An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi

 ,
,  ,
,  and
and 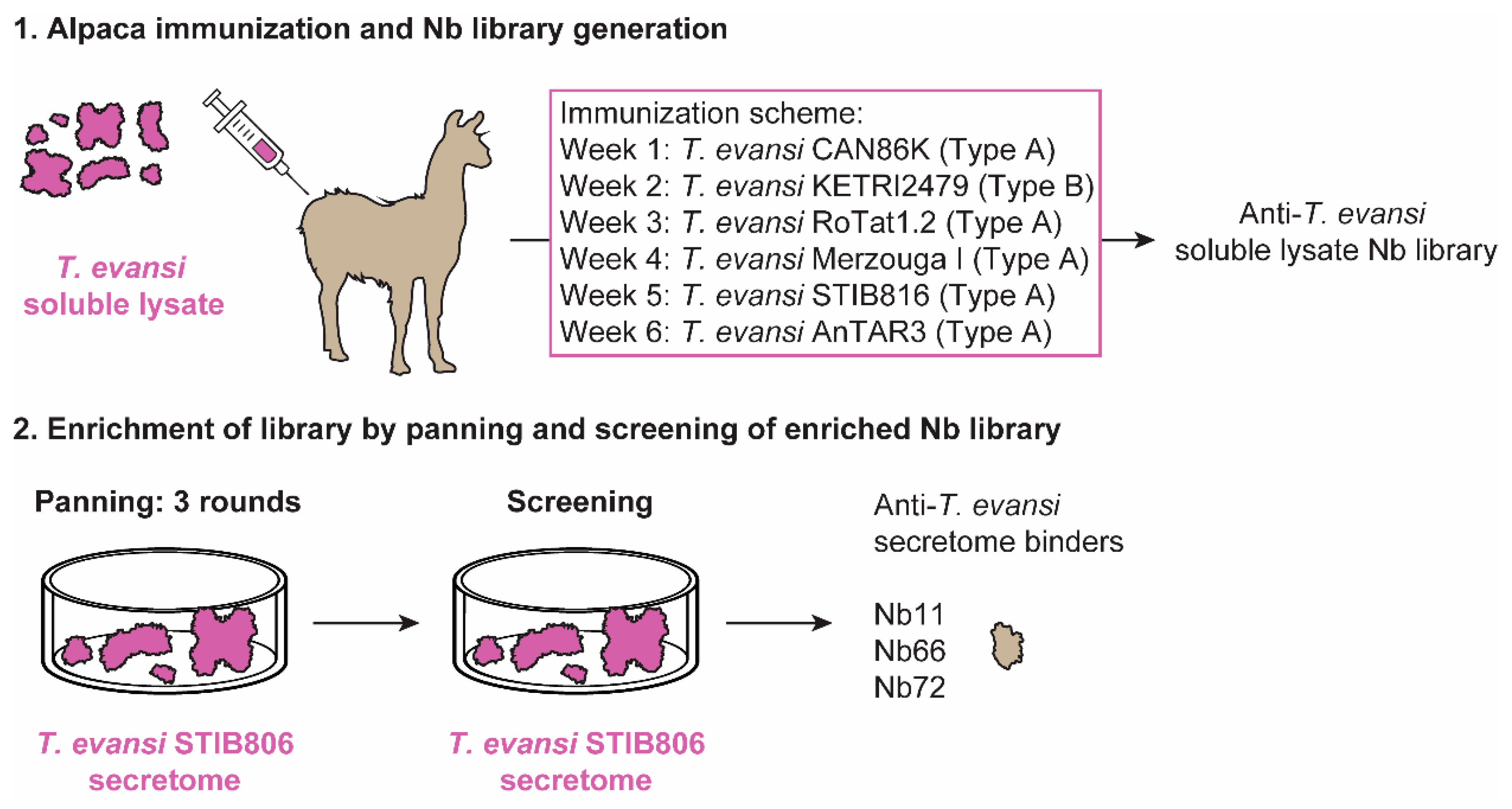{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Secretome and Soluble Lysate Preparation
2.2.1. Secretome Preparation
2.2.2. Soluble Lysate Preparation
2.3. Nb Library Construction and Phage Display
2.3.1. Construction of anti-T. Evansi Lysate and anti-TevENO Nb Libraries
2.3.2. Phage Display and Panning of the anti-T. Evansi Lysate Nb Library
2.3.3. Phage Display and Panning of the anti-TevENO Nb Library
2.4. Production and Purification of Nbs
2.5. Immuno-Capturing and Antigen Identification by LC-MS
2.6. Recombinant Production and Purification of TevENO
2.7. Thermal Stability of TevENO
2.8. ELISA
2.8.1. Indirect ELISA
2.8.2. Sandwich ELISA
2.9. SDS-PAGE and Western Blot
2.10. Circular Dichroism (CD) Spectroscopy
2.11. Enzyme Activity Assay
2.12. Analytical SEC
3. Results
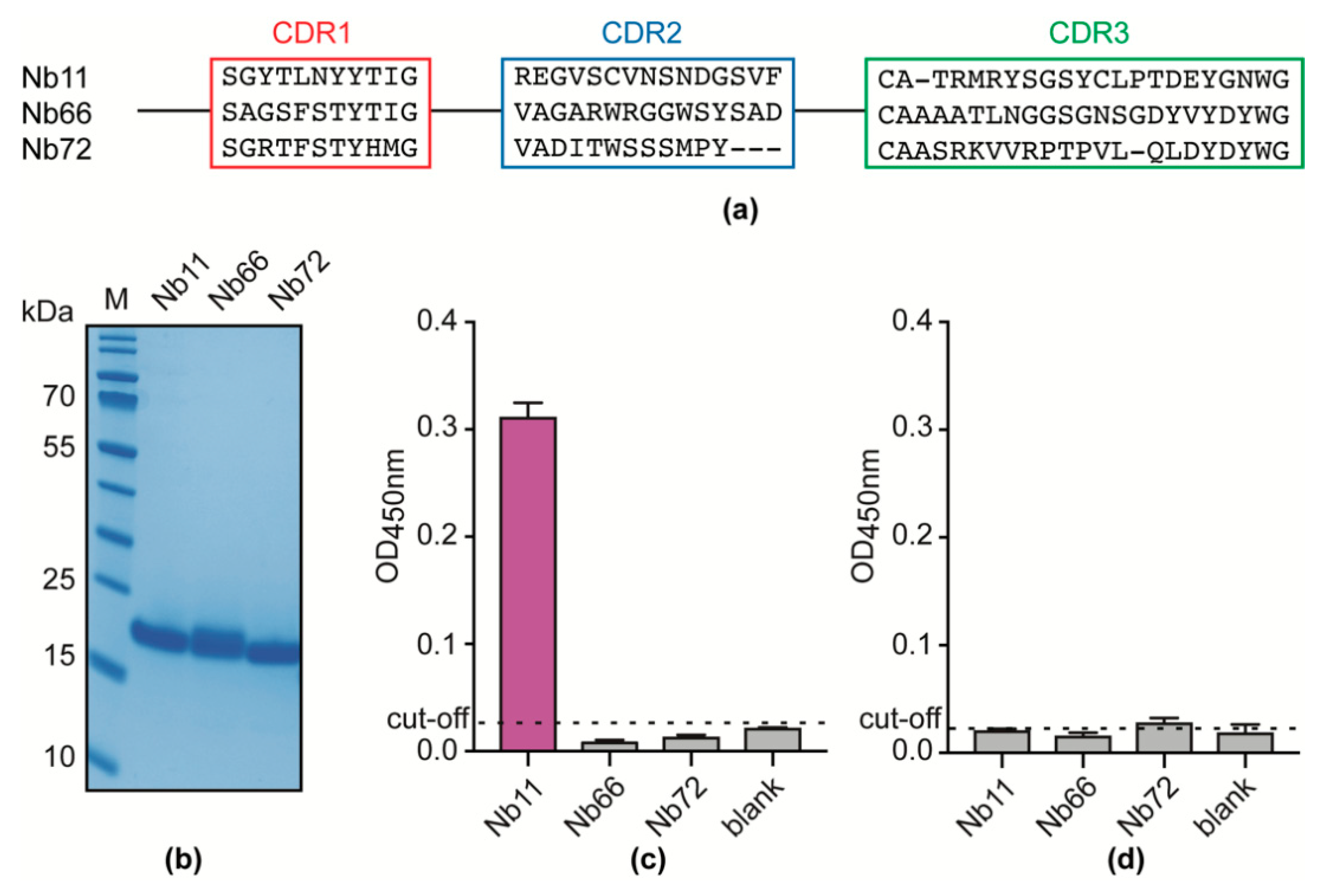
3.1. An Unbiased Alpaca Immunization Strategy Using T. Evansi Soluble Lysates Yields a Single Nb able to Recognize a T. Evansi Secretome Component
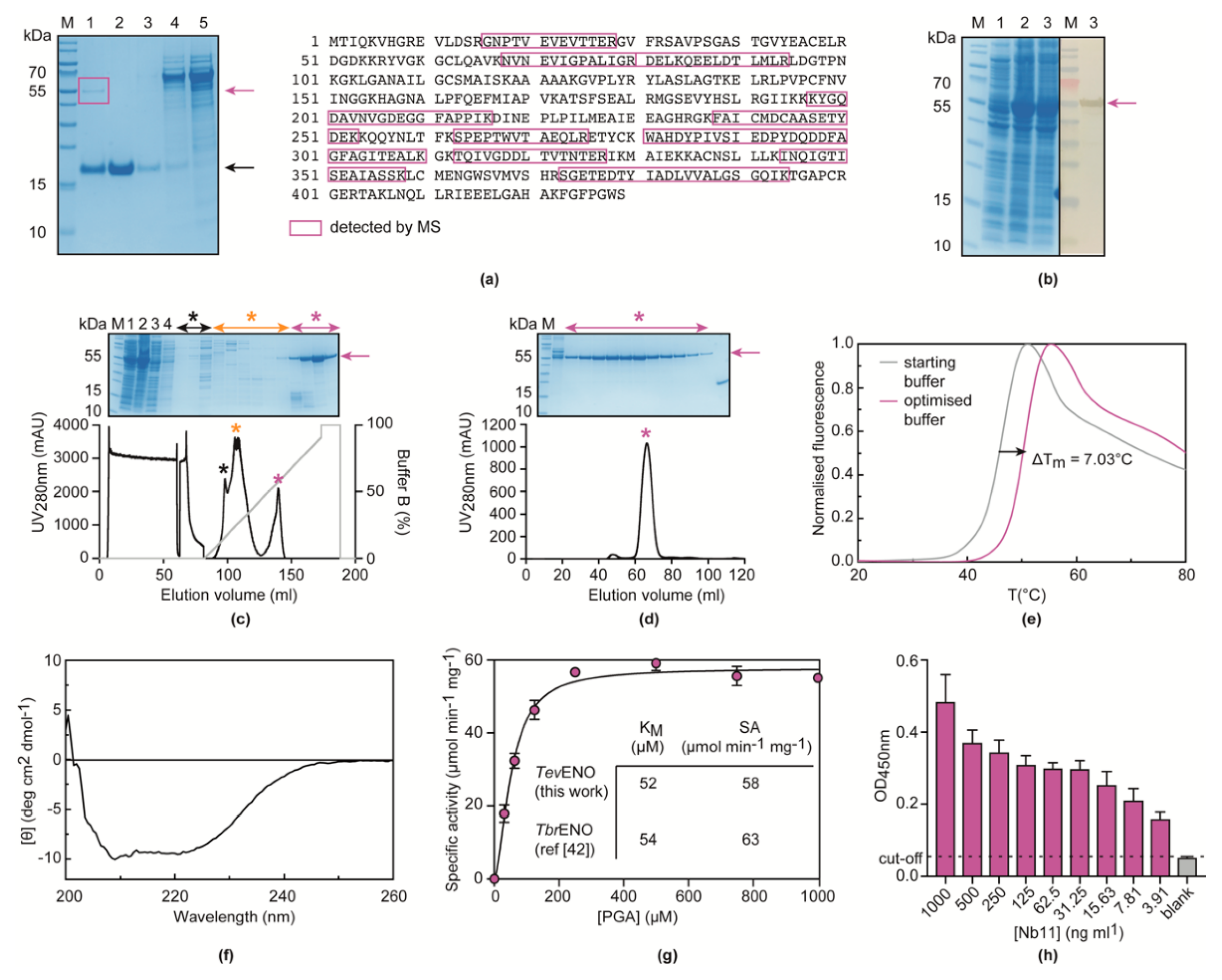
3.2. Nb11 Targets T. Evansi Enolase
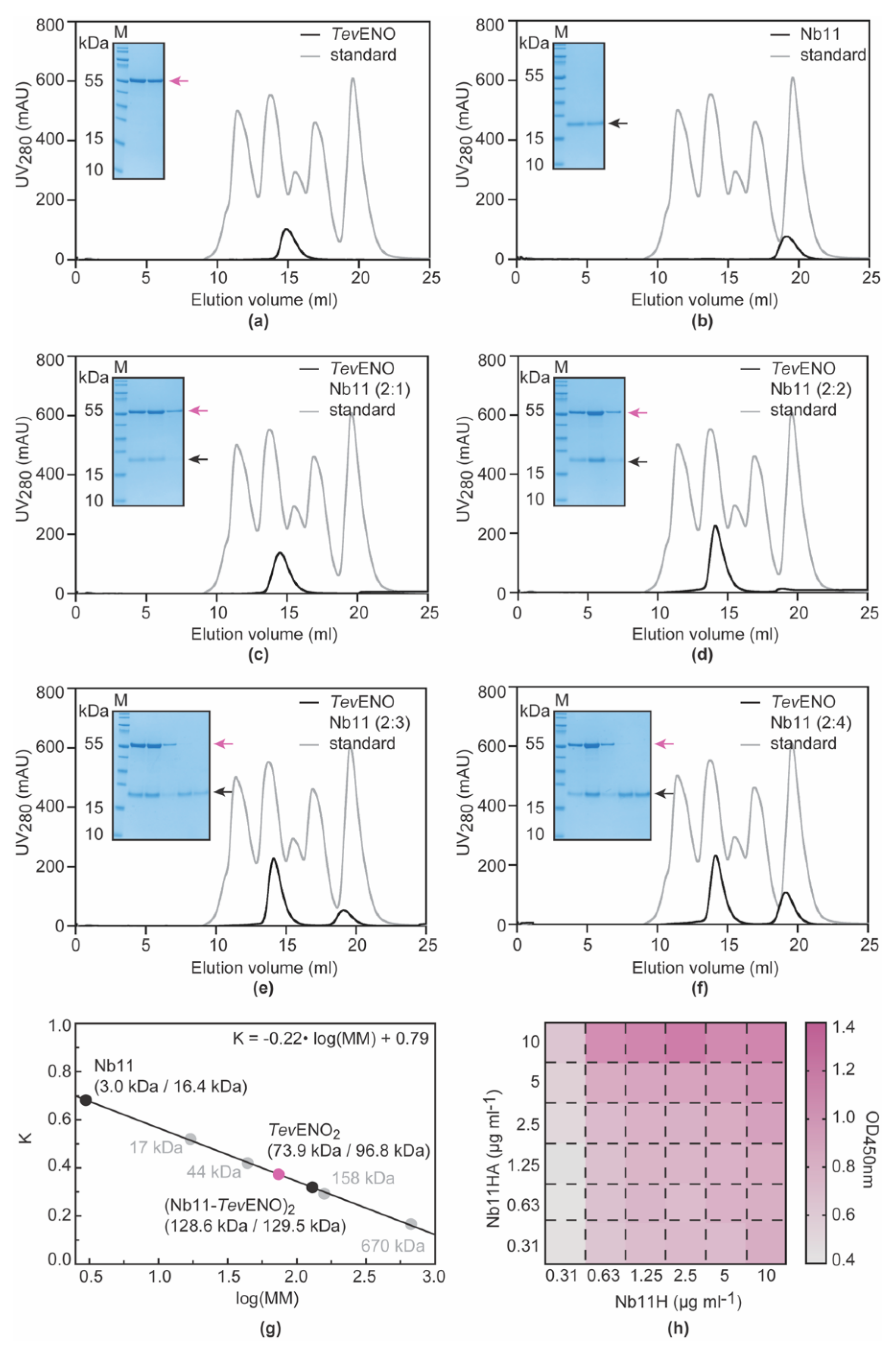
3.3. Nb11 Can Be Employed in a Homologous Sandwich Assay Targeting TevENO
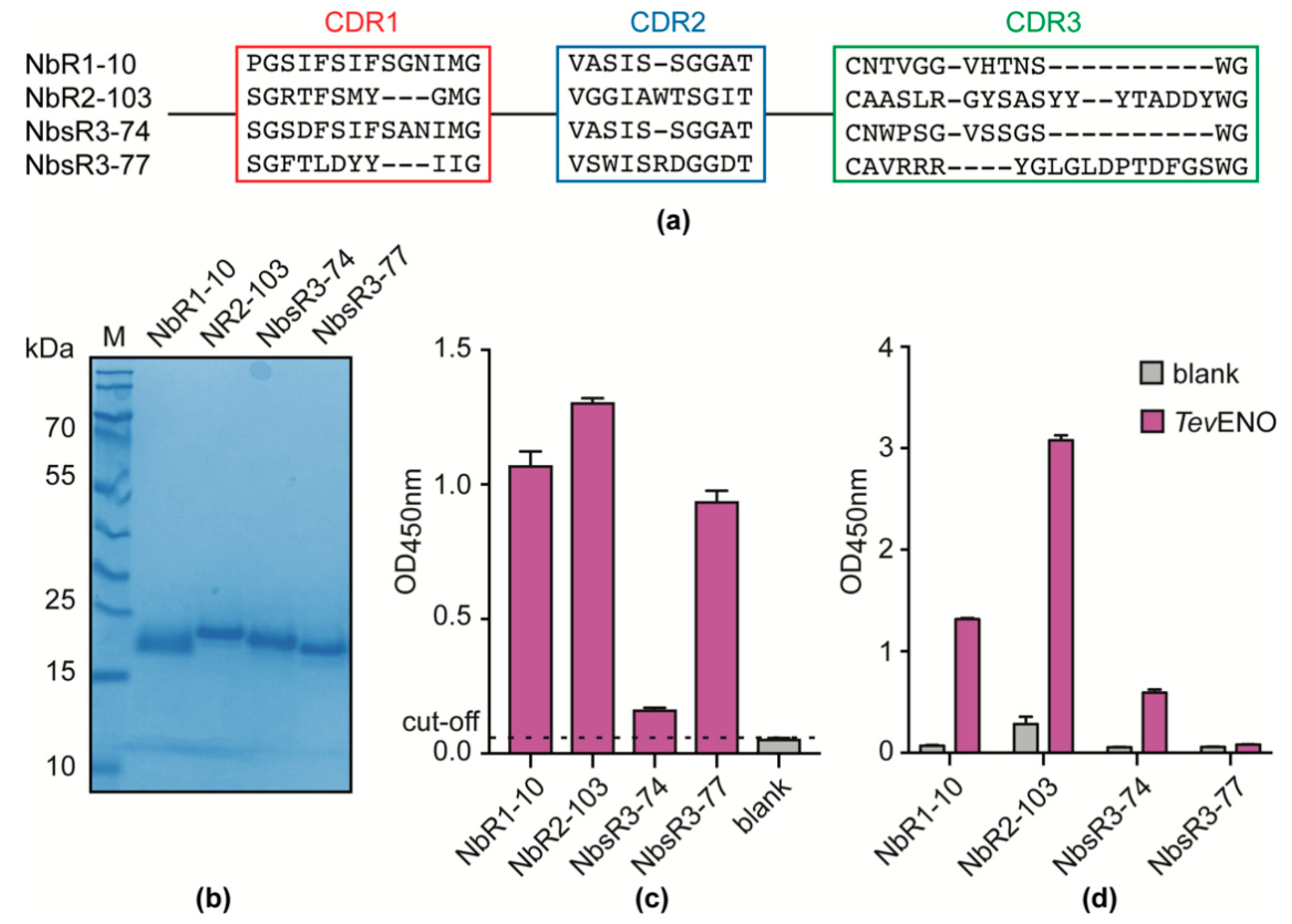
3.4. Alpaca Immunization with Recombinant TevENO Yields Four Additional Nbs That Can Be Paired with Nb11 in Heterologous Sandwich Assays Targeting TevENO
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Franke, C.R.; Greiner, M.; Mehlitz, D. Investigations on naturally occurring Trypanosoma evansi infections in horses, cattle, dogs and capybaras (Hydrochaeris hydrochaeris) in Pantanal de Poconé (Mato Grosso, Brazil). Acta Trop. 1994, 58, 159–169. [Google Scholar] [CrossRef]
- Adrian, M.S.; Sani, R.A.; Hassan, L.; Wong, M.T. Outbreaks of trypanosomiasis and the seroprevalence of T. evansi in a deer breeding centre in Perak, Malaysia. Trop. Anim. Health Prod. 2010, 42, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Thekisoe, O.M.; Inoue, N.; Kuboki, N.; Tuntasuvan, D.; Bunnoy, W.; Borisutsuwan, S.; Igarashi, I.; Sugimoto, C. Evaluation of loop-mediated isothermal amplification (LAMP), PCR and parasitological tests for detection of Trypanosoma evansi in experimentally infected pigs. Vet. Parasitol. 2005, 130, 327–330. [Google Scholar] [CrossRef]
