Intratumoral Gene Electrotransfer of Plasmid DNA Encoding shRNA against Melanoma Cell Adhesion Molecule Radiosensitizes Tumors by Antivascular Effects and Activation of an Immune Response



 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids
2.3. In Vitro Gene Electrotransfer and Irradiation of Cells
2.4. In Vitro Total RNA Extraction and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR) Analysis
2.5. Animals
2.6. In Vivo Gene Electrotransfer and Irradiation of Tumors
2.7. Histology
2.8. Statistical Analysis
3. Results
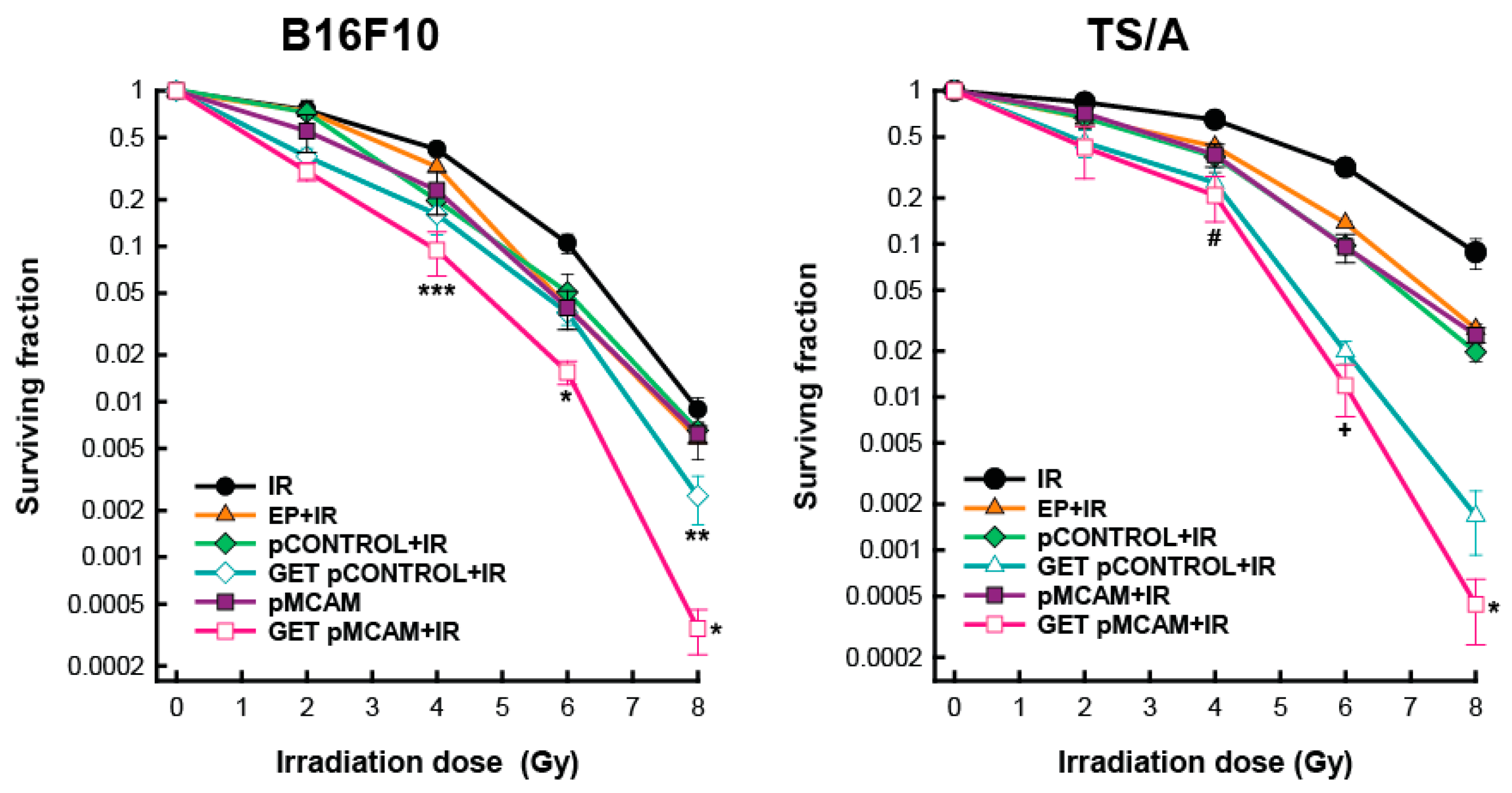
3.1. Radiosensitization of B16F10 Melanoma and TS/A Carcinoma Cells After GET of pMCAM In Vitro
3.2. MCAM Silencing after GET of pMCAM in B16F10 Melanoma and TS/A Carcinoma Cells
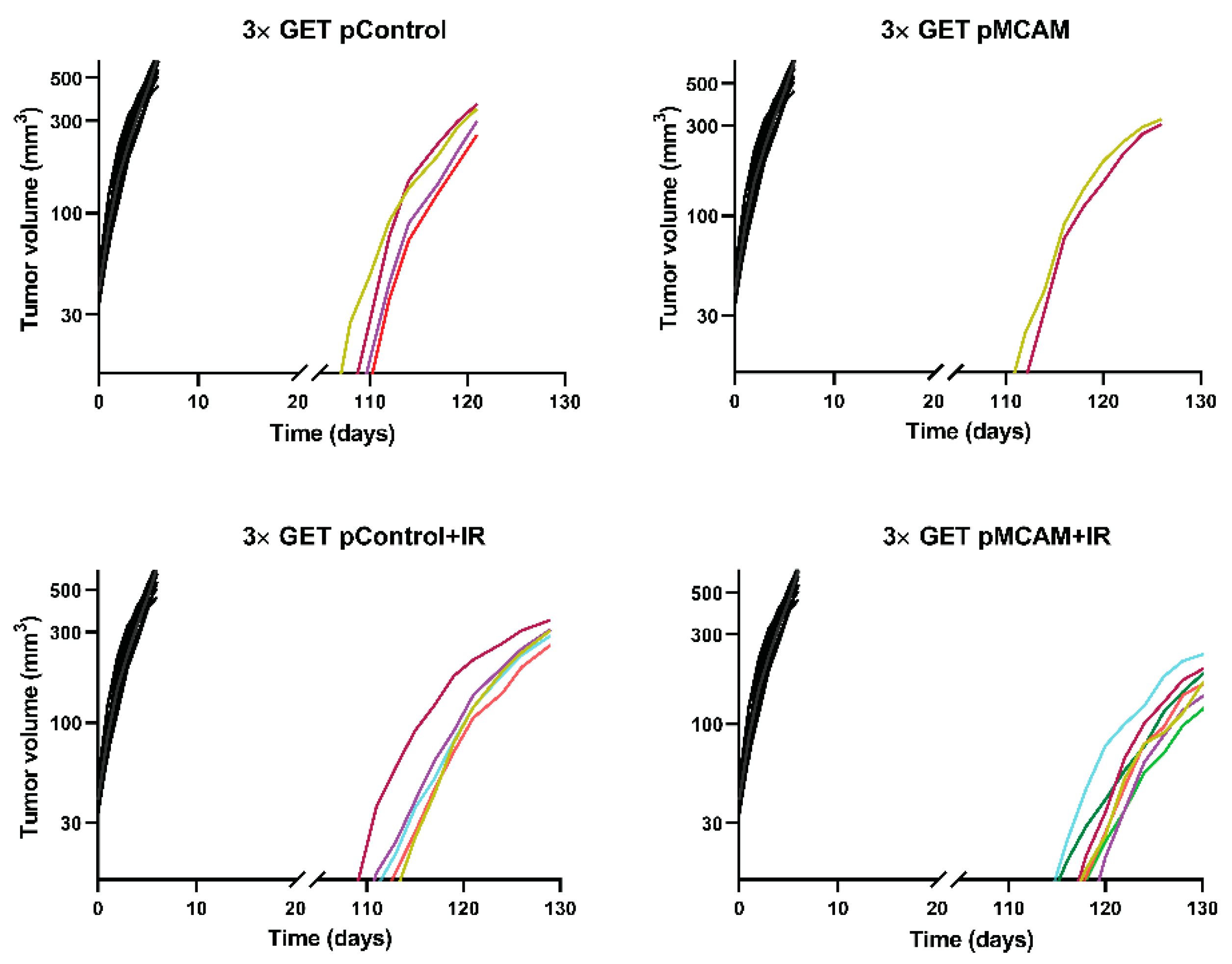
3.3. MCAM Radiosensitization of B16F10 Melanoma and TS/A Carcinoma After GET pMCAM In Vivo
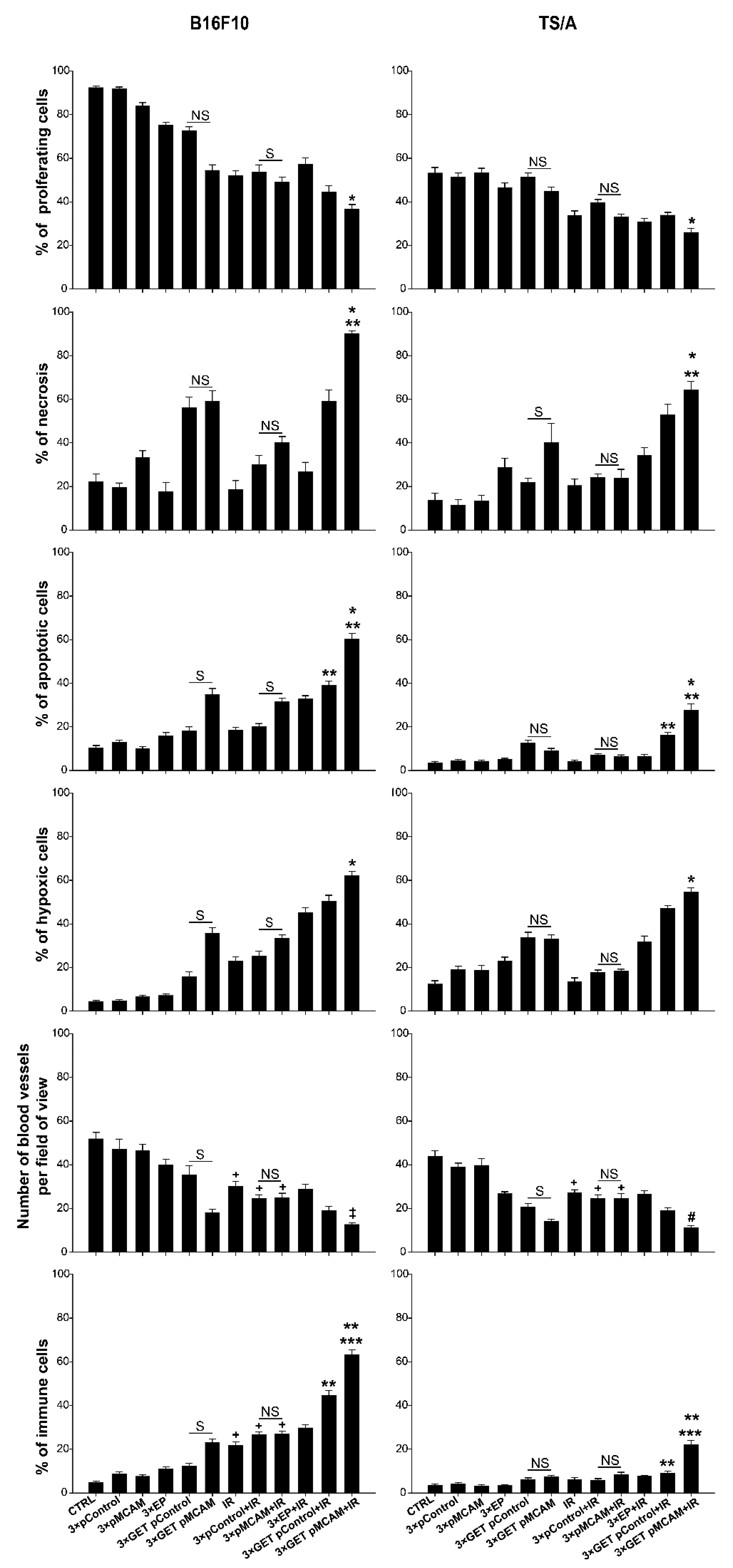
3.4. Histologically Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lehmann, J.M.; Riethmüller, G.; Johnson, J.P. MUC18, a marker of tumor progression in human melanoma, shows sequence similarity to the neural cell adhesion molecules of the immunoglobulin superfamily. Proc. Natl. Acad. Sci. USA 1989, 86, 9891–9895. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Nollet, M.; Ambrosetti, D.; Traboulsi, W.; Viotti, J.; Borchiellini, D.; Grépin, R.; Parola, J.; Giuliano, S.; Helley-Russick, D.; et al. Soluble CD146 is a predictive marker of pejorative evolution and of sunitinib efficacy in clear cell renal cell carcinoma. Theranostics 2018, 8, 2447–2458. [Google Scholar] [CrossRef]
- Nollet, M.; Stalin, J.; Moyon, A.; Traboulsi, W.; Essaadi, A.; Robert, S.; Malissen, N.; Bachelier, R.; Daniel, L.; Foucault-Bertaud, A.; et al. A novel anti-CD146 antibody specifically targets cancer cells by internalizing the molecule. Oncotarget 2017, 8, 112283–112296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xiong, T.; Chen, J.; Li, F.; Qi, T.; Yuan, J. Clinical significance of melanoma cell adhesion molecule CD146 and VEGFA expression in epithelial ovarian cancer. Oncol. Lett. 2019, 17, 2418–2424. [Google Scholar] [CrossRef]
- de Kruijff, I.E.; Timmermans, A.M.; den Bakker, M.A.; MAC Trapman-Jansen, A.; Foekens, R.; Meijer-Van Gelder, M.E.; Oomen-de Hoop, E.; Smid, M.; Hollestelle, A.; van Deurzen, C.H.; et al. The Prevalence of CD146 Expression in Breast Cancer Subtypes and Its Relation to Outcome. Cancers 2018, 10, 134. [Google Scholar] [CrossRef]
- Stalin, J.; Nollet, M.; Garigue, P.; Fernandez, S.; Vivancos, L.; Essaadi, A.; Muller, A.; Bachelier, R.; Foucault-Bertaud, A.; Fugazza, L.; et al. Targeting soluble CD146 with a neutralizing antibody inhibits vascularization, growth and survival of CD146-positive tumors. Oncogene 2016, 35, 5489–5500. [Google Scholar] [CrossRef] [PubMed]
