Development of Tumor Cell-Based Vaccine with IL-12 Gene Electrotransfer as Adjuvant




Abstract
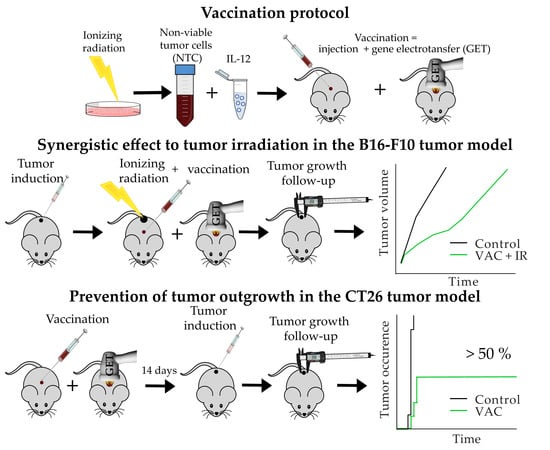
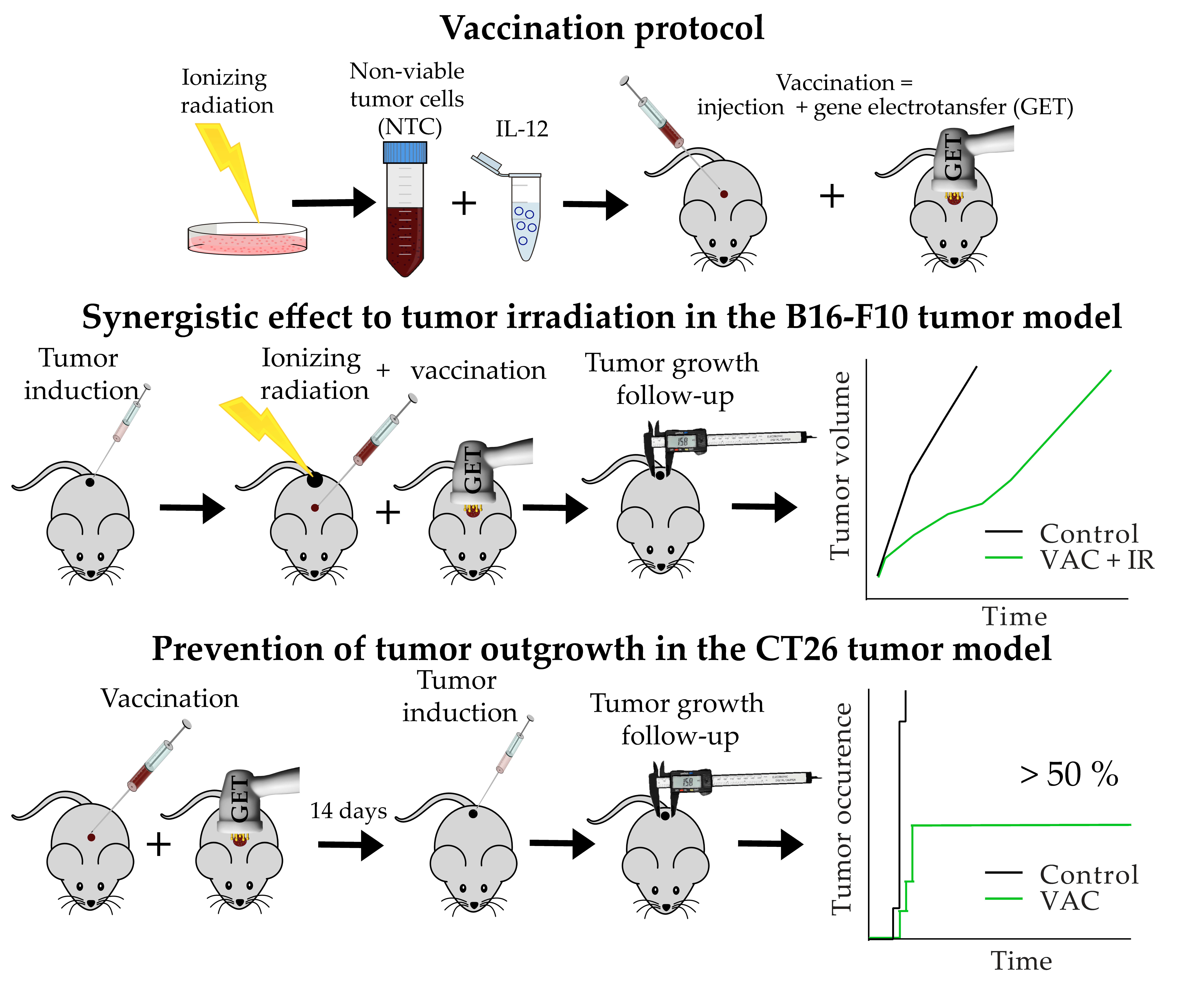{kind=link}
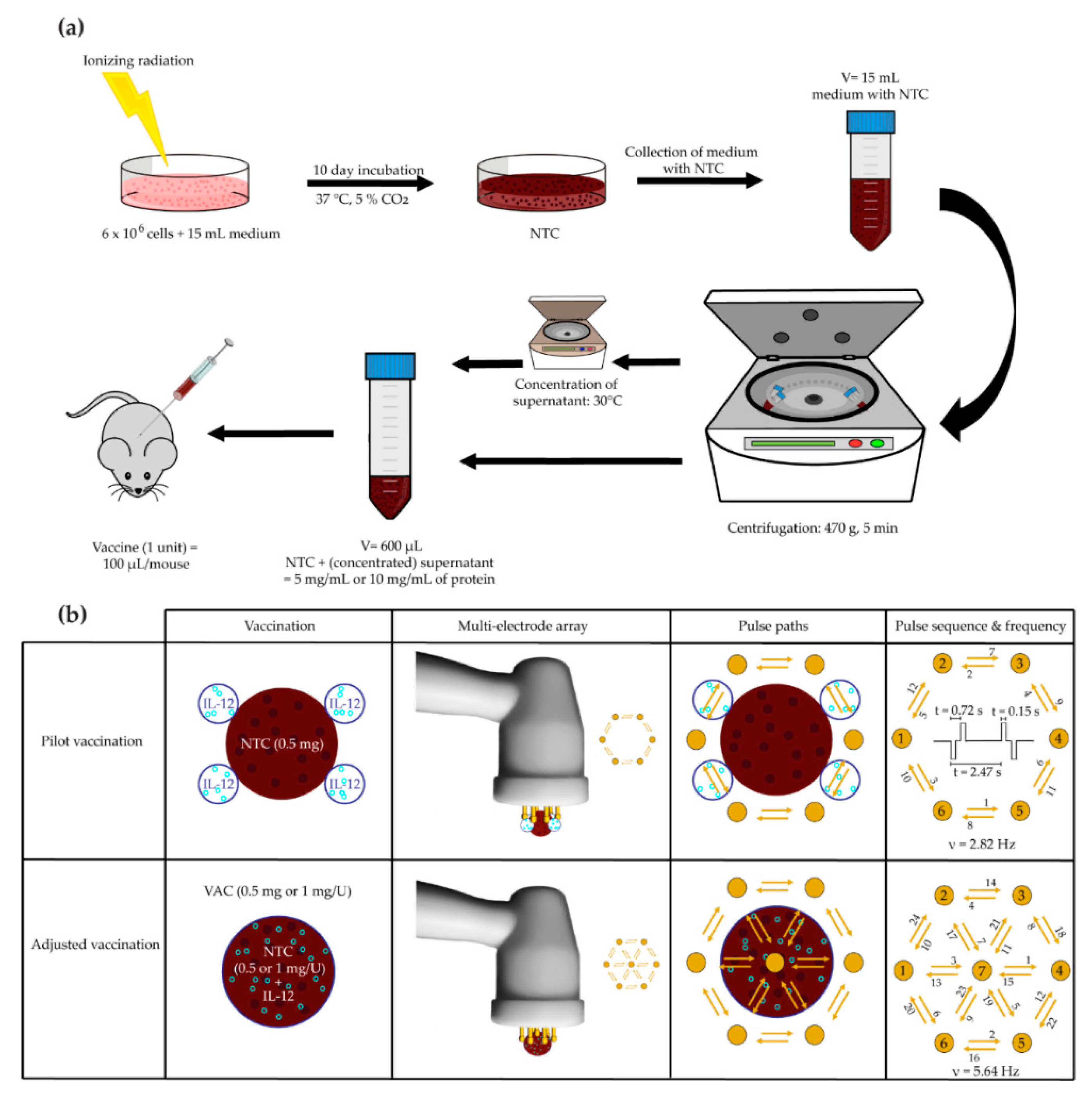{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmid DNA
2.3. Animals
2.4. Irradiation
2.5. Vaccination
2.5.1. Pilot B16-F10 Vaccination
2.5.2. Adjusted B16-F10 and CT26 Vaccine Preparation and Therapeutic Vaccination