- Nunes, V.; Oshiro, E.T. Trypanosoma (Trypanozoon) evansi in the coati from the Pantanal region of Mato Grosso do Sul State, Brazil. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 692. [Google Scholar] [CrossRef]
- Silva, R.; Barros, A.; Herrera, H.M. Trypanosomosis outbreaks due to Trypanosoma evansi in the Pantanal, Brazil. A preliminary approach on risk factors. Revue D’élevage Médecine Vétérinaire Pays Tropicaux 1995, 48, 315–319. [Google Scholar]
- Luckins, A.G.; Dwinger, R.H. Non-tsetse-transmitted animal trypanosomiasis. Trypanosomiases 2004, 269, 281. [Google Scholar]
- Gutierrez, C.; Desquesnes, M.; Touratier, L.; Buscher, P. Trypanosoma evansi: Recent outbreaks in Europe. Vet. Parasitol. 2010, 174, 26–29. [Google Scholar] [CrossRef]
- Aregawi, W.G.; Agga, G.E.; Abdi, R.D.; Büscher, P. Systematic review and meta-analysis on the global distribution, host range, and prevalence of Trypanosoma evansi. Parasites Vectors 2019, 12. [Google Scholar] [CrossRef]
- Joshi, P.P.; Shegokar, V.R.; Powar, R.M.; Herder, S.; Katti, R.; Salkar, H.R.; Dani, V.S.; Bhargava, A.; Jannin, J.; Truc, P.; et al. Human trypanosomiasis caused by Trypanosoma evansi in India: The first case report. Am. J. Trop. Med. Hyg. 2005, 73, 491–495. [Google Scholar] [CrossRef]
- Desquesnes, M.; Holzmuller, P.; Lai, D.-H.; Dargantes, A.; Lun, Z.-R.; Jittaplapong, S. Trypanosoma evansi and surra: A review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, and pathogenic effects. BioMed Res. Int. 2013, 2013, 194176. [Google Scholar] [CrossRef]
- Desquesnes, M.; Dargantes, A.; Lai, D.-H.; Lun, Z.-R.; Holzmuller, P.; Jittapalapong, S. Trypanosoma evansi and surra: A review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. BioMed Res. Int. 2013, 2013, 321237. [Google Scholar] [CrossRef] [PubMed]
- Masiga, D.K.; Gibson, W.C. Specific probes for Trypanosoma (Trypanozoon) evansi based on kinetoplast DNA minicircles. Mol. Biochem. Parasitol. 1990, 40, 279–283. [Google Scholar] [CrossRef]
- Kamidi, C.M.; Saarman, N.P.; Dion, K.; Mireji, P.O.; Ouma, C.; Murilla, G.; Aksoy, S.; Schnaufer, A.; Caccone, A. Multiple evolutionary origins of Trypanosoma evansi in Kenya. PLoS Negl. Trop. Dis. 2017, 11, e0005895. [Google Scholar] [CrossRef] [PubMed]