- Bidlingmaier, S.; He, J.; Wang, Y.; An, F.; Feng, J.; Barbone, D.; Gao, D.; Franc, B.; Broaddus, V.C.; Liu, B. Identification of MCAM/CD146 as the target antigen of a human monoclonal antibody that recognizes both epithelioid and sarcomatoid types of mesothelioma. Cancer Res. 2009, 69, 1570–1577. [Google Scholar] [CrossRef][Green Version]
- Nodomi, S.; Umeda, K.; Saida, S.; Kinehara, T.; Hamabata, T.; Daifu, T.; Kato, I.; Hiramatsu, H.; Watanabe, K.-I.; Kuwahara, Y.; et al. CD146 is a novel marker for highly tumorigenic cells and a potential therapeutic target in malignant rhabdoid tumor. Nat. Publ. Gr. 2016, 35, 5317–5327. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Li, W.; Lu, D.; Wu, Z.; Duan, H.; Luo, Y.; Feng, J.; Yang, D.; Fu, L.; Yan, X. CD146, an epithelial-mesenchymal transition inducer, is associated with triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 1127–1132. [Google Scholar] [CrossRef]
- Bardin, N.; Anfosso, F.; Massé, J.M.; Cramer, E.; Sabatier, F.; Bivic, A.L.; Sampol, J.; Dignat-George, F. Identification of CD146 as a component of the endothelial junction involved in the control of cell-cell cohesion. Blood 2001, 98, 3677–3684. [Google Scholar] [CrossRef]
- Espagnolle, N.; Guilloton, F.; Deschaseaux, F.; Gadelorge, M.; Sensébé, L.; Bourin, P. CD146 expression on mesenchymal stem cells is associated with their vascular smooth muscle commitment. J. Cell. Mol. Med. 2014, 18, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.L.; Shepherd, B.R.; Yi, T.; Pober, J.S. Explant outgrowth, propagation and characterization of human pericytes. Microcirculation 2010, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Liu, F.L.; Sytwu, H.K.; Tsai, C.Y.; Chang, D.M. CD146+ mesenchymal stem cells display greater therapeutic potential than CD146- cells for treating collagen-induced arthritis in mice. Stem Cell Res. Ther. 2016, 7, 23. [Google Scholar] [CrossRef]
- Olajuyin, A.M.; Olajuyin, A.K.; Wang, Z.; Zhao, X.; Zhang, X. CD146 T cells in lung cancer: Its function, detection, and clinical implications as a biomarker and therapeutic target. Cancer Cell Int. 2019, 19, 247. [Google Scholar] [CrossRef] [PubMed]
- Prosen, L.; Markelc, B.; Dolinsek, T.; Music, B.; Cemazar, M.; Sersa, G. Mcam silencing with RNA interference using magnetofection has antitumor effect in murine melanoma. Mol. Ther. Nucleic Acids 2014, 3, e205. [Google Scholar] [CrossRef]
- Kang, Y.; Wang, F.; Feng, J.; Yang, D.; Yang, X.; Yan, X. Knockdown of CD146 reduces the migration and proliferation of human endothelial cells. Cell Res. 2006, 16, 313–318. [Google Scholar] [CrossRef]
