Comparison of Exosomes Derived from Non- and Gamma-Irradiated Melanoma Cancer Cells as a Potential Antigenic and Immunogenic Source for Dendritic Cell-Based Immunotherapeutic Vaccine

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation and Characterization of Melanoma Cancer Cell-Derived Exosome
2.3. Cytotoxicity of Exosomes in Bone Marrow-Derived Dendritic Cells (BMDCs)
2.4. Analysis of Exosomes Incorporated by BMDC
2.5. Analysis of High Mobility Group Box Protein 1 (HMGB1) Level in Exosomes
2.6. Analysis of DC Maturation Induced by Exosomes
2.7. Mixed Lymphocyte Reaction (MLR)
2.8. Antigenicity of Exosomes from Melanoma Tumor-Bearing Mice
2.9. Antitumor Effect and Tumor Antigen-Specific Multifunctional T Cell Induction by Exosomal Vaccination in Melanoma Tumor-Bearing Mice
2.10. Statistical Analysis
3. Results
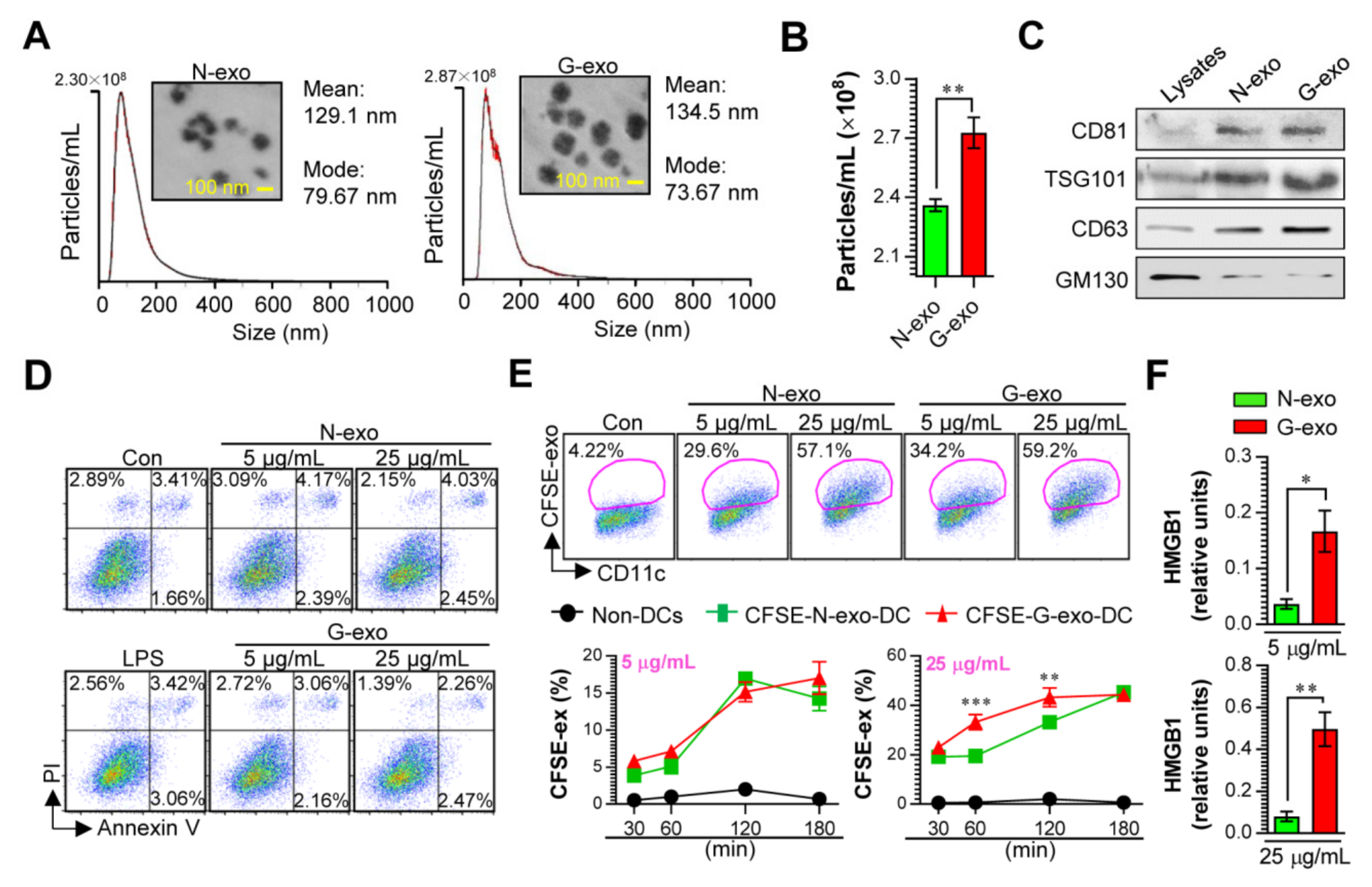
3.1. Cellular Incorporation, Cell Viability, and Immunostimulant Potential of Tumor Cell-Derived Exosomes in Dendritic Cells
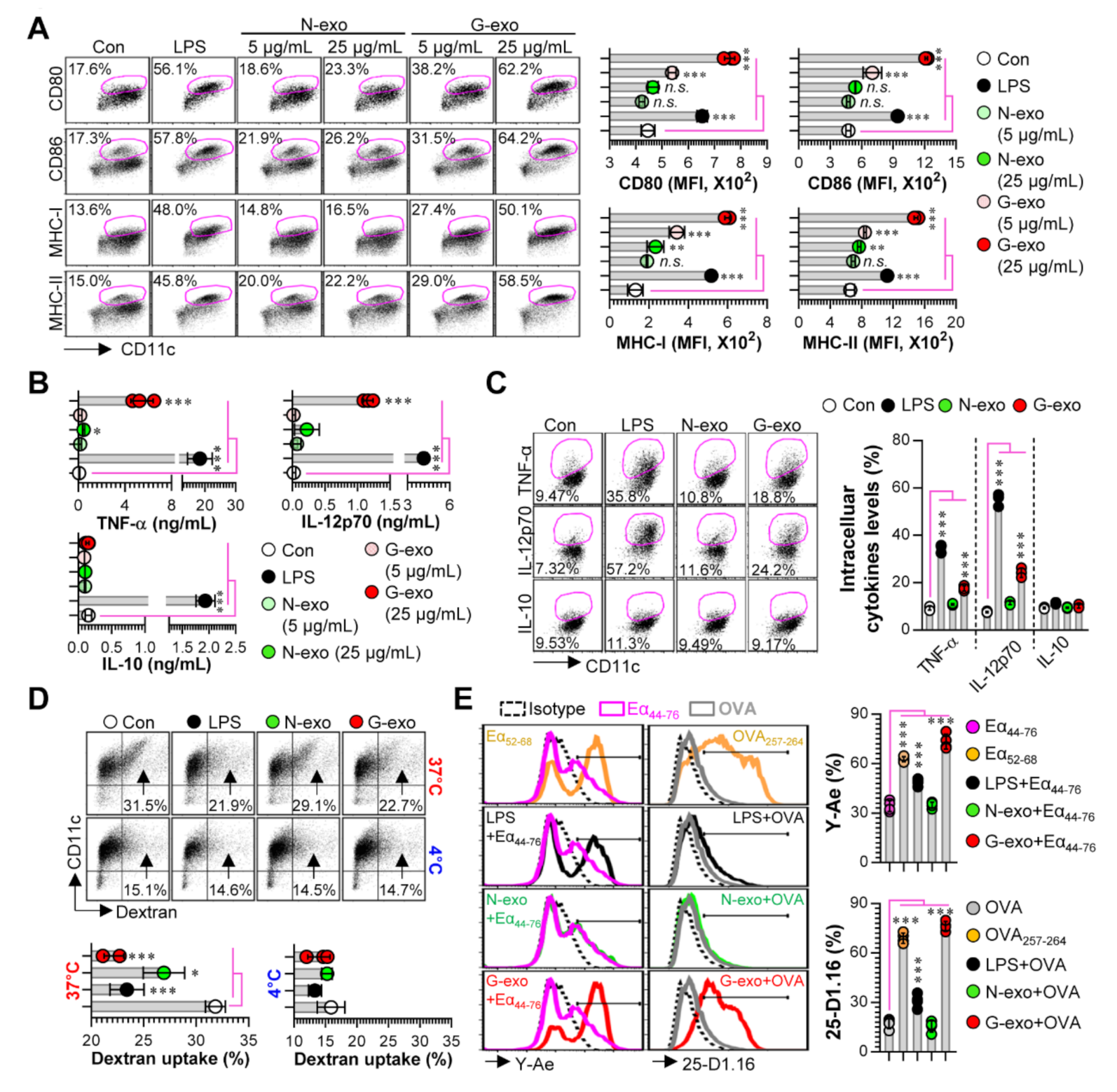
3.2. Comparison of Dendritic Cell Stimulatory Capacity of N-Exo and G-Exo
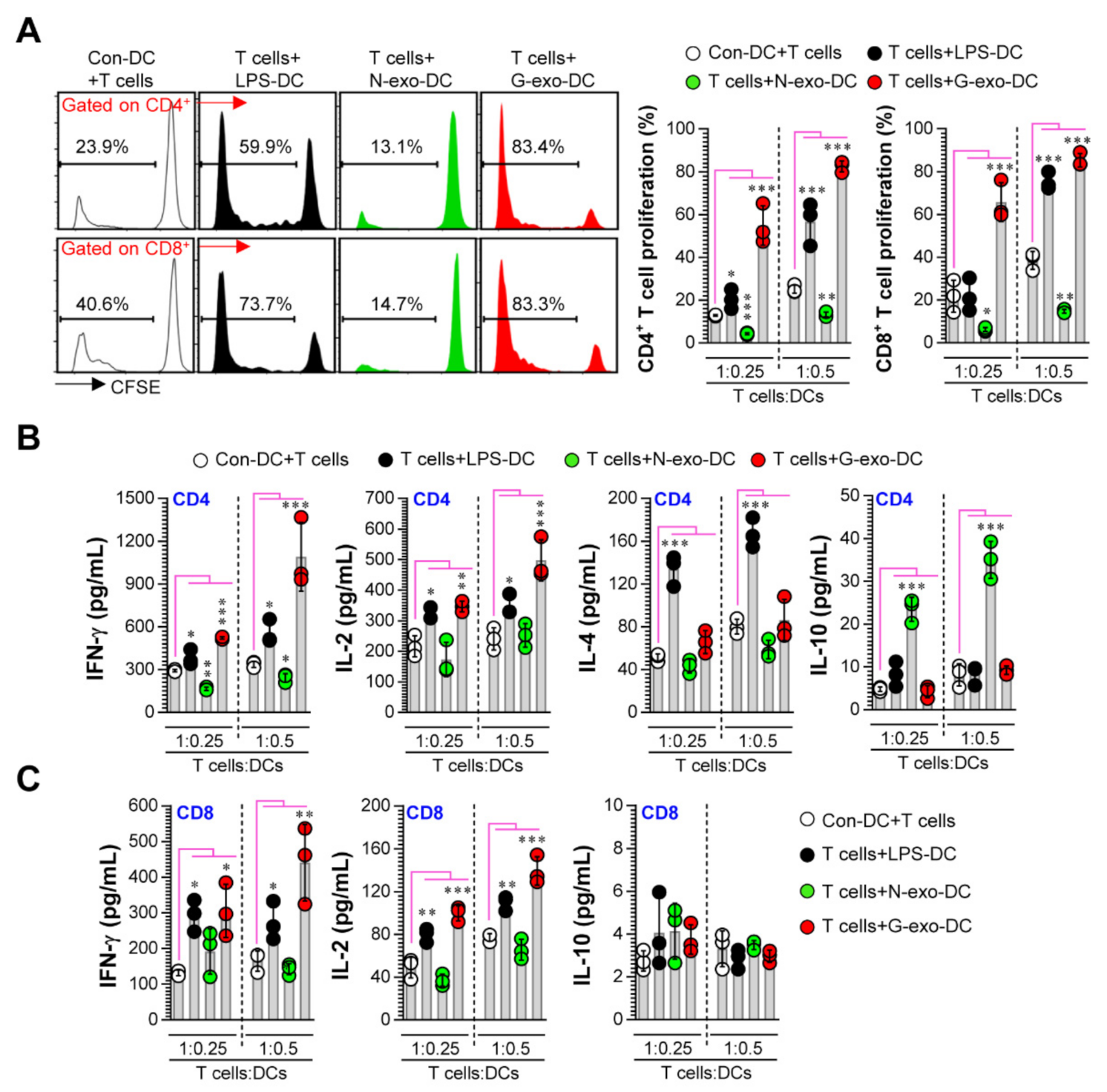
3.3. Comparative Analysis of T Cell Proliferation and Polarization Elicited by N-Exo- and G-Exo-Stimulated DCs upon Interacting with Naive T Cells
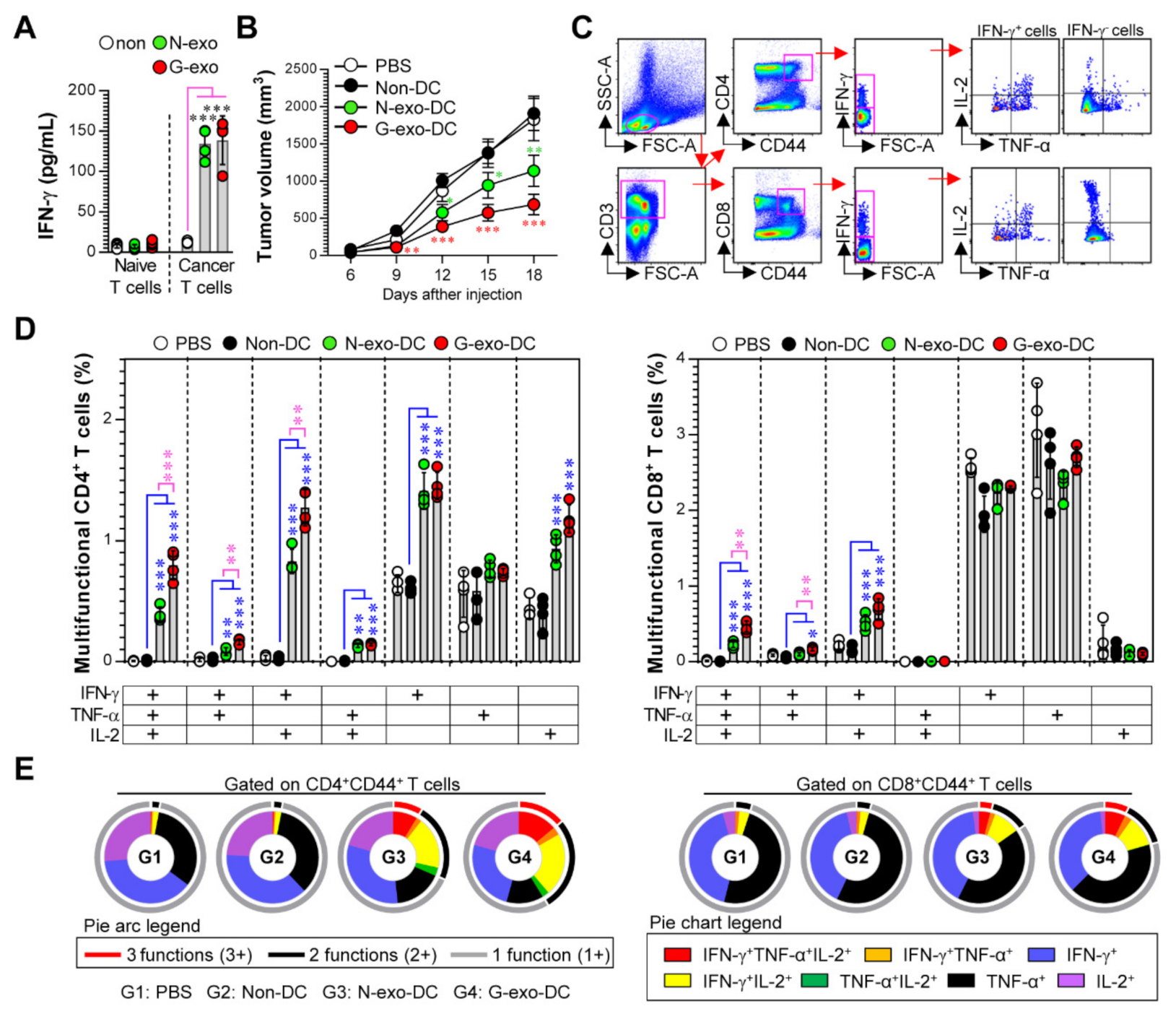
3.4. Protective Effect and Multifunctional T Cell Responses Induced by N-Exo- and G-Exo-Stimulated DC-Based Vaccines in Tumor-Bearing Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dombroski, J.A.; King, M.R. Engineering of exosomes to target cancer metastasis. Cell. Mol. Bioeng. 