeneca | human IgG1 | PD-L1 | 2020 | 19 |
| Avelumab | Bavencio | Merck, Pfizer | human IgG1 | PD-L1 | 2017 | 9 |
| Atezolizumab | Tenetriq | Roche | humanized IgG1 | PD-L1 | 2016 | 20 |
| Drug Combination | Mode of Action of the Combination Agent(s) Other Than Immune Checkpoint Inhibitors | Study Phase | Disease Status | Estimated Study Completion Date | ClinicalTrials.gov Identifier (Other Identifier) |
|---|---|---|---|---|---|
| Nivolumab + R(ituximab)-GemOx compared to R-GemOx | immunochemotherapy gemcitabine + oxaliplatin (GemOx) | 2/3 | R/R elderly B-NHL | November 2024 | NCT03366272 (NIVEAU) |
| Avelumab +/− Utomilumab +/− Rituximab +/− Azacitidine +/− bendamustin +/− Gemcitabine +/− Oxaliplatine | CD137 (4-1BB) antigen agonist antibody utomilumab, anti-CD20 antibody rituximab, epigenetic modulator azacitidine, conventinal chemotherapy GemOx | 1/3 | R/R DLBCL | December 2019 | NCT02951156 (JAVELIN DLBCL) |
| Nivolumab + DA-EPOCH-R + Nivolumab as a consolidation | immunochemotherapy regimen (dose-adjusted EPOCH-R) | 2 | B-NHL | December 2021 | NCT03749018 |
| Nivolumab + Copanlisib | pan-PI3K inhibitor copanlisib | 2 | R/R DLBCL, PMBCL | October 2021 | NCT03484819 |
| Pembrolizumab | 2 | untreated B-NHL | September 2024 | NCT03498612 | |
| Pembrolizumab | 2 | R/R grey-zone lymphoma, R/R PCNSL, R/R DLBCL | July 2022 | NCT03255018 | |
| Pembrolizumab + R-CHOP | R-CHOP immunochemotherapy regimen | 2 | DLBCL, high-grade B-NHL | August 2024 | NCT03995147 |
| Pembrolizumab + Rituximab +/− Lenalidomide | anti-CD20 antibody, immunomodulatory agent lenalidomide | 2 | R/R FL, R/R DLBCL | November 2021 | NCT02446457 |
| Durvalumab + R-CHOP/R2-CHOP | standard immunochemotherapy, immunomodulatory agent lenalidomide | 2 | DLBCL | March 2023 | NCT03003520 |
| Ipilimumab + Lenalidomide | immunomodulatory agent lenalidomide | 2 | NHL (post-HSCT) | June 2021 | NCT01919619 |
| Pembrolizumab + ALX-148 | CD47 antagonist ALX-148 | 1 | NHL, solid tumors | December 2021 | NCT03013218 |
| Full Generic Name | Axicabtagene Ciloleucel | Tisagenlecleucel | Lisocabtagene Maraleucel |
|---|---|---|---|
| Shortened name | Axi-cel | Tisa-cel | Liso-cel |
| Manufacturer | Kite-Gilead | Novartis | Bristol-Myers Squibb |
| Registration study | ZUMA-1 | JULIETT | TRANSCEND |
| Number of patients | |||
| Total | 111 | 165 | 344 |
| Infused | 101 (91% of total) | 111 (67% of total) | 269 (78% of total) |
| DLBCL de novo | 77 (76%) | 88 (79%) | 137 (51%) |
| DLBCL transformed | 16 (16%) | 21 (19%) | 78 (29%) |
| PMBL | 8 (8%) | 0 (0%) | 15 (6%) |
| Double/triple hit | NR | 19 (27%) * | 36 (13%) |
| Other | 0 (0%) | 2 (2%) ** | 3 (1%) # |
| Age | |||
| Median (range) | 58 (23–76) | 56 (22–76) | 63 (54–70) |
| ≥65 years | 24 (24%) | 25 (23%) | 112 (42%) |
| Gender | |||
