Differential Immune Responses to Hemorrhagic Fever-Causing Arenaviruses

{kind=link}
{kind=link}
Abstract
:1. Introduction
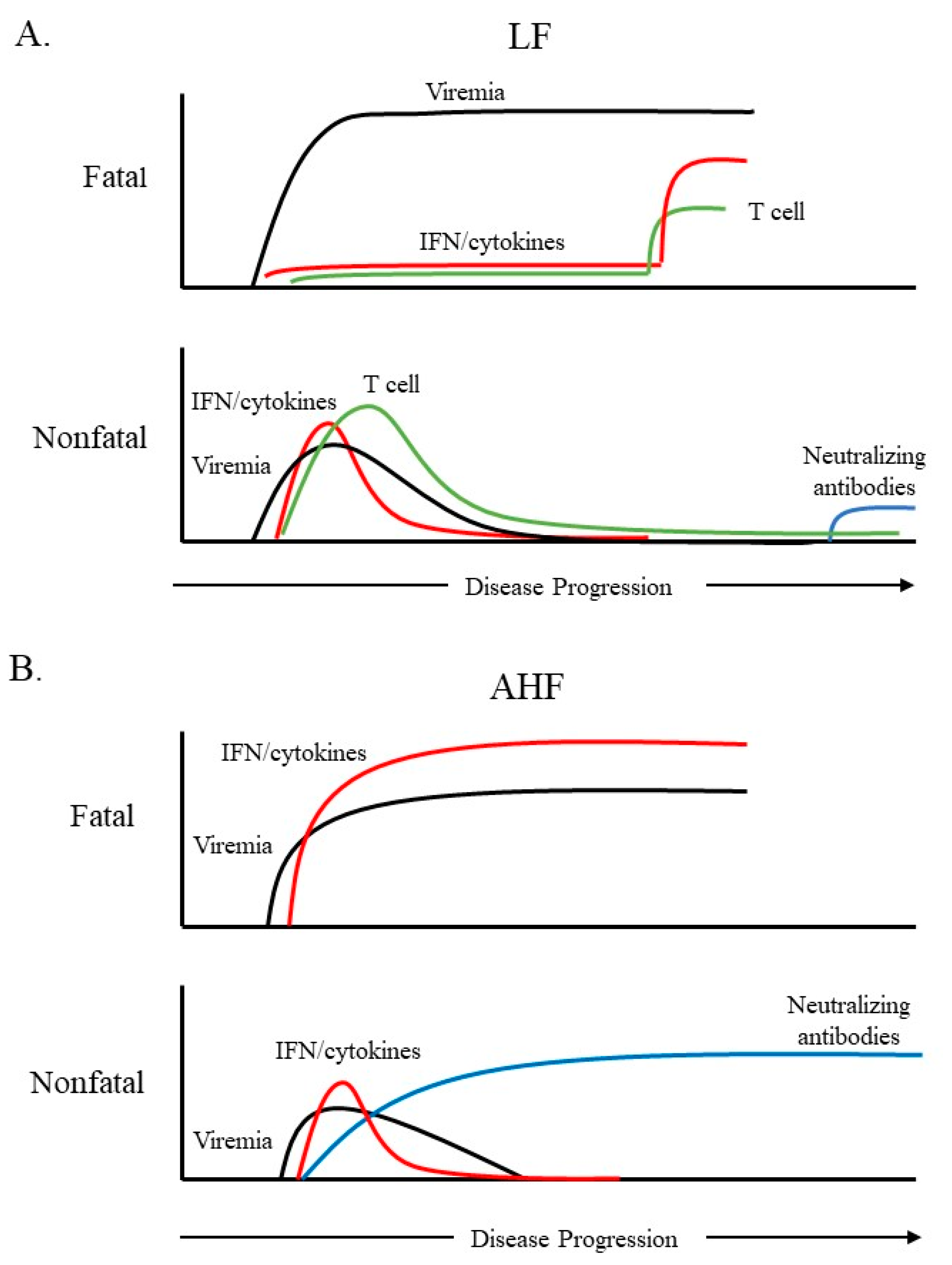
2. Immune Response to Hemorrhagic Fever-Causing Arenaviruses
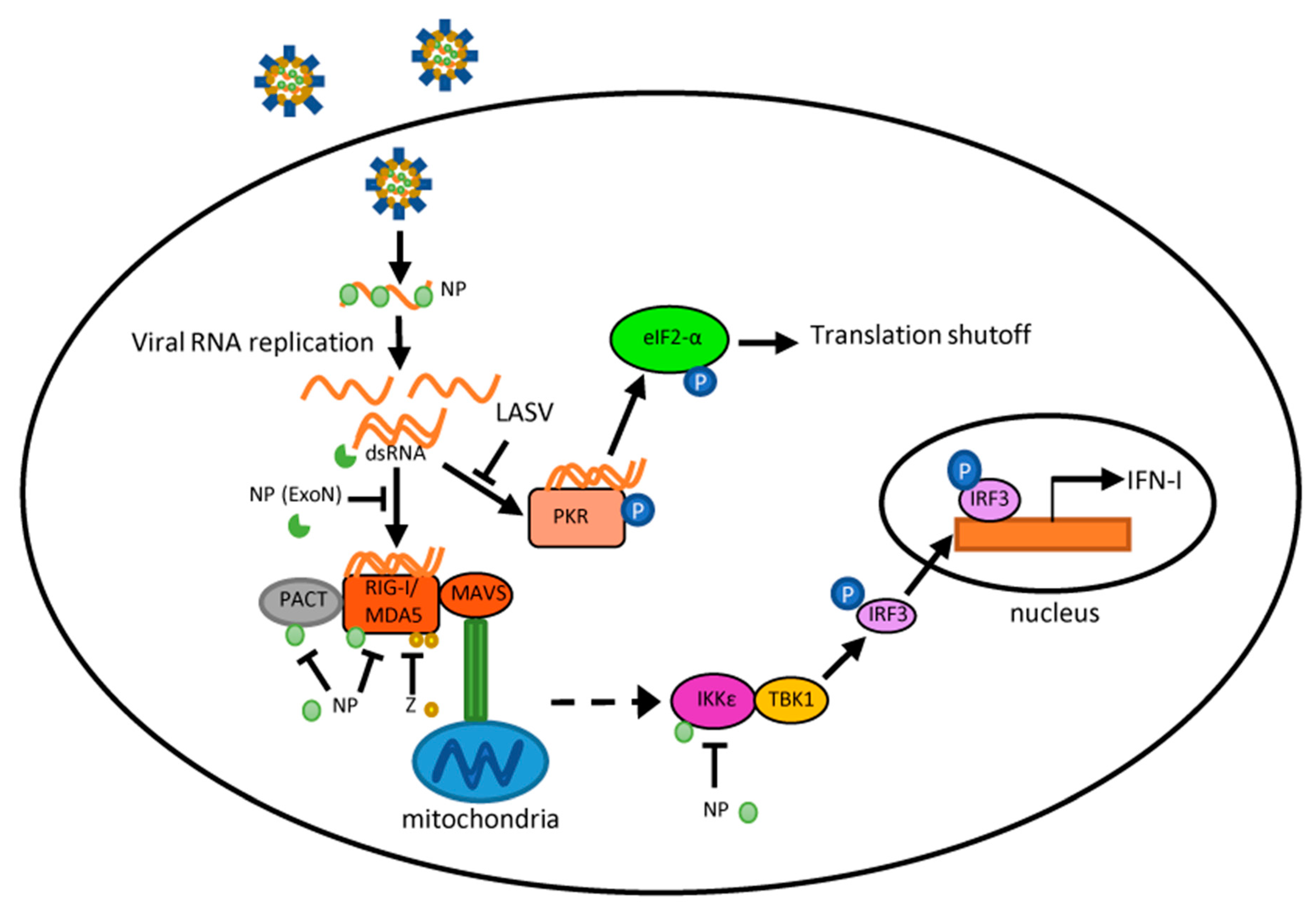
3. Arenavirus Interactions with Pattern Recognition Receptors
4. Molecular Mechanisms for Arenavirus Antagonism of the IFN Response
5. Arenavirus Subversion of Other Host Antiviral Defenses
6. Adaptive Immune Responses Following Successful Arenavirus Vaccination in Animal Models
7. Impacts of the Adaptive Cellular Immune Response on Post-Infection Sequelae
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Buchmeier, M.J.; Bowen, M.D.; Peters, C.J. Arenaviridae: The viruses and their replication. In Fields Virology; Knipe, H.P., Howley, P.M., Eds.; Wolter Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1791–1827. [Google Scholar]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avsic-Zupanc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, E.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate rna viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Albariño, C.G.; Posik, D.M.; Ghiringhelli, P.D.; Lozano, M.E.; Rivera Pomar, R.; Romanowski, V. Arenavirus nucleocapsid protein displays a transcriptional antitermination activity in vivo. Virus Res. 2001, 73, 41–55. [Google Scholar] [CrossRef]
- Meyer, B.J.; Southern, P.J. Sequence heterogeneity in the termini of lymphocytic choriomeningitis virus genomic and antigenomic rnas. J. Virol 1994, 68, 7659–7664. [Google Scholar]
- Kranzusch, P.J.; Schenk, A.D.; Rahmeh, A.A.; Radoshitzky, S.R.; Bavari, S.; Walz, T.; Whelan, S.P. Assembly of a functional machupo virus polymerase complex. Proc. Natl. Acad. Sci. USA 2010, 107, 20069–20074. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; Oldstone, M.B. Protein structure of lymphocytic choriomeningitis virus: Evidence for a cell-associated precursor of the virion glycopeptides. Virology 1979, 99, 111–120. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The lassa virus glycoprotein precursor gp-c is proteolytically processed by subtilase ski-1/s1p. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef]