- Todorovic, V.; Sersa, G.; Cemazar, M. Gene electrotransfer of siRNAs against CD146 inhibits migration and invasion of human malignant melanoma cells SK-MEL28. Cancer Gene Ther. 2013, 20, 208–210. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, Z.; Li, J.; Yang, X.; Wang, Y.; Yu, Y.; Ye, J.; Xu, C.; Qin, W.; Zhang, Z. MCAM is a novel metastasis marker and regulates spreading, apoptosis and invasion of ovarian cancer cells. Tumor Biol. 2012, 33, 1619–1628. [Google Scholar] [CrossRef]
- Zabouo, G.; Imbert, A.M.; Jacquemier, J.; Finetti, P.; Moreau, T.; Esterni, B.; Birnbaum, D.; Bertucci, F.; Chabannon, C. CD146 expression is associated with a poor prognosis in human breast tumors and with enhanced motility in breast cancer cell lines. Breast Cancer Res. 2009, 11, R1. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, H.L.; Jiang, Y.G.; Li, J.H.; Liu, B.L.; Sun, M.Y. Inhibition of CD146 gene expression via RNA interference reduces in vitro perineural invasion on ACC-M cell. J. Oral Pathol. Med. 2009, 38, 198–205. [Google Scholar] [CrossRef]
- Mills, L.; Tellez, C.; Huang, S.; Baker, C.; McCarty, M.; Green, L.; Gudas, J.M.; Feng, X.; Bar-Eli, M. Fully human antibodies to MCAM/MUC18 inhibit tumor growth and metastasis of human melanoma. Cancer Res. 2002, 62, 5106–5114. [Google Scholar] [PubMed]
- McGary, E.C.; Heimberger, A.; Mills, L.; Weber, K.; Thomas, G.W.; Shtivelband, M.; Lev, D.C.; Bar-Eli, M. A Fully Human Antimelanoma Cellular Adhesion Molecule/MUC18 Antibody Inhibits Spontaneous Pulmonary Metastasis of Osteosarcoma Cells in Vivo. Clin. Cancer Res. 2003, 9, 6560–6566. [Google Scholar] [PubMed]
- Yan, X.; Lin, Y.; Yang, D.; Shen, Y.; Yuan, M.; Zhang, Z.; Li, P.; Xia, H.; Li, L.; Luo, D.; et al. A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth. Blood 2003, 102, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Gao, L.; Zhuang, J.; Feng, J.; Yang, D.; Yan, X. Anti-CD146 monoclonal antibody AA98 inhibits angiogenesis via suppression of nuclear factor-κB activation. Mol. Cancer Ther. 2006, 5, 2872–2878. [Google Scholar] [CrossRef] [PubMed]
- Prosen, L.; Hudoklin, S.; Cemazar, M.; Stimac, M.; Lampreht Tratar, U.; Ota, M.; Scancar, J.; Romih, R.; Sersa, G. Magnetic field contributes to the cellular uptake for effective therapy with magnetofection using plasmid DNA encoding against Mcam in B16F10 melanoma in vivo. Nanomedicine 2016, 11, 627–641. [Google Scholar] [CrossRef]
- Savarin, M.; Kamensek, U.; Cemazar, M.; Heller, R.; Sersa, G. Electrotransfer of plasmid DNA radiosensitizes B16F10 tumors through activation of immune response. Radiol. Oncol. 2017, 51, 30–39. [Google Scholar] [CrossRef]
- Bosnjak, M.; Jesenko, T.; Kamensek, U.; Sersa, G.; Lavrencak, J.; Heller, L.; Cemazar, M. Electrotransfer of different control plasmids elicits different antitumor effectiveness in B16.F10 melanoma. Cancers 2018, 10, 37. [Google Scholar] [CrossRef]