| Male | 68 (67%) | NR | 174 (65%) |
| Female | 33 (33%) | NR | 95 (35%) |
| Disease status | |||
| Primary refractory | 2 (2%) | NR | NR |
| Relapse after ASCT | 21 (21%) | 54 (49%) | 94 (35%) |
| ≥3 lines of therapy | 70 (69%) | 57 (52%) | 139 (52%) |
| Bridging therapy administered | 0 | 92% | 159 (59%) |
| Response rate | |||
| ORR | 83% | 52% | 73% |
| CR | 58% | 40% | 53% |
| Survival | |||
| OS | 52% at 18 months | Median, 12 months | Median, 21.1 months |
| PFS | Median, 5.9 months | Median, 2.9 months | Median, 6.8 months |
| DOR | Median, 11.1 months | 65% at 12 months | 55% at 12 months |
| Adverse events | |||
| Cytokine release syndrome | |||
| All grades | 93% | 64 (58%) | 113 (42%) |
| Grade 3–4 | 13% | 24 (22%) | 6 (2%) |
| Neurotoxicity | |||
| All grades | 34% | 23 (21%) | 80 (30%) |
| Grade 3–4 | 21% | 13 (12%) | 27 (10%) |
| Infections | |||
| All grades | NR | 38 (34%) | not reported |
| Grade 3–4 | NR | 22 (20%) | not reported |
| Drug Combination | Mode of Action | Study Phase | Disease Status | Estimated Study Completion Date | ClinicalTrials.gov Identifier (Other Identifier) |
|---|---|---|---|---|---|
| Axi-cel | Anti-CD19 CAR T-cells versus ASCT (2nd line therapy) | 3 | R/R hgB-NHL | January 2022 | NCT03391466 (ZUMA-7) |
| Liso-cel | Anti-CD19 CAR T-cells versus ASCT (2nd line therapy) | 3 | R/R hgB-NHL | January 2024 | NCT03575351 (TRANSFORM) |
| Tisa-cel | Anti-CD19 CAR T-cells versus ASCT (2nd line therapy) | 3 | R/R hgB-NHL | December 2025 | NCT03570892 (BELINDA) |
| Axi-cel | Anti-CD19 CAR T-cells | 2 | R/R FL, R/R MZL | February 2022 | NCT03105336 (ZUMA-5) |
| Liso-cel | Anti-CD19 CAR T-cells | 2 | R/R B-NHL ineligible for ASCT | April 2021 | NCT03483103 (TRANSCEND-PILOT-017006) |
| KTE-X19 | Anti-CD19 CAR T-cells | 1 | R/R SLL/CLL | August 2021 | NCT03624036 |
| Liso-cel + ibrutinib | Anti-CD19 CAR T-cells + BTK inhibitor ibrutinib | 1/2 | R/R CLL/SLL | October 2021 | NCT03331198 |
| Axi-cel + acalabrutinib | BTK inhibitor acalabrutinib administered before leukapheresis | 1/2 | R/R hgB-NHL | March 2024 | NCT04257578 |
| CD30.CAR T cells | Anti-CD30 CAR T-cells | 1 | R/R HL, CD30+ NHL | April 2021 | NCT02917083 (RELY-30) |
| AUTO4 | Anti-TRBC1 CAR T-cells | 1/2 | R/R T-NHL | July 2021 | NCT03590574 |
| CD4CAR | Anti-CD4 CAR T-cells | 1 | R/R T-NHL | December 2022 | NCT03829540 |
| Axi-cel | Anti-CD19 CAR T-cells | 1 | DLBCL (PET+ after 2 cycles of therapy) | June 2021 | NCT03761056 (ZUMA-12) |
| ALTCAR.CD30 | ASCT followed by anti-CD30 CAR T-cells | 1 | R/R HL, CD30+ NHL | September 2021 | NCT02663297 |
| AlloSCT + CAR-T | T-cell depleted alloSCT + donor anti-CD19 CAR T-cell-based consolidation | 1 | B-ALL, CLL, NHL | September 2023 | NCT04556266 |
| CAR 20/19 | Bispecific anti-CD20/anti-CD19 CAR T-cells | 1/2 | R/R B-NHL | May 2023 | NCT04186520 |