2020, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-mediated metastasis: From epithelial-mesenchymal transition to escape from immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, C.; Harikumar, K.B. The origin and functions of exosomes in cancer. Front. Oncol. 2018, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Lasser, C. Exosomes in diagnostic and therapeutic applications: Biomarker, vaccine and rna interference delivery vehicle. Expert Opin. Biol. Ther. 2015, 15, 103–117. [Google Scholar] [CrossRef]
- Tan, A.; De La Pena, H.; Seifalian, A.M. The application of exosomes as a nanoscale cancer vaccine. Int. J. Nanomed. 2010, 5, 889–900. [Google Scholar]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release Off. J. Control. Release Soc. 2015, 199, 145–155. [Google Scholar] [CrossRef]
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnology 2018, 16, 81. [Google Scholar] [CrossRef]
- Ailawadi, S.; Wang, X.; Gu, H.; Fan, G.C. Pathologic function and therapeutic potential of exosomes in cardiovascular disease. Biochim. Biophys. Acta 2015, 1852, 1–11. [Google Scholar] [CrossRef]
- Chen, T.; Guo, J.; Yang, M.; Zhu, X.; Cao, X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J. Immunol. 2011, 186, 2219–2228. [Google Scholar] [CrossRef]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes shuttle trex1-sensitive ifn-stimulatory dsdna from irradiated cancer cells to dcs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Correction: Hypoxic tumor-derived exosomal mir-301a mediates m2 macrophage polarization via pten/pi3kgamma to promote pancreatic cancer metastasis. Cancer Res. 2020, 80, 922. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microrna-940 to induce macrophage m2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver micrornas to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kim, K.; Byun, E.B.; Song, H.Y.; Han, J.M.; Park, W.Y.; Yuk, J.M.; Byun, E.H. Rm, a novel resveratrol derivative, attenuates inflammatory responses induced by lipopolysaccharide via selectively increasing the tollip protein in macrophages: A partial mechanism with therapeutic potential in an inflammatory setting. Int. Immunopharmacol. 2020, 78, 106072. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Jung, I.D.; Kim, J.S.; Kim, H.M.; Kwon, K.W.; Park, Y.M.; Shin, S.J. Mycobacterium tuberculosis grpe, a heat-shock stress responsive chaperone, promotes th1-biased t cell immune response via tlr4-mediated activation of dendritic cells. Front. Cell. Infect. Microbiol. 2018, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Han, J.M.; Song, H.Y.; Byun, E.H.; Lim, S.T.; Byun, E.B. Annona muricata l.-derived polysaccharides as a potential adjuvant to a dendritic cell-based vaccine in a thymoma-bearing model. Nutrients 2020, 12, 1602. [Google Scholar] [CrossRef]
- De Sanctis, F.; Sandri, S.; Martini, M.; Mazzocco, M.; Fiore, A.; Trovato, R.; Garetto, S.; Brusa, D.; Ugel, S.; Sartoris, S. Hyperthermic treatment at 56 degrees c induces tumour-specific immune protection in a mouse model of prostate cancer in both prophylactic and therapeutic immunization regimens. Vaccine 2018, 36, 3708–3716. [Google Scholar] [CrossRef]
- DeVito, N.C.; Plebanek, M.P.; Theivanthiran, B.; Hanks, B.A. Role of tumor-mediated dendritic cell tolerization in immune evasion. Front. Immunol. 2019, 10, 2876. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New insights into il-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 directly inhibits cd8(+) t cell function by enhancing n-glycan branching to decrease antigen sensitivity. Immunity 2018, 48, 299–312.e295. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, semi-mature, and fully mature dendritic cells: Toward a dc-cancer cells interface that augments anticancer immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef]