- Znidar, K.; Bosnjak, M.; Semenova, N.; Pakhomova, O.; Heller, L.; Cemazar, M. Tumor cell death after electrotransfer of plasmid DNA is associated with cytosolic DNA sensor upregulation. Oncotarget 2018, 9, 18665–18681. [Google Scholar] [CrossRef]
- Semenova, N.; Bosnjak, M.; Markelc, B.; Znidar, K.; Cemazar, M.; Heller, L. Multiple cytosolic DNA sensors bind plasmid DNA after transfection. Nucleic Acids Res. 2019, 47, 10235–10246. [Google Scholar] [CrossRef]
- Nanni, P.; De Giovanni, C.; Lollini, P.L.; Nicoletti, G.; Prodi, G. TS/A: A new metastasizing cell line from a BALB/c spontaneous mammary adenocarcinoma. Clin. Exp. Metastasis 1983, 1, 373–380. [Google Scholar] [CrossRef]
- De Giovanni, C.; Nicoletti, G.; Landuzzi, L.; Palladini, A.; Lollini, P.L.; Nanni, P. Bioprofiling TS/A murine mammary cancer for a functional precision experimental model. Cancers 2019, 11, 1889. [Google Scholar] [CrossRef] [PubMed]
- Mosely, S.I.S.; Prime, J.E.; Sainson, R.C.A.; Koopmann, J.O.; Wang, D.Y.Q.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Douin-Echinard, V.; Bornes, S.; Rochaix, P.; Tilkin, A.F.; Peron, J.M.; Bonnet, J.; Favre, G.; Couderc, B. The expression of CD70 and CD80 by gene-modified tumor cells induces an antitumor response depending on the MHC status. Cancer Gene Ther 2000, 7, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.; Dalla Santa, S.; Zoso, A.; Giacomelli, S.; Milan, G.; Macino, B.; Tosello, V.; Dellabona, P.; Lollini, P.L.; De Giovanni, C.; et al. The cytotoxic T-lymphocyte response against a poorly immunogenic mammary adenocarcinoma is focused on a single immunodominant class I epitope derived from the gp70 Env product of an endogenous retrovirus. Cancer Res. 2003, 63, 2158–2163. [Google Scholar] [PubMed]
- Falcioni, R.; Sacchi, A.; Giacomini, P.; Zupi, G.; Kennel, S.J. Expression of tumor antigen correlated with metastatic potential of lewis lung carcinoma and b16 melanoma clones in mice. Cancer Res. 1986, 46, 5772–5778. [Google Scholar] [PubMed]
- Stimac, M.; Kamensek, U.; Cemazar, M.; Kranjc, S.; Coer, A.; Sersa, G. Tumor radiosensitization by gene therapy against endoglin. Cancer Gene Ther. 2016, 23, 214–220. [Google Scholar] [CrossRef]
- Sedlar, A.; Kranjc, S.; Dolinsek, T.; Cemazar, M.; Coer, A.; Sersa, G. Radiosensitizing effect of intratumoral interleukin-12 gene electrotransfer in murine sarcoma. BMC Cancer 2013, 13, 38. [Google Scholar] [CrossRef]
- Kamensek, U.; Cemazar, M.; Lampreht Tratar, U.; Ursic, K.; Sersa, G. Antitumor in situ vaccination effect of TNFα and IL-12 plasmid DNA electrotransfer in a murine melanoma model. Cancer Immunol. Immunother. 2018, 67, 785–795. [Google Scholar] [CrossRef]