| Liso-cel + avadomide, iberdomide, ibrutinib, or durvalumab | Anti-CD19 CAR T-cells in combination with immunomodulatory drugs avadomide/iberdomide, BTK inhibitor ibrutinib or anti PD-L1 checkpoint durvalumab | 1/2 | R/R hgB-NHL | August 2023 | NCT03310619 (PLATFORM) |
| AUTO3 + pembrolizumab | Dual anti-CD19/anti-CD22 CAR T-cells + anti PD-1 immune checkpoint inhibitor pembrolizumab | 1/2 | R/R hgB-NHL | March 2021 | NCT03287817 (ALEXANDER) |
| CD19-PD1-CART | Anti-CD19 CAR T-cells with PD-1/CD28 co-stimulation | 1 | R/R B-NHL | July 2021 | NCT04163302 |
| CD7.CAR | Anti-CD7 CAR T-cells with CD7 deletion | 1 | R/R CD7+ T-cell malignancies | May 2023 | NCT03690011 (CRIMSON) |
| iC9/CAR.19/IL15-Transduced CB-NK Cells | Cord blood-derived allogeneic anti-CD19 NK-cells with IL-15 and inducible caspase 9 | 1/2 | B-ALL, CLL, B-NHL | June 2022 | NCT03056339 |
| CD19.CAR-aNKT | Anti-CD19 allo CAR NK/T cells with IL-15 | 1 | R/R ALL, CLL, hgB-NHL | April 2023 | NCT03774654 |
| Drug Combination | Mode of Action of the Drug Combination | Study Phase | Target Population | Estimated Study Completion Date | GovTrial Denominator |
|---|---|---|---|---|---|
| Brentuximab vedotin + lenalidomide + rituximab | anti-CD30 antibody-drug conjugate brentuximab-vedotin, anti-CD20 rituximab | 3 | R/R DLBCL | December 2025 | NCT04404283 |
| Lenalidomide + R-ICE | anti-CD20 rituximab, chemotherapy ifosfamide, carboplatin, etoposide (ICE) | 1/2 | R/R DLBCL | December 2025 | NCT02628405 |
| lenalidomide + R-CHOP | chemotherapy regimen R-CHOP | 2 | DLBCL (CNS prophylaxis) | October 2024 | NCT04544059 |
| Lenalidomide + R-GemOx | anti-CD20 rituximab, gemcitabine, oxaliplatin | 1 | Newly dg. elderly DLBCL | December 2024 | NCT04432402 |
| Lenalidomide + Rituximab + Dexamethasone | anti-CD20 rituximab | 2 | Newly dg. elderly DLBCL | March 2021 | NCT02955823 |
| Axicabtagene Ciloleucel + rituximab or lenalidomide | anti-CD20 rituximab | 1 | R/R DLBCL | September 2036 | NCT04002401 (Zuma-14) |
| Brentuximab vedotin + lenalidomide + rituximab | anti-CD30 antibody-drug conjugate brentuximab-vedotin, anti-CD20 rituximab | 3 | R/R DLBCL | December 2025 | NCT04404283 |
| lenalidomide + obinutuzumab + venetoclax | anti-CD20 obinutuzumab, BCL2 inhibitor venetoclax | 1/2 | Newly dg. FL | November 2026 | NCT03980171 (LEVERAGE) |
| Lenalidomide + Acalabrutinib + Rituximab | BTK inhibitor acalabrutinib, anti-CD20 rituximab | 2 | Newly dg. FL | March 2025 | NCT04404088 |
| Venetoclax, Lenalidomide and Rituximab | BCL2 inhibitor venetoclax, anti-CD20 antibody rituximab | 1 | Newly dg. MCL | July 2022 | NCT03523975 |
| Lenalidomide + Acalabrutinib + Rituximab | BTK inhibitor acalabrutinib, anti-CD20 rituximab | 2 | Newly dg. MCL | November 2024 | NCT03863184 |
