Intranasal Delivery of a Chitosan-Hydrogel Vaccine Generates Nasal Tissue Resident Memory CD8+ T Cells That Are Protective against Influenza Virus Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Infections
2.2. Chitosan Vaccine Solution
2.3. In Vitro Activation of OT-I Cells
2.4. Study Design
2.5. Flow Cytometry and Cytometric Bead Array
2.6. Influenza Virus Plaque Assay
2.7. Histological Staining
2.8. Detection of Serum Antibodies by ELISA
3. Results and Discussion
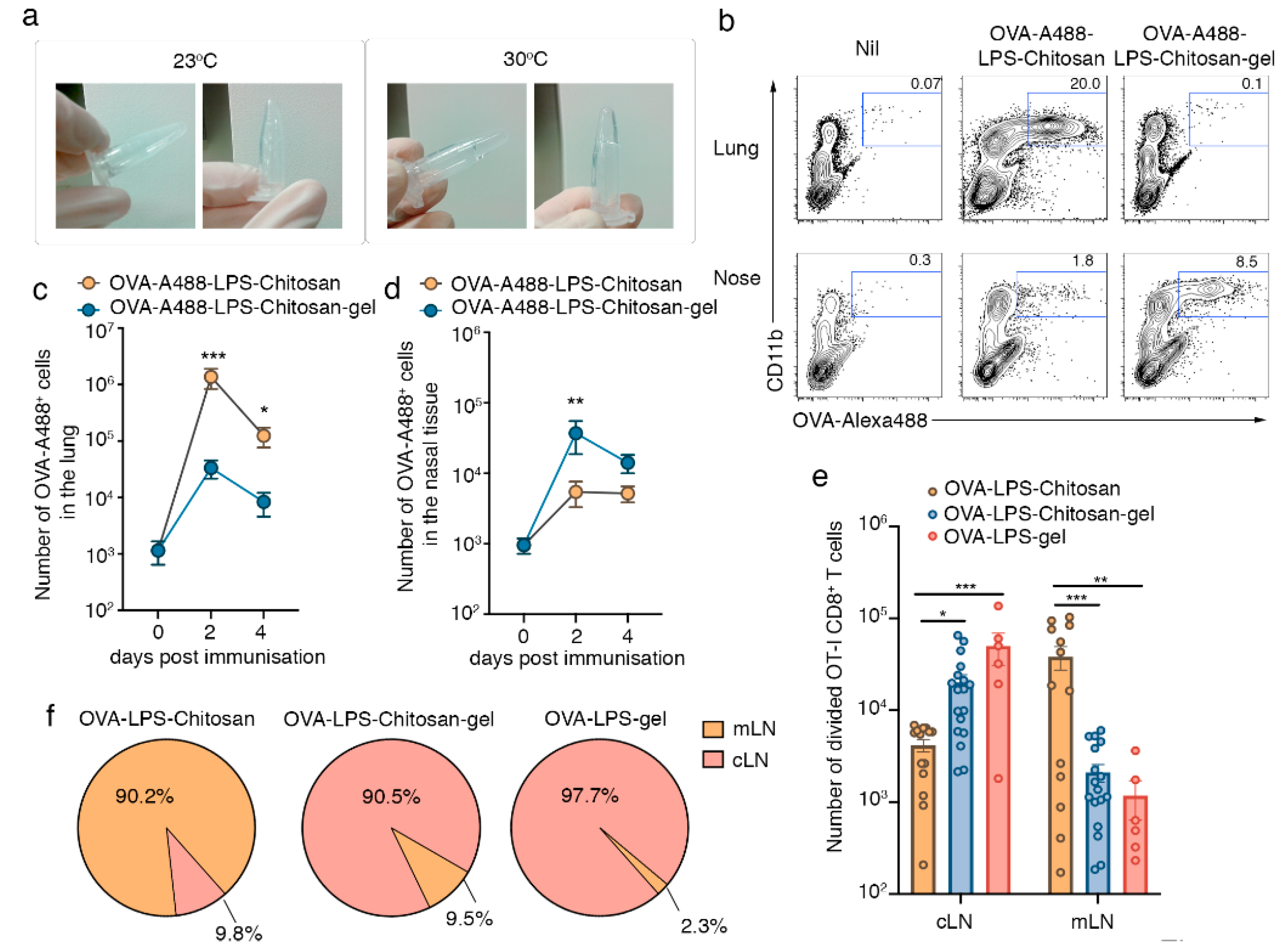
3.1. Intranasal Immunization with a Chitosan-Hydrogel Vaccine Retains Antigen in the Nasal Mucosa and Results in CD8+ T Cell Priming in the Lymph Nodes that Drain the Upper Respiratory Tract
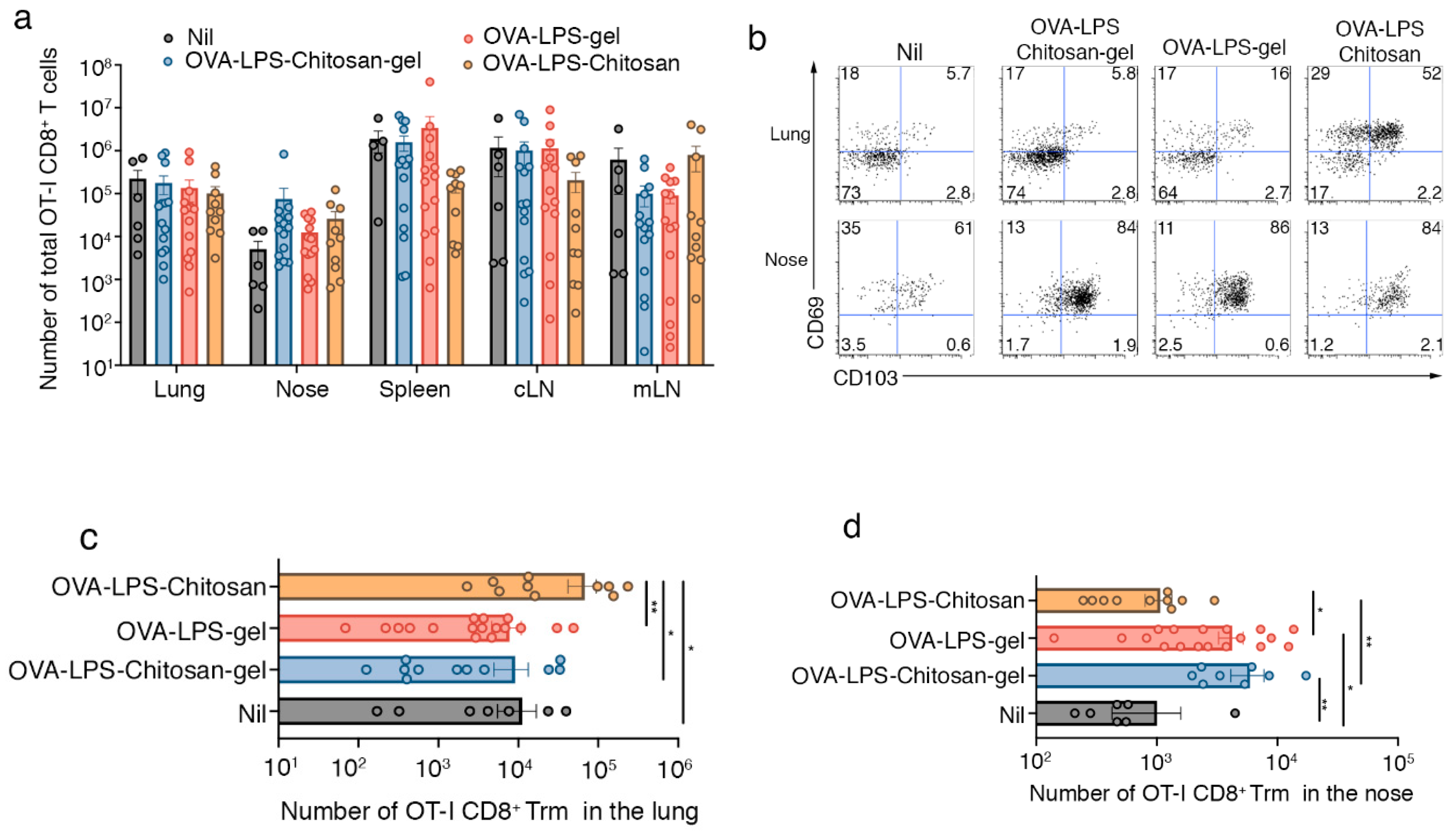
3.2. Intranasal Immunization with a Chitosan-Hydrogel Vaccine Results in the Development of Nasal Trm
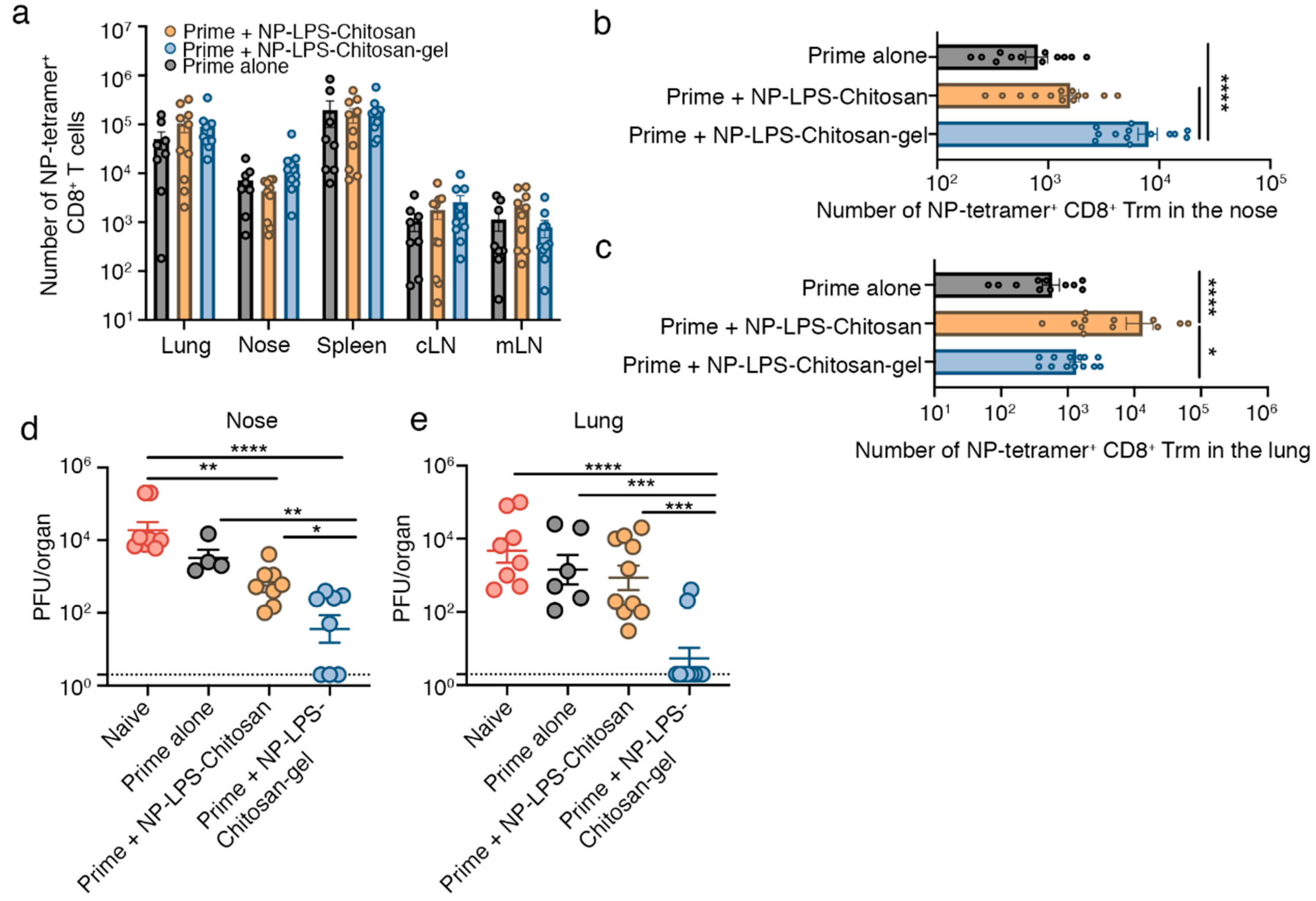
3.3. Intranasal Immunization with an Influenza Virus NP-peptide Loaded Chitosan Hydrogel Vaccine Results in the Development of Nasal Trm that are Protective against Influenza Virus Infection
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Martau, G.A.; Mihai, M.; Vodnar, D.C. The Use of Chitosan, Alginate, and Pectin in the Biomedical and Food Sector-Biocompatibility, Bioadhesiveness, and Biodegradability. Polymers 2019, 11, 1837. [Google Scholar] [CrossRef] [PubMed]