- Njiru, Z.K.; Constantine, C.C.; Masiga, D.K.; Reid, S.A.; Thompson, R.C.; Gibson, W.C. Characterization of Trypanosoma evansi type B. Infect. Genet. Evol. 2006, 6, 292–300. [Google Scholar] [CrossRef]
- Masiga, D.K.; Smyth, A.J.; Hayes, P.; Bromidge, T.J.; Gibson, W.C. Sensitive detection of trypanosomes in tsetse flies by DNA amplification. Int. J. Parasitol. 1992, 22, 909–918. [Google Scholar] [CrossRef]
- Dávila, A.; Herrera, H.; Schlebinger, T.; Souza, S.; Traub-Cseko, Y.M. Using PCR for unraveling the cryptic epizootiology of livestock trypanosomosis in the Pantanal, Brazil. Vet. Parasitol. 2003, 117, 1–13. [Google Scholar] [CrossRef]
- Tong, Q.; Chen, R.; Kong, Q.; Goossens, J.; Radwanska, M.; Lou, D.; Ding, J.; Zheng, B.; Fu, Y.; Wang, T.; et al. DNA detection of Trypanosoma evansi: Diagnostic validity of a new assay based on loop-mediated isothermal amplification (LAMP). Vet. Parasitol. 2018, 250, 1–6. [Google Scholar] [CrossRef]
- Li, Z.; Torres, J.E.P.; Goossens, J.; Stijlemans, B.; Sterckx, Y.G.-J.; Magez, S. Development of a recombinase polymerase amplification lateral flow assay for the detection of active Trypanosoma evansi infections. PLoS Negl. Trop. Dis. 2020, 14, e0008044. [Google Scholar] [CrossRef]
- Davison, H.; Thrusfield, M.; Muharsini, S.; Husein, A.; Partoutomo, S.; Rae, P.; Masake, R.; Luckins, A.G. Evaluation of antigen detection and antibody detection tests for Trypanosoma evansi infections of buffaloes in Indonesia. Epidemiol. Infect. 1999, 123, 149–155. [Google Scholar] [CrossRef]
- Ngaira, J.; Bett, B.; Karanja, S.; Njagi, E.N.M. Evaluation of antigen and antibody rapid detection tests for Trypanosoma evansi infection in camels in Kenya. Vet. Parasitol. 2003, 114, 131–141. [Google Scholar] [CrossRef]
- Laha, R.; Sasmal, N.K. Detection of Trypanosoma evansi infection in clinically ill cattle, buffaloes and horses using various diagnostic tests. Epidemiol. Infect. 2009, 137, 1583–1585. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, S.Y.; Tabatabai, J.; Schnitzler, P.; Farah, C.; Rameil, S.; Sander, P.; Koromyslova, A.; Hansman, G.S. Development of a nanobody-based lateral flow immunoassay for detection of human norovirus. J. Clin. Epidemiol. 2016, 1, e00219-16. [Google Scholar] [CrossRef] [PubMed]