- York, J.; Romanowski, V.; Lu, M.; Nunberg, J.H. The signal peptide of the junín arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature g1-g2 complex. J. Virol. 2004, 78, 10783–10792. [Google Scholar] [CrossRef]
- Agnihothram, S.S.; York, J.; Nunberg, J.H. Role of the stable signal peptide and cytoplasmic domain of g2 in regulating intracellular transport of the junín virus envelope glycoprotein complex. J. Virol. 2006, 80, 5189–5198. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small ring finger protein z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef]
- Djavani, M.; Lukashevich, I.S.; Sanchez, A.; Nichol, S.T.; Salvato, M.S. Completion of the lassa fever virus sequence and identification of a ring finger open reading frame at the l rna 5’ end. Virology 1997, 235, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Strecker, T.; Eichler, R.; Meulen, J.; Weissenhorn, W.; Dieter Klenk, H.; Garten, W.; Lenz, O. Lassa virus z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J. Virol. 2003, 77, 10700–10705. [Google Scholar] [CrossRef] [PubMed]
- Wolff, H.; Lange, J.V.; Webb, P.A. Interrelationships among arenaviruses measured by indirect immunofluorescence. Intervirology 1978, 9, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Bào, Y.; Buchmeier, M.J.; Charrel, R.N.; Clawson, A.N.; Clegg, C.S.; DeRisi, J.L.; Emonet, S.; Gonzalez, J.P.; Kuhn, J.H.; et al. Past, present, and future of arenavirus taxonomy. Arch. Virol. 2015, 160, 1851–1874. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Hesse, R.A.; Rhoderick, J.B.; Elwell, M.A.; Moe, J.B. Pathogenesis of a pichinde virus strain adapted to produce lethal infections in guinea pigs. Infect. Immun. 1981, 32, 872–880. [Google Scholar] [PubMed]