2.5.3. B16-F10 and CT26 Preventative Vaccination
2.6. Treatment Effectiveness Assessment
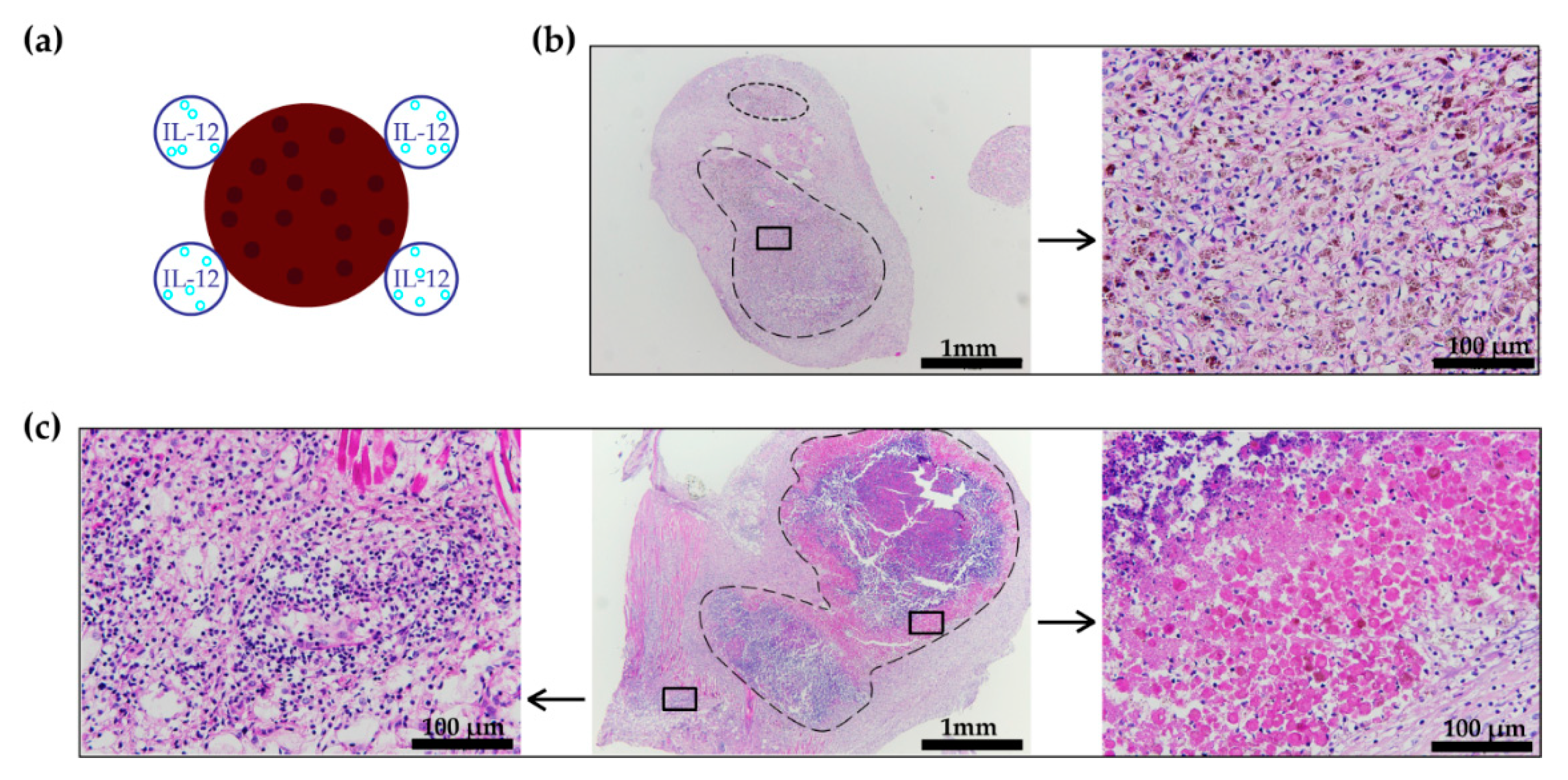
2.7. Histology
2.8. Statistical Analysis
3. Results
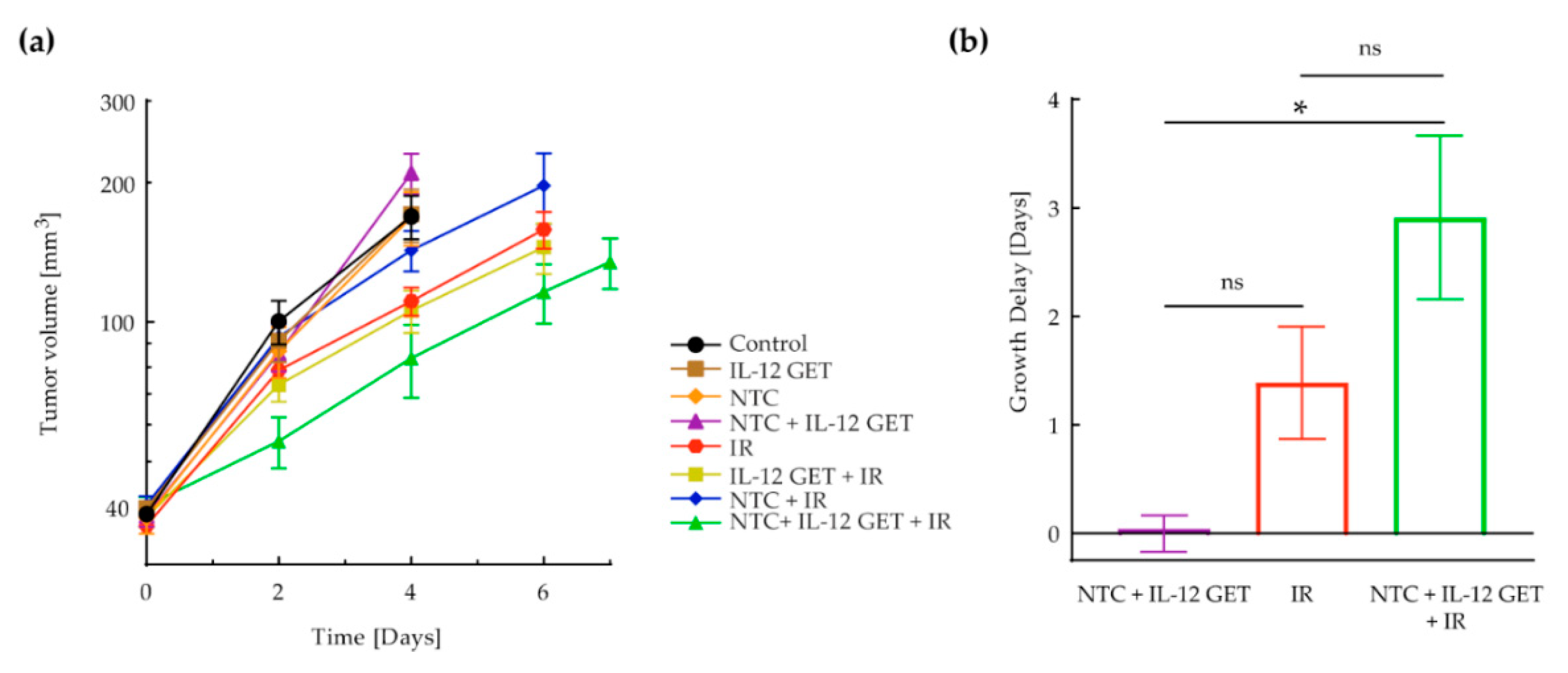
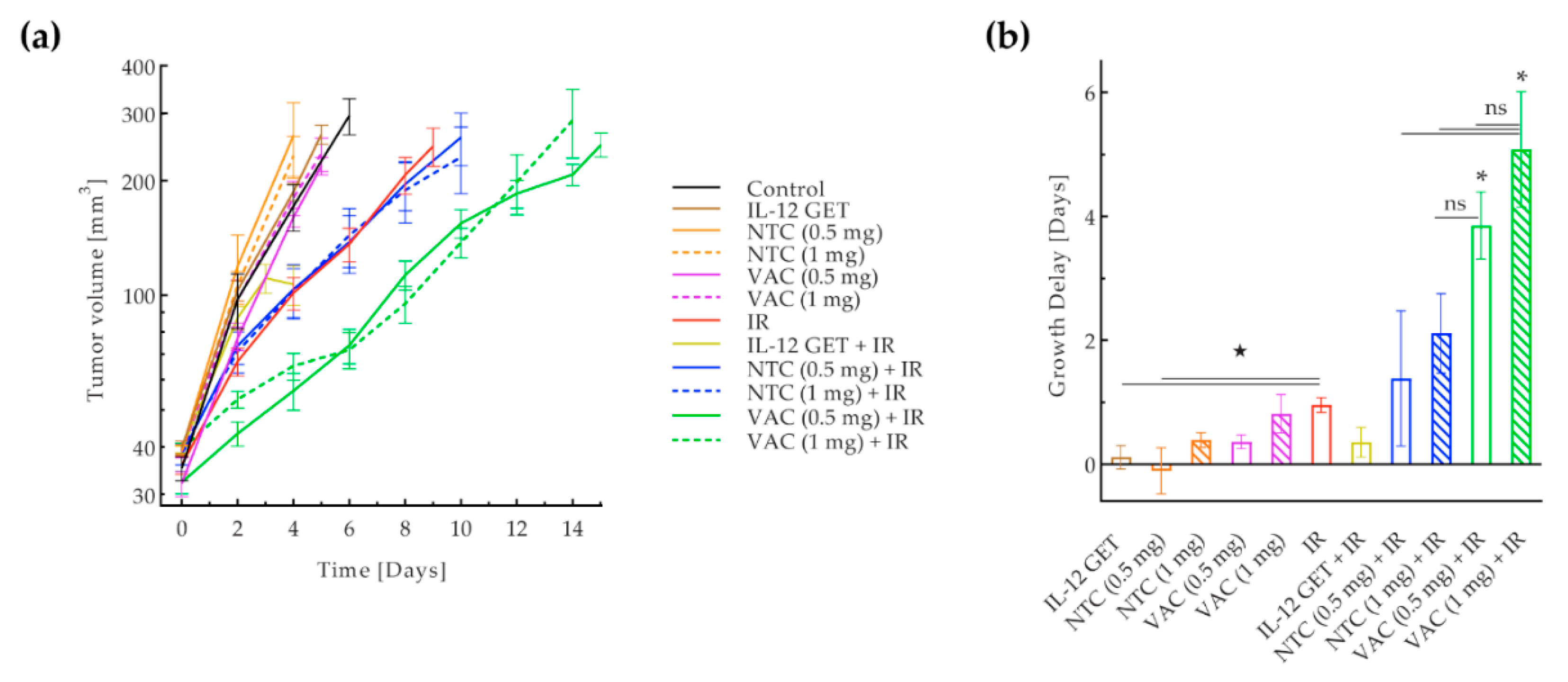
3.1. Pilot B16-F10 Vaccination
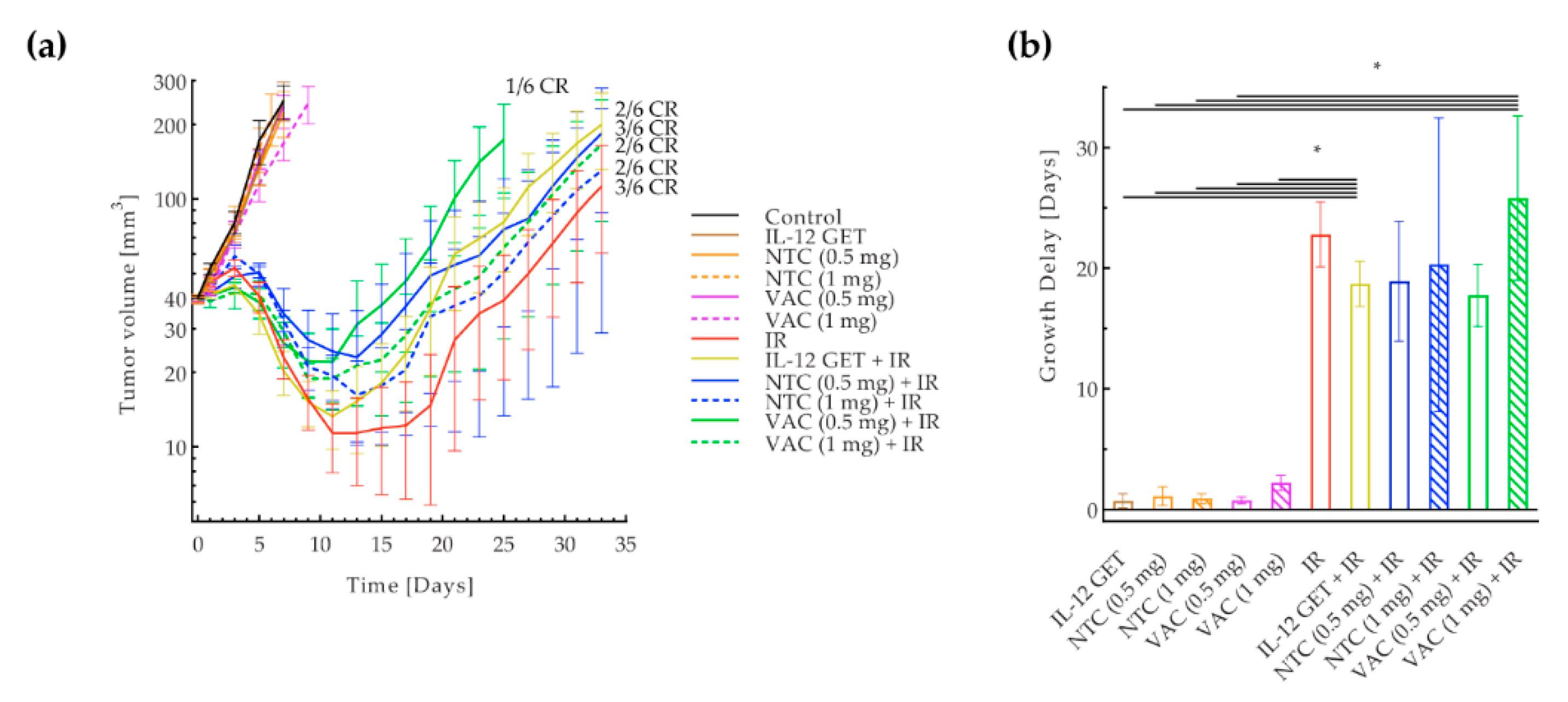
3.2. Adjusted B16-F10 and CT26 Vaccination
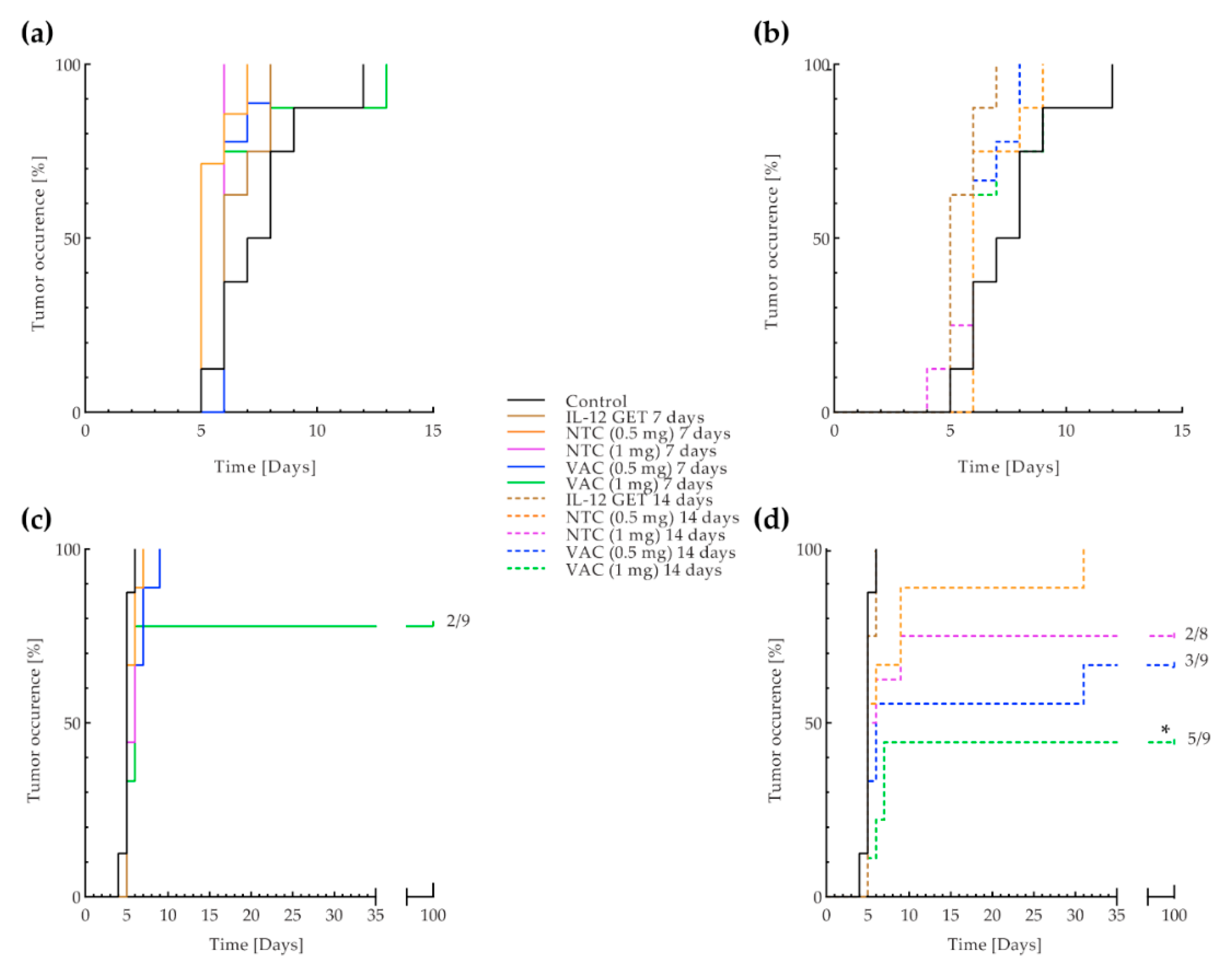
3.3. B16-F10 and CT26 Preventative Vaccination