- Mu, M.; Li, X.; Tong, A.; Guo, G. Multi-functional chitosan-based smart hydrogels mediated biomedical application. Expert Opin. Drug Deliv. 2019, 16, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Han, I.K.; Kim, Y.B.; Kang, H.S.; Sul, D.; Jung, W.W.; Cho, H.J.; OH, Y.K. Thermosensitive and mucoadhesive delivery systems of mucosal vaccines. Methods 2006, 38, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Woodward-Davis, A.; Bevan, M.J. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl. Acad. Sci. USA 2010, 107, 17872–17879. [Google Scholar] [CrossRef] [PubMed]
- Pizzolla, A.; Nguyen, T.H.O.; Smith, J.M.; Brooks, A.G.; Kedzieska, K.; Heath, W.R.; Reading, P.C.; Wakim, L.M. Resident memory CD8+ T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Pizzolla, A.; Wang, Z.; Groom, J.R.; Kedzierska, K.; Brooks, A.G.; Reading, P.C. Nasal-associated lymphoid tissues (NALTs) support the recall but not priming of influenza virus-specific cytotoxic T cells. Proc. Natl. Acad. Sci. USA 2017, 114, 5225–5230. [Google Scholar] [CrossRef]
- Lahoud, M.H.; Ahmet, F.; Kitsoulis, S.; Wan, S.S.; Vremec, D.; Lee, C.N.; Phipson, B.; Shi, W.; Smyth, G.K.; Lew, A.M.; et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol. 2011, 187, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Oja, A.E.; Piet, B.; Helbig, C.; Stark, R.; Zwan, D.; Blaauwgeers, H.; Remmerswaal, E.B.M.; Amsen, D.; Jonkers, R.E.; Moerland, P.D.; et al. Trigger-happy resident memory CD4(+) T cells inhabit the human lungs. Mucosal. Immunol. 