- Connolly, B.M.; Jenson, A.B.; Peters, C.J.; Geyer, S.J.; Barth, J.F.; McPherson, R.A. Pathogenesis of pichinde virus infection in strain 13 guinea pigs: An immunocytochemical, virologic, and clinical chemistry study. Am. J. Trop. Med. Hyg. 1993, 49, 10–24. [Google Scholar] [CrossRef]
- Demby, A.H.; Inapogui, A.; Kargbo, K.; Koninga, J.; Kourouma, K.; Kanu, J.; Coulibaly, M.; Wagoner, K.D.; Ksiazek, T.G.; Peters, C.J.; et al. Lassa fever in guinea: Ii. Distribution and prevalence of lassa virus infection in small mammals. Vector Borne Zoonotic Dis. 2001, 1, 283–297. [Google Scholar] [CrossRef]
- Gryseels, S.; Baird, S.J.; Borremans, B.; Makundi, R.; Leirs, H.; Goüy de Bellocq, J. When viruses don’t go viral: The importance of host phylogeographic structure in the spatial spread of arenaviruses. PLoS Pathog. 2017, 13, e1006073. [Google Scholar] [CrossRef]
- McCormick, J.B. Epidemiology and control of lassa fever. Curr. Top. Microbiol. Immunol. 1987, 134, 69–78. [Google Scholar]
- Monath, T.P.; Mertens, P.E.; Patton, R.; Moser, C.R.; Baum, J.J.; Pinneo, L.; Gary, G.W.; Kissling, R.E. A hospital epidemic of lassa fever in zorzor, liberia, march-april 1972. Am. J. Trop. Med. Hyg. 1973, 22, 773–779. [Google Scholar] [CrossRef]
- Stinebaugh, B.J.; Schloeder, F.X.; Johnson, K.M.; Mackenzie, R.B.; Entwisle, G.; De Alba, E. Bolivian hemorrhagic fever. A report of four cases. Am. J. Med. 1966, 40, 217–230. [Google Scholar] [CrossRef]
- McCormick, J.B.; Webb, P.A.; Krebs, J.W.; Johnson, K.M.; Smith, E.S. A prospective study of the epidemiology and ecology of lassa fever. J. Infect. Dis. 1987, 155, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Gunther, S.; Lenz, O. Lassa virus. Crit. Rev. Clin. Lab. Sci. 2004, 41, 339–390. [Google Scholar] [CrossRef] [PubMed]
- Enria, D.A.; Briggiler, A.M.; Sánchez, Z. Treatment of argentine hemorrhagic fever. Antiviral. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef]
- Patterson, M.; Grant, A.; Paessler, S. Epidemiology and pathogenesis of bolivian hemorrhagic fever. Curr. Opin. Virol. 2014, 5, 82–90. [Google Scholar] [CrossRef]