- Petyovka, N.; Lyach, L.; Voitenok, N.N. Homologous ELISA for detection of oligomeric human TNF: Properties of the assay. J. Immunol. Methods 1995, 186, 161–170. [Google Scholar] [CrossRef]
- Odongo, S.; Sterckx, Y.G.; Stijlemans, B.; Pillay, D.; Baltz, T.; Muyldermans, S.; Magez, S. An Anti-proteome Nanobody Library Approach Yields a Specific Immunoassay for Trypanosoma congolense Diagnosis Targeting Glycosomal Aldolase. PLoS Negl. Trop. Dis. 2016, 10, e0004420. [Google Scholar] [CrossRef]
- Lambert, P.; Berney, M.; Kazyumba, G. Immune complexes in serum and in cerebrospinal fluid in African trypanosomiasis: Correlation with polyclonal B cell activation and with intracerebral immunoglobulin synthesis. J. Clin. Investig. 1981, 67, 77–85. [Google Scholar] [CrossRef]
- Süsal, C.; Lewin, I.; Stanworth, D.; Terness, P.; Daniel, V.; Oberg, H.H.; Huth-Kuhne, A.; Zimmermann, R.; Opelz, G. Anti-IgG autoantibodies in HIV-infected hemophilia patients. Vox Sang. 1992, 62, 224–229. [Google Scholar] [CrossRef]
- Muyldermans, S.; Baral, T.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunop. 2009, 128, 178–183. [Google Scholar] [CrossRef]
- Obishakin, E.; Stijlemans, B.; Santi-Rocca, J.; Vandenberghe, I.; Devreese, B.; Muldermans, S.; Bastin, P.; Magez, S. Generation of a nanobody targeting the paraflagellar rod protein of trypanosomes. PLoS ONE 2014, 9, e115893. [Google Scholar] [CrossRef]
- Skottrup, P.D.; Leonard, P.; Kaczmarek, J.Z.; Veillard, F.; Enghild, J.J.; O’Kennedy, R.; Sroka, A.; Clausen, R.P.; Potempa, J.; Riise, E. Diagnostic evaluation of a nanobody with picomolar affinity toward the protease RgpB from Porphyromonas gingivalis. Anal. Biochem. 2011, 415, 158–167. [Google Scholar] [CrossRef]
- Zhu, M.; Gong, X.; Hu, Y.; Ou, W.; Wan, Y. Streptavidin-biotin-based directional double Nanobody sandwich ELISA for clinical rapid and sensitive detection of influenza H5N1. J. Transl. Med. 2014, 12, 352. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Stijlemans, B.; Conrath, K.; Cortez-Retamozo, V.; Van Xong, H.; Wyns, L.; Senter, P.; Revets, H.; De Baetselier, P.; Muyldermans, S.; Magez, S. Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies African trypanosomes as paradigm. J. Biol. Chem. 2004, 279, 1256–1261. [Google Scholar] [CrossRef]