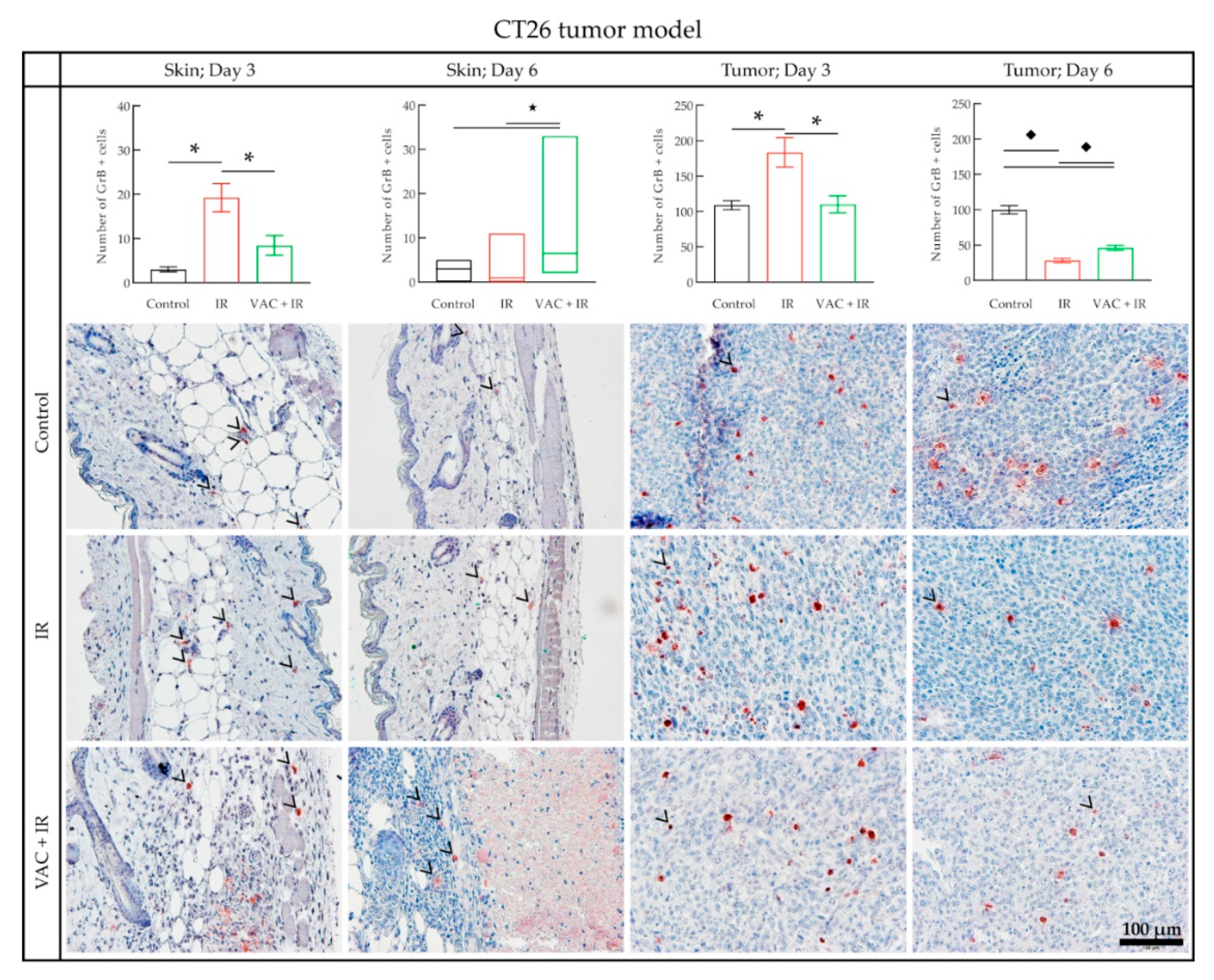
3.4. Histology
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Lai-Lai Chiang, C.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: A Phase 2 Study. J. Gastrointest. Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine ClinicalTrail.Gov. Search of: GVAX-List Results-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=GVAX&cntry=&state=&city=&dist= (accessed on 15 December 2019).