- Buchmeier, M.J. Lassa fever: Central nervous system manifestations. J. Trop. Geo. Neurol. 1991, 1, 23–30. [Google Scholar]
- Mateer, E.J.; Huang, C.; Shehu, N.Y.; Paessler, S. Lassa fever-induced sensorineural hearing loss: A neglected public health and social burden. PLoS Negl. Trop. Dis. 2018, 12, e0006187. [Google Scholar] [CrossRef]
- Cummins, D.; McCormick, J.B.; Bennett, D.; Samba, J.A.; Farrar, B.; Machin, S.J.; Fisher-Hoch, S.P. Acute sensorineural deafness in lassa fever. JAMA 1990, 264, 2093–2096. [Google Scholar] [CrossRef]
- Yun, N.E.; Walker, D.H. Pathogenesis of lassa fever. Viruses 2012, 4, 2031–2048. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Falcoff, E.; Feuillade, M.R.; Enria, D.A.; Maiztegui, J.I.; Falcoff, R. Endogenous interferon in argentine hemorrhagic fever. J. Infect. Dis. 1984, 149, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Feuillade, M.R.; Enria, D.A.; Maiztegui, J.I.; Falcoff, R. Correlation between endogenous interferon and the clinical evolution of patients with argentine hemorrhagic fever. J. Interferon. Res. 1985, 5, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Pozner, R.G.; Ure, A.E.; Jaquenod de Giusti, C.; D’Atri, L.P.; Italiano, J.E.; Torres, O.; Romanowski, V.; Schattner, M.; Gómez, R.M. Junín virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type i ifn signaling. PLoS Pathog. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar] [CrossRef]
- Schaeffer, J.; Carnec, X.; Reynard, S.; Mateo, M.; Picard, C.; Pietrosemoli, N.; Dillies, M.A.; Baize, S. Lassa virus activates myeloid dendritic cells but suppresses their ability to stimulate t cells. PLoS Pathog. 2018, 14, e1007430. [Google Scholar] [CrossRef]
- Pannetier, D.; Faure, C.; Georges-Courbot, M.C.; Deubel, V.; Baize, S. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to mopeia virus infection. J. Virol 2004, 78, 10516–10524. [Google Scholar] [CrossRef]
- Schaeffer, J.; Reynard, S.; Carnec, X.; Pietrosemoli, N.; Dillies, M.A.; Baize, S. Non-pathogenic mopeia virus induces more robust activation of plasmacytoid dendritic cells than lassa virus. Viruses 2019, 11, 287. [Google Scholar] [CrossRef]