- Lauwereys, M.; Ghahroudi, M.A.; Desmyter, A.; Kinne, J.; Hölzer, W.; De Genst, E.; Wyns, L.; Muyldermans, S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998, 17, 3512–3520. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, C.; Muyldermans, S. Nanobody-based delivery systems for diagnosis and targeted tumor therapy. Front. Immunol. 2017, 8, 1442. [Google Scholar] [CrossRef]
- Deckers, N.; Saerens, D.; Kanobana, K.; Conrath, K.; Victor, B.; Wernery, U.; Vercruysse, J.; Muyldermans, S.; Dorny, P. Nanobodies, a promising tool for species-specific diagnosis of Taenia solium cysticercosis. Int. J. Parasitol. 2009, 39, 625–633. [Google Scholar] [CrossRef]
- Rasoulinejad, S.; Gargari, S.L.M. Aptamer-nanobody based ELASA for specific detection of Acinetobacter baumannii isolates. J. Biotechnol. 2016, 231, 46–54. [Google Scholar] [CrossRef]
- Pinto Torres, J.E.; Goossens, J.; Ding, J.; Li, Z.; Lu, S.; Vertommen, D.; Naniima, P.; Chen, R.; Muyldermans, S.; Sterckx, Y.G.; et al. Development of a Nanobody-based lateral flow assay to detect active Trypanosoma congolense infections. Sci. Rep. 2018, 8, 9019. [Google Scholar] [CrossRef]
- Holzmuller, P.; Grébaut, P.; Peltier, J.B.; Brizard, J.P.; Perrone, T.; Gonzatti, M.; Bengaly, Z.; Rossignol, M.; Aso, P.M.; Vincendeau, P. Secretome of animal trypanosomes. Anim. Biodivers. Emerg. Dis. 2008, 1149, 337–342. [Google Scholar] [CrossRef]
- Lanham, S.M.; Godfrey, D. Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Exp. Parasitol. 1970, 28, 521–534. [Google Scholar] [CrossRef]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.; Wohlkonig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef]
- Reinhard, L.; Mayerhofer, H.; Geerlof, A.; Mueller-Dieckmann, J.; Weiss, M.S. Optimization of protein buffer cocktails using Thermofluor. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Hannaert, V.; Albert, M.-A.; Rigden, D.J.; Theresa da Silva Giotto, M.; Thiemann, O.; Garratt, R.C.; Van Roy, J.; Opperdoes, F.R.; Michels, P.A.M. Kinetic characterization, structure modelling studies and crystallization of Trypanosoma brucei enolase. Eur. J. Biochem. 2003, 270, 3205–3213. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Giotto, M.T.; Hannaert, V.; Vertommen, D.; Navarro, M.V.d.A.S.; Rider, M.H.; Michels, P.A.M.; Garratt, R.C.; Rigden, D.J. The Crystal Structure of Trypanosoma brucei Enolase: Visualisation of the Inhibitory Metal Binding Site III and Potential as Target for Selective, Irreversible Inhibition. J. Mol. Biol. 2003, 331, 653–665. [Google Scholar] [CrossRef]