- Zoni, E.; Astrologo, L.; Ng, C.K.Y.; Piscuoglio, S.; Melsen, J.; Grosjean, J.; Klima, I.; Chen, L.; Snaar-Jagalska, E.B.; Flanagan, K.; et al. Therapeutic targeting of CD146/MCAM reduces bone metastasis in prostate cancer. Mol. Cancer Res. 2019, 17, 1049–1062. [Google Scholar] [CrossRef]
- Grosel, A.; Sersa, G.; Kranjc, S.; Cemazar, M. Electrogene therapy with p53 of murine sarcomas alone or combined with electrochemotherapy using cisplatin. DNA Cell Biol. 2006, 25, 674–683. [Google Scholar] [CrossRef]
- Marrero, B.; Shirley, S.; Heller, R. Delivery of interleukin-15 to B16 melanoma by electroporation leads to tumor regression and long-term survival. Technol. Cancer Res. Treat. 2014, 13, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.L.; Heller, L.; Coppola, D.; Heller, R. IL-12 plasmid delivery by in Vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol. Ther. 2002, 5, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.; Lundberg, C.; Li, F.; Burcus, N.; Heller, R. Controlled Gene Delivery Can Enhance Therapeutic Outcome for Cancer Immune Therapy for Melanoma. Curr. Gene Ther. 2014, 15, 32–43. [Google Scholar] [CrossRef] [PubMed]
- McCray, A.N.; Ugen, K.E.; Muthumani, K.; Kim, J.J.; Weiner, D.B.; Heller, R. Complete Regression of Established Subcutaneous B16 Murine Melanoma Tumors after Delivery of an HIV-1 Vpr-Expressing Plasmid by in Vivo Electroporation. Mol. Ther. 2006, 14, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Deharvengt, S.; Rejiba, S.; Wack, S.; Aprahamian, M.; Hajri, A. Efficient electrogene therapy for pancreatic adenocarcinoma treatment using the bacterial purine nucleoside phosphorylase suicide gene with fludarabine. Int. J. Oncol. 2007, 30, 1397–1406. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.S.; Tsang, Y.W.; Chi, C.H.; Chang, C.C.; Chu, R.M.; Chi, K.H. Synergistic anti-tumor effect of combination radio- and immunotherapy by electro-gene therapy plus intra-tumor injection of dendritic cells. Cancer Lett. 2008, 266, 275–285. [Google Scholar] [CrossRef]
- Radkevich-Brown, O.; Piechocki, M.P.; Back, J.B.; Weise, A.M.; Pilon-Thomas, S.; Wei, W.Z. Intratumoral DNA electroporation induces anti-tumor immunity and tumor regression. Cancer Immunol. Immunother. 2010, 59, 409–417. [Google Scholar] [CrossRef]
- Znidar, K.; Bosnjak, M.; Cemazar, M.; Heller, L.C. Cytosolic DNA Sensor Upregulation Accompanies DNA Electrotransfer in B16.F10 Melanoma Cells. Mol. Ther. Nucleic Acids 2016, 5, e322. [Google Scholar] [CrossRef]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479, 146–152. [Google Scholar] [CrossRef]
- Heller, L.; Coppola, D. Electrically mediated delivery of vector plasmid DNA elicits an antitumor effect. Gene Ther. 2002, 9, 1321–1325. [Google Scholar] [CrossRef]