2018, 11, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Monk, I.R.; Pizzolla, A.; Wang, N.; Bedford, J.G.; Stinear, T.P.; Westall, G.P.; Wakim, L.M. Bystander Activation of Pulmonary Trm Cells Attenuates the Severity of Bacterial Pneumonia by Enhancing Neutrophil Recruitment. Cell Rep. 2019, 29, 4236–4244. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Shieh, W.J.; Blau, D.M.; Denison, A.M.; Deleon-Carnes, M.; Adem, P.; Bhatnagar, J.; Sumner, G.; Liu, L.; Patel, M.; Batten, B.; et al. 2009 pandemic influenza A (H1N1): Pathology and pathogenesis of 100 fatal cases in the United States. Am. J. Pathol. 2010, 177, 166–175. [Google Scholar] [CrossRef] [PubMed]
- McMaster, S.R.; Wein, A.N.; Dunbar, P.R.; Hayward, S.L.; Cartwright, E.K.; Denning, T.L.; Kohlmeier, G.E. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal. Immunol. 2018, 11, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Muschaweckh, A.; Buchholz, V.R.; Fellenzer, A.; Hessel, C.; Konig, P.A.; Tao, S.; Tao, A.; Heikenwälder, M.; Busch, D.H.; Korn, T.; et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J. Exp. Med. 2016, 213, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, Y.; Lee, Y.T.; Bouchard, K.R.; Benechet, A.; Khanna, K.; Cauley, L.S. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 2014, 95, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Killingley, B.; Greatorex, J.; Digard, P.; Wise, H.; Garcia, F.; Varsani, H.; Cauchemez, S.; Enstone, J.E.; Hayward, A.; Curran, M.D.; et al. The environmental deposition of influenza virus from patients infected with influenza A(H1N1)pdm09: Implications for infection prevention and control. J. Infect. Public