- Tehseen, S.; Jahan, N.; Qamar, M.F.; Desquesnes, M.; Shahzad, M.I.; Deborggraeve, S.; Buscher, P. Parasitological, serological and molecular survey of Trypanosoma evansi infection in dromedary camels from Cholistan Desert, Pakistan. Parasit Vectors 2015, 8, 415. [Google Scholar] [CrossRef] [PubMed]
- Espino, A.M.; Millán, J.C.; Finlay, C.M. Detection of antibodies and circulating excretory-secretory antigens for assessing cure in patients with fascioliasis. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 649. [Google Scholar] [CrossRef]
- Lejon, V.; Claes, F.; Verloo, D.; Maina, M.; Urakawa, T.; Majiwa, P.; Büscher, P. Recombinant RoTat 1.2 variable surface glycoprotein as antigen for diagnosis of Trypanosoma evansi in dromedary camels. Int. J. Parasitol. 2005, 35, 455–460. [Google Scholar] [CrossRef]
- Rebeski, D.E.; Winger, E.M.; Van Rooij, E.M.; Schöchl, R.; Schuller, W.; Dwinger, R.H.; Crowther, J.R.; Wright, P. Pitfalls in the application of enzyme-linked immunoassays for the detection of circulating trypanosomal antigens in serum samples. Parasitol. Res. 1999, 85, 550–556. [Google Scholar] [CrossRef]
- Meyer, T.; Schirrmann, T.; Frenzel, A.; Miethe, S.; Stratmann-Selke, J.; Gerlach, G.F.; Strutzberg-Minder, K.; Dübel, S.; Hust, M. Identification of immunogenic proteins and generation of antibodies against Salmonella Typhimurium using phage display. BMC Biotechnol. 2012, 12, 29. [Google Scholar] [CrossRef]
- Byrnes, S.A.; Weigl, B.H. Selecting analytical biomarkers for diagnostic applications: A first principles approach. Expert Rev. Mol. Diagn. 2018, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Nantulya, V.M. Trypanosomiasis in domestic animals: The problems of diagnosis. Rev. Sci. Tech. Off. Int. Epiz. 1990, 9, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Rebeski, D.; Winger, E.; Rogovic, B.; Robinson, M.; Crowther, J.; Dwinger, R.H. Improved methods for the diagnosis of African trypanosomosis. Mem. Inst. Oswaldo Cruz. 1999, 94, 249–253. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashiwazaki, Y.; Pholpark, M.; Polsar, C.; Pholpark, S. Haemoparasite infections in newly introduced dairy cattle in Loei Province, Thailand: Trypanosoma evansi antigen levels by ELISA referring to abortion. Vet. Parasitol. 1998, 80, 99–109. [Google Scholar] [CrossRef]
- Olaho-Mukani, W.; Munyua, W.; Mutugi, M.; Njogu, A. Comparison of antibody-and antigen-detection enzyme immunoassays for the diagnosis of Trypanosoma evansi infections in camels. Vet. Parasitol. 1993, 45, 231–240. [Google Scholar] [CrossRef]
- Szempruch, A.J.; Sykes, S.E.; Kieft, R.; Dennison, L.; Becker, A.C.; Gartrell, A.; Martin, W.J.; Nakayasu, E.S.; Almeida, I.C.; Hajduk, S.L.; et al. Extracellular Vesicles from Trypanosoma brucei Mediate Virulence Factor Transfer and Cause Host Anemia. Cell 2016, 164, 246–257. [Google Scholar] [CrossRef]
- Grébaut, P.; Chuchana, P.; Brizard, J.-P.; Demettre, E.; Seveno, M.; Bossard, G.; Jouin, P.; Vincendeau, P.; Bengaly, Z.; Boulangé, A. Identification of total and differentially expressed excreted–secreted proteins from Trypanosoma congolense strains exhibiting different virulence and pathogenicity. Int. J. Parasitol. 2009, 39, 1137–1150. [Google Scholar] [CrossRef]
- Geiger, A.; Hirtz, C.; Becue, T.; Bellard, E.; Centeno, D.; Gargani, D.; Rossignol, M.; Cuny, G.; Peltier, J.B. Exocytosis and protein secretion in Trypanosoma. BMC Microbiol. 2010, 10, 20. [Google Scholar] [CrossRef]
- Atyame Nten, C.M.; Sommerer, N.; Rofidal, V.; Hirtz, C.; Rossignol, M.; Cuny, G.; Peltier, J.-B.; Geiger, A. Excreted/secreted proteins from trypanosome procyclic strains. J. Biomed. Biotechnol. 2009, 2010. [Google Scholar] [CrossRef]
- Dzakah, E.E.; Kang, K.; Ni, C.; Wang, H.; Wu, P.; Tang, S.; Wang, J.; Wang, J.; Wang, X. Plasmodium vivax aldolase-specific monoclonal antibodies and its application in clinical diagnosis of malaria infections in China. Malar. J. 2013, 12, 199. [Google Scholar] [CrossRef]
- Krause, R.G.; Hurdayal, R.; Choveaux, D.; Przyborski, J.M.; Coetzer, T.H.; Goldring, J.P.D. Plasmodium glyceraldehyde-3-phosphate dehydrogenase: A potential malaria diagnostic target. Exp. Parasitol. 2017, 179, 7–19. [Google Scholar] [CrossRef]