- Shi, G.; Edelblute, C.; Arpag, S.; Lundberg, C.; Heller, R. IL-12 gene electrotransfer triggers a change in immune response within mouse tumors. Cancers 2018, 10, 498. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.; Babiuk, S.; Babiuk, L.A.; Griebel, P. A single DNA immunization in combination with electroporation prolongs the primary immune response and maintains immune memory for six months. Vaccine 2007, 25, 5485–5494. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, P.; De Santis, S.; Fazio, V.M.; Signori, E. Hyaluronidase contributes to early inflammatory events induced by electrotransfer in mouse skeletal muscle. Hum. Gene Ther. 2013, 24, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Babiuk, S.; Baca-Estrada, M.E.; Foldvari, M.; Middleton, D.M.; Rabussay, D.; Widera, G.; Babiuk, L.A. Increased gene expression and inflammatory cell infiltration caused by electroporation are both important for improving the efficacy of DNA vaccines. J. Biotechnol. 2004, 110, 1–10. [Google Scholar] [CrossRef]
- Lampreht Tratar, U.; Loiacono, L.; Cemazar, M.; Kamensek, U.; Fazio, V.M.; Sersa, G.; Signori, E. Gene Electrotransfer of Plasmid-Encoding IL-12 Recruits the M1 Macrophages and Antigen-Presenting Cells Inducing the Eradication of Aggressive B16F10 Murine Melanoma. Mediat. Inflamm. 2017, 2017, 5285890. [Google Scholar] [CrossRef]
- Markelc, B.; Tevz, G.; Cemazar, M.; Kranjc, S.; Lavrencak, J.; Zegura, B.; Teissie, J.; Sersa, G. Muscle gene electrotransfer is increased by the antioxidant tempol in mice. Gene Ther. 2012, 19, 312–320. [Google Scholar] [CrossRef]
- Bonnafous, P.; Vernhes, M.C.; Teissié, J.; Gabriel, B. The generation of reactive-oxygen species associated with long-lasting pulse-induced electropermeabilisation of mammalian cells is based on a non-destructive alteration of the plasma membrane. Biochim. Biophys. Acta Biomembr. 1999, 1461, 123–134. [Google Scholar] [CrossRef]
- Muralidharan, S.; Mandrekar, P. Cellular stress response and innate immune signaling: Integrating pathways in host defense and inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8 + T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Crokart, N.; Danhier, F.; Daugimont, L.; Gonçalves, N.; Jordan, B.F.; Grégoire, V.; Feron, O.; Bouquet, C.; Gallez, B.; Préat, V. Potentiation of radiotherapy by a localized antiangiogenic gene therapy. Radiother. Oncol. 2013, 107, 252–258. [Google Scholar] [CrossRef]
- Maio, M. Melanoma as a model tumour for immuno-oncology. Proc. Ann. Oncol. 2012, 23 (Suppl. 8), viii10–viii14. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | DT (Days) AM ± SE | GD (Days) AM ± SE | CR | SC (Secondary Challenge) | ||
|---|---|---|---|---|---|---|---|
| n | % | n | % | ||||
| Control | 18 | 1.2 ± 0.1 | 0.0 ± 0.1 | 0 | 0 | 0 | 0 |
| 3× EP | 12 | 3.3 ± 0.3 | 2.1 ± 0.3 | 0 | 0 | 0 | 0 |
| 3× pControl | 12 | 2.1 ± 0.2 | 0.9 ± 0.2 | 0 | 0 | 0 | 0 |
| 3× pMCAM | 12 | 2.1 ± 0.3 | 0.9 ± 0.3 | 0 | 0 | 0 | 0 |