| Venetoclax, Lenalidomide and Rituximab | BCL2 inhibitor venetoclax, anti-CD20 antibody rituximab | 1/2 | R/R MCL | June 2022 | NCT03505944 (VALERIA) |
| Carfilzomib + Lenalidomide + Dexamethasone | Irreversible proteasome inhibitor carfilzomib | 1 | R/R MCL (ibrutinib refractory) | April 2022 | NCT03891355 (FIL_KLIMT) |
| Lenalidomide maintenance | 2 | R/R T-NHL (after salvage therapy) | November 2023 | NCT03730740 (Lemon-T) | |
| Lenalidomide + Brentuximab Vedotin | anti-CD30 antibody-drug conjugate brentuximab-vedotin | 1 | R/R PTCL | August 2021 | NCT03302728 |
| Lenalidomide + Ibrutinib + Rituximab | BTK inhibitor ibrutinib, anti-CD20 rituximab | 1 | primary/secondary CNS lymphoma | November 2021 | NCT03703167 |
| Class of Agents/Agent | Targeted Structure | Effective in NHL Subtypes |
|---|---|---|
| Monospecific monoclonal antibodies | ||
| Rituximab | CD20 | All B-NHL [11,12] |
| Obinutuzumab | CD20 | CLL/SLL [13], FL frontline [14], R/R FL [15] |
| Tafasitamab | CD19 | R/R B-NHL [26], R/R DLBCL [27] |
| Alemtuzumab | CD52 | Mycosis fungoides [33], T-PLL [34] |
| Mogamulizumab | CCR4 | Adult T-cel leukemia/lymphoma [37,38] |
| Bispecific monoclonal antibodies | ||
| Blinatumomab | CD3-CD19 | R/R B-NHL [48] R/R DLBCL [49,50] |
| Mosunetuzumab | CD3-CD20 | R/R B-NHL [52] |
| Glofitamab | CD3-CD20 | R/R B-NHL [56,57] |
| Checkpoint inhibitors | ||
| Pembrolizumab | PD-1 | R/R PMBCL [80,81], Richter’s syndrome [93], mycosis fungoides [95] |
| Nivolumab | PD-1 | R/R PMBCL [83], PCNSL and PTL [77] |
| Pidilizumab | PD-1 | DLBCL after autologous SCT [75] |
| CAR-T cells | ||
| Tisagenlecleucel | CD19 | R/R aggressive NHL [141] |
| Axicabtagene ciloleucel | CD19 | R/R aggressive NHL [142,143] |
| Lisocabtagene maraleucel | CD19 | R/R aggressive NHL [144] |
| Brexucabtagene autoleucel | CD19 | R/R MCL [150] |
| Immunomodulatory agents | ||
| Lenalidomide | Cereblon | R/R FL and MZL [180] MCL frontline [182,183], R/R PCNSL [184], R/R DLBCL [185] |
| Avadomide | Cereblon | R/R DLBCL [194] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pytlik, R.; Polgarova, K.; Karolova, J.; Klener, P. Current Immunotherapy Approaches in Non-Hodgkin Lymphomas. Vaccines 2020, 8, 708. https://doi.org/10.3390/vaccines8040708
Pytlik R, Polgarova K, Karolova J, Klener P. Current Immunotherapy Approaches in Non-Hodgkin Lymphomas. Vaccines. 2020; 8(4):708. https://doi.org/10.3390/vaccines8040708
Chicago/Turabian StylePytlik, Robert, Kamila Polgarova, Jana Karolova, and Pavel Klener. 2020. "Current Immunotherapy Approaches in Non-Hodgkin Lymphomas" Vaccines 8, no. 4: 708. https://doi.org/10.3390/vaccines8040708
APA StylePytlik, R., Polgarova, K., Karolova, J., & Klener, P. (2020). Current Immunotherapy Approaches in Non-Hodgkin Lymphomas. Vaccines, 8(4), 708. https://doi.org/10.3390/vaccines8040708