Health 2016, 9, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Carroll, E.C.; Jin, L.; Mori, A.; Munoz-Wolf, N.; Oleszycka, E.; Moran, H.B.T.; Mansouri, S.; McEntee, C.P.; Lambe, E.; Agger, E.M.; et al. The Vaccine Adjuvant Chitosan Promotes Cellular Immunity via DNA Sensor cGAS-STING-Dependent Induction of Type I Interferons. Immunity 2016, 44, 597–608. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedford, J.G.; Caminschi, I.; Wakim, L.M. Intranasal Delivery of a Chitosan-Hydrogel Vaccine Generates Nasal Tissue Resident Memory CD8+ T Cells That Are Protective against Influenza Virus Infection. Vaccines 2020, 8, 572. https://doi.org/10.3390/vaccines8040572
Bedford JG, Caminschi I, Wakim LM. Intranasal Delivery of a Chitosan-Hydrogel Vaccine Generates Nasal Tissue Resident Memory CD8+ T Cells That Are Protective against Influenza Virus Infection. Vaccines. 2020; 8(4):572. https://doi.org/10.3390/vaccines8040572
Chicago/Turabian StyleBedford, James G., Irina Caminschi, and Linda M. Wakim. 2020. "Intranasal Delivery of a Chitosan-Hydrogel Vaccine Generates Nasal Tissue Resident Memory CD8+ T Cells That Are Protective against Influenza Virus Infection" Vaccines 8, no. 4: 572. https://doi.org/10.3390/vaccines8040572
APA StyleBedford, J. G., Caminschi, I., & Wakim, L. M. (2020). Intranasal Delivery of a Chitosan-Hydrogel Vaccine Generates Nasal Tissue Resident Memory CD8+ T Cells That Are Protective against Influenza Virus Infection. Vaccines, 8(4), 572. https://doi.org/10.3390/vaccines8040572