- Hardt, P.; Ngoumou, B.; Rupp, J.; Schnell-Kretschmer, H.; Kloer, H. Tumor M2-pyruvate kinase: A promising tumor marker in the diagnosis of gastro-intestinal cancer. Anticancer Res. 2000, 20, 4965–4968. [Google Scholar] [PubMed]
- Lee, K.H.; Chung, H.S.; Kim, H.S.; Oh, S.H.; Ha, M.K.; Baik, J.H.; Lee, S.; Bang, D. Human alpha-enolase from endothelial cells as a target antigen of anti-endothelial cell antibody in Behcet’s disease. Arthritis Rheum. 2003, 48, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I.; Bodemer, M.; Räcker, S.; Grosche, S.; Poser, S.; Weber, T.; Kretzschmar, H. Cerebrospinal fluid concentration of neuron-specific enolase in diagnosis of Creutzfeldt-Jakob disease. Lancet 1995, 345, 1609–1610. [Google Scholar] [CrossRef]
- Duarte, M.C.; Lage, D.P.; Martins, V.T.; Costa, L.E.; Salles, B.C.S.; Carvalho, A.; de Oliveira Santos, T.T.; Dias, D.S.; Ribeiro, P.A.F.; Chavez-Fumagalli, M.A.; et al. Performance of Leishmania braziliensis enolase protein for the serodiagnosis of canine and human visceral leishmaniosis. Vet. Parasitol. 2017, 238, 77–81. [Google Scholar] [CrossRef]
- Pinto, J.; Odongo, S.; Lee, F.; Gaspariunaite, V.; Muyldermans, S.; Magez, S.; Sterckx, Y.G. Structural basis for the high specificity of a Trypanosoma congolense immunoassay targeting glycosomal aldolase. PLoS Negl. Trop. Dis. 2017, 11, e0005932. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Pinto Torres, J.E.; Goossens, J.; Vertommen, D.; Caljon, G.; Sterckx, Y.G.-J.; Magez, S. An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi. Vaccines 2020, 8, 415. https://doi.org/10.3390/vaccines8030415
Li Z, Pinto Torres JE, Goossens J, Vertommen D, Caljon G, Sterckx YG-J, Magez S. An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi. Vaccines. 2020; 8(3):415. https://doi.org/10.3390/vaccines8030415
Chicago/Turabian StyleLi, Zeng, Joar Esteban Pinto Torres, Julie Goossens, Didier Vertommen, Guy Caljon, Yann G.-J. Sterckx, and Stefan Magez. 2020. "An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi" Vaccines 8, no. 3: 415. https://doi.org/10.3390/vaccines8030415
APA StyleLi, Z., Pinto Torres, J. E., Goossens, J., Vertommen, D., Caljon, G., Sterckx, Y. G.-J., & Magez, S. (2020). An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi. Vaccines, 8(3), 415. https://doi.org/10.3390/vaccines8030415