- U.S. National Library of Medicine ClinicalTrail.Gov. Search of: Algenpantucel-L-List Results-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Algenpantucel-L&cntry=&state=&city=&dist= (accessed on 15 December 2019).
- Maier, T.; Tun-Kyi, A.; Tassis, A.; Jungius, K.-P.; Burg, G.; Dummer, R.; Nestle, F.O. Vaccination of patients with cutaneous T-cell lymphoma using intranodal injection of autologous tumor-lysate-pulsed dendritic cells. Blood 2003, 102, 2338–2344. [Google Scholar] [CrossRef]
- Herr, W.; Ranieri, E.; Olson, W.; Zarour, H.; Gesualdo, L.; Storkus, W.J. Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4(+) and CD8(+) T lymphocyte responses. Blood 2000, 96, 1857–1864. [Google Scholar] [CrossRef]
- Weiss, E.M.; Meister, S.; Janko, C.; Ebel, N.; Schlücker, E.; Meyer-Pittroff, R.; Fietkau, R.; Herrmann, M.; Gaipl, U.S.; Frey, B. High hydrostatic pressure treatment generates inactivated mammalian tumor cells with immunogeneic features. J. Immunotoxicol. 2010, 7, 194–204. [Google Scholar] [CrossRef]
- Galili, U.; Chen, Z.C.; DeGeest, K. Expression of α-gal epitopes on ovarian carcinoma membranes to be used as a novel autologous tumor vaccine. Gynecol. Oncol. 2003, 90, 100–108. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Provenge (sipuleucel-T)|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/provenge-sipuleucel-t (accessed on 26 June 2019).