| IR | 12 | 1.7 ± 0.2 | 0.5 ± 0.2 | 0 | 0 | 0 | 0 |
| 3× GET pControl | 16 | 11.3 ± 2.3 | 10.1 ± 2.3* | 5 | 31.2 | 1 | 20 |
| 3× GET pMCAM | 14 | 14.4 ± 2.5 | 13.2 ± 2.5* | 5 | 35.7 | 3 | 60 |
| 3× EP + IR | 17 | 6.1 ± 1.0 | 4.9 ± 1.0 | 2 | 11.8 | 0 | 0 |
| pControl + IR | 16 | 2.9 ± 0.5 | 1.7 ± 0.5 | 1 | 6.3 | 1 | 100 |
| pMCAM + IR | 16 | 6.8 ± 1.9 | 5.6 ± 1.9 | 2 | 12.5 | 2 | 100 |
| 3× GET pControl + IR | 18 | 31.0 ± 5.9 | 29.8 ± 5.9* | 13 | 72.2 | 7 | 53.8 |
| 3× GET pMCAM + IR | 21 | 19.7 ± 4.3 | 18.5 ± 4.3* | 17 | 81.0 | 8 | 47.1 |
| Group | n | DT (Days) AM ± SE | GD (Days) AM ± SE | CR | SC (Secondary Challenge) | ||
|---|---|---|---|---|---|---|---|
| n | % | n | % | ||||
| Control | 16 | 2.2 ± 0.2 | 0.0 | 0 | 0 | 0 | 0 |
| 3× EP | 11 | 2.8 ± 0.2 | 0.6 ± 0.2 | 0 | 0 | 0 | 0 |
| 3× pControl | 11 | 2.9 ± 0.7 | 0.7 ± 0.7 | 0 | 0 | 0 | 0 |
| 3× pMCAM | 9 | 2.1 ± 0.2 | −0.1 ± 0.2 | 0 | 0 | 0 | 0 |
| IR | 16 | 3.0 ± 0.3 | 0.8 ± 0.3 | 0 | 0 | 0 | 0 |
| 3× GET pControl | 14 | 6.3 ± 0.9 | 4.1 ± 0.9 | 0 | 0 | 0 | 0 |
| 3× GET pMCAM | 13 | 11.1 ± 0.8 | 8.9 ± 0.8 | 0 | 0 | 0 | 0 |
| 3× EP+IR | 16 | 13.9 ± 2.8 | 11.7 ± 2.8* | 3 | 18.8 | 0 | 0 |
| pControl+IR | 15 | 2.8 ± 0.2 | 0.6 ± 0.2 | 0 | 0 | 0 | 0 |
| pMCAM+IR | 16 | 3.6 ± 1.4 | 1.4 ± 1.4 | 0 | 0 | 0 | 0 |
| 3× GET pControl+IR | 13 | 24.4 ± 3.7 | 22.2 ± 3.7** | 1 | 7.7 | 0 | 0 |
| 3× GET pMCAM+IR | 15 | 30.9 ± 2.9 | 28.7 ± 2.9** | 4 | 26.7 | 0 | 0 |
| Group | % of Proliferative Cells (AM ± SE) | % of Necrosis (AM ± SE) | %Apoptosis (AM ± SE) | % of Hypoxic Cells (AM ± SE) | Number of Blood Vessels/Field of View (AM ± SE) | % of Immune Cells (AM ± SE) |
|---|---|---|---|---|---|---|
| Control | 92.3 ± 0.8 | 22.2 ± 3.6 | 10.4 ± 1.1 | 4.5 ± 0.5 | 51.8 ± 3.0 | 6.8 ± 0.6 |
| 3× pControl | 92.2 ± 0.8 | 19.5 ± 2.1 | 12.9 ± 0.9 | 5.3 ± 07 | 47.1 ± 4.7 | 9.0 ± 0.8 |
| 3× pMCAM | 83.9 ± 1.6 | 33.3 ± 3.1 | 9.8 ± 0.9 | 6.4 ± 0.5 | 46.5 ± 2.9 | 7.9 ± 0.7 |
| 3× EP | 75.6 ± 1.2 | 17.5 ± 4.2 | 17.2 ± 1.6 | 7.5 ± 0.5 | 39.9 ± 2.6 | 11.0 ± 1.1 |
| 3× GETpControl | 72.7 ± 1.7 | 56.2 ± 4.9 | 18.6 ± 1.9 | 15.6 ± 2.0 | 35.3 ± 4.2 | 13.6 ± 1.2 |
| 3× GET pMCAM | 54.4 ± 2.6 | 59.2 ± 4.9 | 34.7 ± 2.9 | 35.4 ± 2.6 | 18.1 ± 1.7 | 23.5 ± 1.9 |
| IR | 51.8 ± 2.3 | 18.7 ± 4.0 | 18.4 ± 1.3 | 22.9 ± 2.0 | 30.1 ± 2.3 | 23.6 ± 1.7 |
| 3× pControl+IR | 53.8 ± 3.2 | 30.0 ± 4.3 | 20.2 ± 1.4 | 25.3 ± 2.1 | 24.5 ± 1.8 | 26.5 ± 1.3 |
| 3× pMCAM+IR | 49.2 ± 2.3 | 40.0 ± 2.9 | 31.6 ± 1.5 | 33.9 ± 1.7 | 25.0 ± 2.0 | 27.0 ± 1.3 |
| 3× EP+IR | 57.3 ± 2.9 | 26.7 ± 4.4 | 32.9 ± 1.4 | 45.1 ± 2.2 | 28.9 ± 2.3 | 29.7 ± 1.6 |
| 3× GET pControl+IR | 44.3 ± 3.1 | 59.2 ± 5.2 | 39.0 ± 1.9 | 50.2 ± 2.9 | 18.9 ± 2.1 | 44.5 ± 2.3 |
| 3× GET pMCAM+IR | 36.7 ± 2.1* | 90.0 ± 1.3* | 60.1 ± 2.9* | 62.0 ± 2.0* | 12.6 ± 0.9* | 63.4 ± 2.5* |