- Xi, J.; Xu, M.; Song, Z.; Li, H.; Xu, S.; Wang, C.; Song, H.; Bai, J. Stimulatory role of interleukin 10 in cd8(+) t cells through stats in gastric cancer. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317706209. [Google Scholar] [CrossRef]
- Schneider, T.; Hoffmann, H.; Dienemann, H.; Schnabel, P.A.; Enk, A.H.; Ring, S.; Mahnke, K. Non-small cell lung cancer induces an immunosuppressive phenotype of dendritic cells in tumor microenvironment by upregulating b7-h3. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 1162–1168. [Google Scholar] [CrossRef]
- Tietze, J.K.; Wilkins, D.E.; Sckisel, G.D.; Bouchlaka, M.N.; Alderson, K.L.; Weiss, J.M.; Ames, E.; Bruhn, K.W.; Craft, N.; Wiltrout, R.H.; et al. Delineation of antigen-specific and antigen-nonspecific cd8(+) memory t-cell responses after cytokine-based cancer immunotherapy. Blood 2012, 119, 3073–3083. [Google Scholar] [CrossRef]
- Kaech, S.M.; Ahmed, R. Memory cd8+ t cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef]
- De Groot, R.; Van Loenen, M.M.; Guislain, A.; Nicolet, B.P.; Freen-Van Heeren, J.J.; Verhagen, O.; Van Den Heuvel, M.M.; De Jong, J.; Burger, P.; Van Der Schoot, C.E.; et al. Polyfunctional tumor-reactive t cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged t cell profile. Oncoimmunology 2019, 8, e1648170. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.C.; Huang, L.; Blazar, B.R.; Yagita, H.; Mellor, A.L.; Munn, D.H.; Zhou, G. Polyfunctional cd4(+) t cells are essential for eradicating advanced b-cell lymphoma after chemotherapy. Blood 2012, 120, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Yun, C.H.; Han, S.H. Enhanced anti-cancer activity of human dendritic cells sensitized with gamma-irradiation-induced apoptotic colon cancer cells. Cancer Lett. 2013, 335, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Strome, S.E.; Voss, S.; Wilcox, R.; Wakefield, T.L.; Tamada, K.; Flies, D.; Chapoval, A.; Lu, J.; Kasperbauer, J.L.; Padley, D.; et al. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res. 2002, 62, 1884–1889. [Google Scholar]
- Vandenberk, L.; Garg, A.D.; Verschuere, T.; Koks, C.; Belmans, J.; Beullens, M.; Agostinis, P.; De Vleeschouwer, S.; Van Gool, S.W. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (dcs), potentiates dc vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2016, 5, e1083669. [Google Scholar] [CrossRef]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.L. Modulating both tumor cell death and innate immunity is essential for improving radiation therapy effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming anti-tumor immunity by radiotherapy: Dying tumor cell-derived damps trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology 2019, 8, e1523097. [Google Scholar] [CrossRef]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the t cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Mao, Y. The role of exosomes in tumor immunity. Ann. Transl. Med. 2018, 6, S116. [Google Scholar] [CrossRef]
- Gu, X.; Erb, U.; Buchler, M.W.; Zoller, M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, C.; Wei, W.; Shen, C.; Deng, X.; Chen, L.; Ma, L.; Hao, S. Dendritic cells pulsed with leukemia cell-derived exosomes more efficiently induce antileukemic immunities. PLoS ONE 2014, 9, e91463. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Aarntzen, E.H.; Duiveman-deBoer, T.; Figdor, C.G.; Jacobs, J.F.; Tel, J.; de Vries, I.J. Long-lasting multifunctional cd8(+) t cell responses in end-stage melanoma patients can be induced by dendritic cell vaccination. Oncoimmunology 2016, 5, e1067745. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, W.S.; Choi, D.; Park, J.M.; Song, H.-Y.; Seo, H.S.; Lee, D.-E.; Byun, E.-B. Comparison of Exosomes Derived from Non- and Gamma-Irradiated Melanoma Cancer Cells as a Potential Antigenic and Immunogenic Source for Dendritic Cell-Based Immunotherapeutic Vaccine. Vaccines 2020, 8, 699. https://doi.org/10.3390/vaccines8040699
Kim WS, Choi D, Park JM, Song H-Y, Seo HS, Lee D-E, Byun E-B. Comparison of Exosomes Derived from Non- and Gamma-Irradiated Melanoma Cancer Cells as a Potential Antigenic and Immunogenic Source for Dendritic Cell-Based Immunotherapeutic Vaccine. Vaccines. 2020; 8(4):699. https://doi.org/10.3390/vaccines8040699
Chicago/Turabian StyleKim, Woo Sik, DaeSeong Choi, Ji Min Park, Ha-Yeon Song, Ho Seong Seo, Dong-Eun Lee, and Eui-Baek Byun. 2020. "Comparison of Exosomes Derived from Non- and Gamma-Irradiated Melanoma Cancer Cells as a Potential Antigenic and Immunogenic Source for Dendritic Cell-Based Immunotherapeutic Vaccine" Vaccines 8, no. 4: 699. https://doi.org/10.3390/vaccines8040699
APA StyleKim, W. S., Choi, D., Park, J. M., Song, H.-Y., Seo, H. S., Lee, D.-E., & Byun, E.-B. (2020). Comparison of Exosomes Derived from Non- and Gamma-Irradiated Melanoma Cancer Cells as a Potential Antigenic and Immunogenic Source for Dendritic Cell-Based Immunotherapeutic Vaccine. Vaccines, 8(4), 699. https://doi.org/10.3390/vaccines8040699