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N.; Czapla, J.; Matuszczak, S.; Szala, S. Combined tumor cell-based vaccination and interleukin-12 gene therapy polarizes the tumor microenvironment in mice. Arch. Immunol. Ther. Exp. 2015, 63, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Young, J.L.; Dean, D.A. Electroporation-mediated gene delivery. In Adv. Genet., 1st ed.; Huang, L., Liu, D., Wagner, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 89, pp. 49–88. [Google Scholar] [CrossRef]
- Forde, P.F.; Hall, L.J.; Sadadcharam, M.; De Kruijf, M.; O’Sullivan, G.C.; Soden, D.M. Development and Characterization of an enhanced nonviral expression vector for electroporation cancer treatment. Mol. Ther. Methods Clin. Dev. 2014, 1, 14012. [Google Scholar] [CrossRef] [PubMed]
- Kos, S.; Blagus, T.; Cemazar, M.; Lampreht Tratar, U.; Stimac, M.; Prosen, L.; Dolinsek, T.; Kamensek, U.; Kranjc, S.; Steinstraesser, L.; et al. Electrotransfer parameters as a tool for controlled and targeted gene expression in skin. Mol. Ther. Nucleic Acids 2016, 5, e356. [Google Scholar] [CrossRef] [PubMed]
- Kos, S.; Vanvarenberg, K.; Dolinsek, T.; Cemazar, M.; Jelenc, J.; Préat, V.; Sersa, G.; Vandermeulen, G. Gene electrotransfer into skin using noninvasive multi-electrode array for vaccination and wound healing. Bioelectrochemistry 2017, 114, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kos, S.; Tesic, N.; Kamensek, U.; Blagus, T.; Cemazar, M.; Kranjc, S.; Lavrencak, J.; Sersa, G. Improved Specificity of Gene Electrotransfer to Skin Using pDNA Under the Control of Collagen Tissue-Specific Promoter. J. Membr. Biol. 2015, 248, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Sersa, G.; Teissie, J.; Cemazar, M.; Signori, E.; Kamensek, U.; Marshall, G.; Miklavcic, D. Electrochemotherapy of tumors as in situ vaccination boosted by immunogene electrotransfer. Cancer Immunol. Immunother. 2015, 64, 1315–1327. [Google Scholar] [CrossRef]
- Salvadori, C.; Svara, T.; Rocchigiani, G.; Millanta, F.; Pavlin, D.; Cemazar, M.; Lampreht Tratar, U.; Sersa, G.; Tozon, N.; Poli, A. Effects of electrochemotherapy with cisplatin and peritumoral IL-12 gene electrotransfer on canine mast cell tumors: A histopathologic and immunohistochemical study. Radiol. Oncol. 2017, 51, 286–294. [Google Scholar] [CrossRef]
- Cemazar, M.; Jarm, T.; Sersa, G. Cancer Electrogene Therapy with Interleukin-12. Curr. Gene. Ther. 2010, 10, 300–311. [Google Scholar] [CrossRef]
- Kamensek, U.; Tesic, N.; Sersa, G.; Cemazar, M. Clinically usable interleukin 12 plasmid without an antibiotic resistance gene: Functionality and toxicity study in murine melanoma model. Cancers 2018, 10, 60. [Google Scholar] [CrossRef]
- Lambricht, L.; Lopes, A.; Kos, S.; Sersa, G.; Préat, V.; Vandermeulen, G. Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert. Opin. Drug Deliv. 2016, 13, 295–310. [Google Scholar] [CrossRef]
- Canton, D.A.; Shirley, S.; Wright, J.; Connolly, R.; Burkart, C.; Mukhopadhyay, A.; Twitty, C.; Qattan, K.E.; Campbell, J.S.; Le, M.H.; et al. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid). Immunotherapy 2017, 9, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine ClinicalTrail.Gov. Search of: IL-12 AND Electroporation-List Results-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=IL-12+AND+electroporation&cntry=&state=&city=&dist=&Search=Search (accessed on 10 December 2019).