| Group | % of Proliferative Cells (AM ± SE) | % of Necrosis (AM ± SE) | % Apoptosis (AM ± SE) | % of Hypoxic Cells (AM ± SE) | Number of Blood Vessels/Field of View (AM ± SE) | % of Immune Cells (AM ± SE) |
|---|---|---|---|---|---|---|
| Control | 53.1 ± 2.7 | 11.2 ± 2.9 | 3.5 ± 0.7 | 12.3 ± 1.7 | 43.9 ± 2.5 | 3.4 ± 0.6 |
| 3× pControl | 51.4 ± 1.8 | 11.5 ± 2.5 | 4.4 ± 0.6 | 18.8 ± 1.7 | 38.9 ± 1.8 | 4.0 ± 0.7 |
| 3× pMCAM | 53.4 ± 2.1 | 13.3 ± 2.5 | 4.2 ± 0.5 | 18.6 ± 2.3 | 39.7 ± 3.2 | 3.2 ± 0.6 |
| 3× EP | 46.6 ± 2.1 | 28.8 ± 4.3 | 5.0 ± 0.6 | 22.9 ± 1.8 | 26.7 ± 1.0 | 3.3 ± 0.6 |
| 3× GETpControl | 51.2 ± 2.0 | 21.7 ± 2.1 | 12.6 ± 1.2 | 33.5 ± 2.6 | 20.5 ± 1.7 | 6.1 ± 0.8 |
| 3× GET pMCAM | 44.9 ± 1.9 | 40.0 ± 8.9 | 9.1 ± 1.1 | 33.1 ± 1.7 | 13.9 ± 1.1 | 7.3 ± 0.8 |
| IR | 33.6 ± 2.2 | 20.3 ± 3.0 | 4.2 ± 0.6 | 13.4 ± 1.8 | 27.0 ± 1.6 | 6.1 ± 0.9 |
| 3× pControl+IR | 39.5 ± 1.6 | 24.2 ± 1.5 | 6.9 ± 0.9 | 17.6 ± 1.2 | 24.7 ± 1.6 | 5.7 ± 0.8 |
| 3× pMCAM+IR | 32.9 ± 1.4 | 23.8 ± 4.0 | 6.4 ± 0.7 | 18.3 ± 0.9 | 24.6 ± 2.3 | 8.4 ± 1.1 |
| 3× EP+IR | 30.8 ± 1.5 | 34.2 ± 3.7 | 6.5 ± 0.8 | 31.8 ± 2.7 | 26.5 ± 1.6 | 7.6 ± 0.6 |
| 3× GET pControl+IR | 33.8 ± 1.3 | 53.0 ± 4.8 | 16.1 ± 1.4 | 47.0 ± 1.5 | 19.1 ± 1.2 | 8.8 ± 1.0 |
| 3× GET pMCAM+IR | 25.7 ± 2.1* | 64.2 ± 4.0** | 27.6 ± 2.9** | 54.4 ± 2.1** | 11.0 ± 1.2** | 21.9 ± 2.1** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kranjc Brezar, S.; Mrak, V.; Bosnjak, M.; Savarin, M.; Sersa, G.; Cemazar, M. Intratumoral Gene Electrotransfer of Plasmid DNA Encoding shRNA against Melanoma Cell Adhesion Molecule Radiosensitizes Tumors by Antivascular Effects and Activation of an Immune Response. Vaccines 2020, 8, 135. https://doi.org/10.3390/vaccines8010135
Kranjc Brezar S, Mrak V, Bosnjak M, Savarin M, Sersa G, Cemazar M. Intratumoral Gene Electrotransfer of Plasmid DNA Encoding shRNA against Melanoma Cell Adhesion Molecule Radiosensitizes Tumors by Antivascular Effects and Activation of an Immune Response. Vaccines. 2020; 8(1):135. https://doi.org/10.3390/vaccines8010135
Chicago/Turabian StyleKranjc Brezar, Simona, Valter Mrak, Masa Bosnjak, Monika Savarin, Gregor Sersa, and Maja Cemazar. 2020. "Intratumoral Gene Electrotransfer of Plasmid DNA Encoding shRNA against Melanoma Cell Adhesion Molecule Radiosensitizes Tumors by Antivascular Effects and Activation of an Immune Response" Vaccines 8, no. 1: 135. https://doi.org/10.3390/vaccines8010135
APA StyleKranjc Brezar, S., Mrak, V., Bosnjak, M., Savarin, M., Sersa, G., & Cemazar, M. (2020). Intratumoral Gene Electrotransfer of Plasmid DNA Encoding shRNA against Melanoma Cell Adhesion Molecule Radiosensitizes Tumors by Antivascular Effects and Activation of an Immune Response. Vaccines, 8(1), 135. https://doi.org/10.3390/vaccines8010135