- Klein, B.; Loven, D.; Lurie, H.; Rakowsky, E.; Nyska, A.; Levin, I.; Klein, T. The effect of irradiation on expression of HLA class I antigens in human brain tumors in culture. J. Neurosurg. 2009, 80, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Hiserodt, J.C.; Chiriva-Internati, M.; Woodliff, J.; Theus, J.W.; Barclay, D.; Pecorelli, S.; Parham, G.P. Effects of irradiation on the expression of major histocompatibility complex class I antigen and adhesion costimulation molecules ICAM-1 in human cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 1997, 39, 737–742. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Formenti, S.C. Radiation as an immunological adjuvant: Current evidence on dose and fractionation. Front. Oncol. 2012, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Helen Barcellos-Hoff, M.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014, 3, e28518. [Google Scholar] [CrossRef]
- Yu, J.W.; Bhattacharya, S.; Yanamandra, N.; Kilian, D.; Shi, H.; Yadavilli, S.; Katlinskaya, Y.; Kaczynski, H.; Conner, M.; Benson, W.; et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS ONE 2018, 13, e0206223. [Google Scholar] [CrossRef]
- Mosely, S.I.S.; Prime, J.E.; Sainson, R.C.A.; Koopmann, J.O.; Wang, D.Y.Q.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2016, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Lampreht Tratar, U.; Loiacono, L.; Cemazar, M.; Kamensek, U.; Fazio, V.M.; Sersa, G.; Signori, E. Gene electrotransfer of plasmid-encoding IL-12 recruits the M1 macrophages and antigen-presenting cells inducing the eradication of aggressive B16F10 murine melanoma. Mediat. Inflamm. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Savarin, M.; Kamensek, U.; Cemazar, M.; Heller, R.; Sersa, G. Electrotransfer of plasmid DNA radiosensitizes B16F10 tumors through activation of immune response. Radiol. Oncol. 2017, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Tyndall, M.; Kelley, E. Effects of acyclovir combined with other antiviral agents on human cytomegalovirus. Am. J. Med. 1982, 73, 36–39. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Curry, W.T., Jr.; Gorrepati, R.; Piesche, M.; Sasada, T.; Agarwalla, P.; Jones, P.S.; Gerstner, E.R.; Golby, A.J.; Batchelor, T.T.; Wen, P.Y.; et al. Vaccination with irradiated autologous tumor cells mixed with irradiated GM-K562 cells stimulates antitumor immunity and t lymphocyte activation in patients with recurrent malignant glioma. Clin. Cancer Res. 2016, 22, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef]
- Cadena, A.; Cushman, T.R.; Anderson, C.; Barsoumian, H.B.; Welsh, J.W.; Angelica Cortez, M. Radiation and anti-cancer vaccines: A winning combination. Vaccines 2018, 6, 9. [Google Scholar] [CrossRef]
- Barnes, T.A.; Amir, E. Hype or hope: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Solito, S.; Pinton, L.; Damuzzo, V.; Mandruzzato, S. Highlights on molecular mechanisms of MDSC-mediated immune suppression: Paving the way for new working hypotheses. Immunol. Investig. 2012, 41, 722–737. [Google Scholar] [CrossRef]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector t-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.-F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chen, Y.H.; Huang, K.W.; Cheng, H.W.; Chan, S.F.; Tai, K.F.; Hwang, L.H. Combined GM-CSF and IL-12 gene therapy synergistically suppresses the growth of orthotopic liver tumors. Hepatology 2007, 45, 746–754. [Google Scholar] [CrossRef]
- Kishida, T.; Asada, H.; Satoh, E.; Tanaka, S.; Shinya, M.; Hirai, H.; Iwai, M.; Tahara, H.; Imanishi, J.; Mazda, O. In vivo electroporation-mediated transfer of interleukin-12 and interleukin-18 genes induces significant antitumor effects against melanoma in mice. Gene Ther. 2001, 8, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Tanemura, M.; Miyoshi, E.; Eguchi, H.; Nagano, H.; Matsunami, K.; Nagaoka, S.; Yamada, D.; Asaoka, T.; Noda, T.; et al. A practical approach to pancreatic cancer immunotherapy using resected tumor lysate vaccines processed to express α-gal epitopes. PLoS ONE 2017, 12, e0184901. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J. Vaccines in cancer: GVAX®, a GM-CSF gene vaccine. Expert. Rev. Vaccines 2005, 4, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Fong, T.; Robbins, J.M.; Edelman, G.; Edwards, W.; Paulson, R.S.; Bruce, J.; Ognoskie, N.; Wynne, D.; Pike, M.; et al. Phase I trial of interferon-γ (IFN-γ) retroviral vector administered intratumorally to patients with metastatic melanoma. Cancer Gene Ther. 1999, 6, 322–330. [Google Scholar] [CrossRef]
- Soiffer, R.; Lynch, T.; Mihm, M.; Jung, K.; Rhuda, C.; Schmollinger, J.C.; Hodi, F.S.; Liebster, L.; Lam, P.; Mentzer, S.; et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 1998, 95, 13141–13146. [Google Scholar] [CrossRef]
- Timothy Qiu, J.; Alson, D.; Lee, T.H.; Tsai, C.C.; Yu, T.W.; Chen, Y.S.; Lin, C.C.; Schuyler, S.C. Effect of multiple vaccinations with tumor cell-based vaccine with codon-modified GM-CSF on tumor growth in a mouse model. Cancers 2019, 11, 368. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rakhit, A.; Schwartz, L.H.; Olencki, T.; Malone, T.M.; Sandstrom, K.; Nadeau, R.; Parmar, H.; Bukowski, R. Phase I trial of subcutaneous recombinant human interleukin-12 in patients with advanced renal cell carcinoma. Clin. Cancer Res. 1998, 4, 1183–1191. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remic, T.; Sersa, G.; Ursic, K.; Cemazar, M.; Kamensek, U. Development of Tumor Cell-Based Vaccine with IL-12 Gene Electrotransfer as Adjuvant. Vaccines 2020, 8, 111. https://doi.org/10.3390/vaccines8010111
Remic T, Sersa G, Ursic K, Cemazar M, Kamensek U. Development of Tumor Cell-Based Vaccine with IL-12 Gene Electrotransfer as Adjuvant. Vaccines. 2020; 8(1):111. https://doi.org/10.3390/vaccines8010111
Chicago/Turabian StyleRemic, Tinkara, Gregor Sersa, Katja Ursic, Maja Cemazar, and Urska Kamensek. 2020. "Development of Tumor Cell-Based Vaccine with IL-12 Gene Electrotransfer as Adjuvant" Vaccines 8, no. 1: 111. https://doi.org/10.3390/vaccines8010111
APA StyleRemic, T., Sersa, G., Ursic, K., Cemazar, M., & Kamensek, U. (2020). Development of Tumor Cell-Based Vaccine with IL-12 Gene Electrotransfer as Adjuvant. Vaccines, 8(1), 111. https://doi.org/10.3390/vaccines8010111