- McElroy, A.K.; Akondy, R.S.; Harmon, J.R.; Ellebedy, A.H.; Cannon, D.; Klena, J.D.; Sidney, J.; Sette, A.; Mehta, A.K.; Kraft, C.S.; et al. A case of human lassa virus infection with robust acute t-cell activation and long-term virus-specific t-cell responses. J. Infect. Dis. 2017, 215, 1862–1872. [Google Scholar] [CrossRef]
- Prescott, J.B.; Marzi, A.; Safronetz, D.; Robertson, S.J.; Feldmann, H.; Best, S.M. Immunobiology of ebola and lassa virus infections. Nat. Rev. Immunol. 2017, 17, 195–207. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Frame, J.D.; Rhoderick, J.B.; Monson, M.H. Endemic lassa fever in liberia. Iv. Selection of optimally effective plasma for treatment by passive immunization. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 380–384. [Google Scholar] [CrossRef]
- Grant, A.; Seregin, A.; Huang, C.; Kolokoltsova, O.; Brasier, A.; Peters, C.; Paessler, S. Junín virus pathogenesis and virus replication. Viruses 2012, 4, 2317–2339. [Google Scholar] [CrossRef] [PubMed]
- Maiztegui, J.I.; Fernandez, N.J.; de Damilano, A.J. Efficacy of immune plasma in treatment of argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet 1979, 2, 1216–1217. [Google Scholar] [CrossRef]
- Hensley, L.E.; Smith, M.A.; Geisbert, J.B.; Fritz, E.A.; Daddario-DiCaprio, K.M.; Larsen, T.; Geisbert, T.W. Pathogenesis of lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in lassa virus-infected cynomolgus monkeys. J. Virol 2009, 83, 5890–5903. [Google Scholar] [CrossRef]
- Oestereich, L.; Lüdtke, A.; Ruibal, P.; Pallasch, E.; Kerber, R.; Rieger, T.; Wurr, S.; Bockholt, S.; Pérez-Girón, J.V.; Krasemann, S.; et al. Chimeric mice with competent hematopoietic immunity reproduce key features of severe lassa fever. PLoS Pathog 2016, 12, e1005656. [Google Scholar] [CrossRef]
- Dejean, C.B.; Ayerra, B.L.; Teyssié, A.R. Interferon response in the guinea pig infected with junín virus. J. Med. Virol 1987, 23, 83–91. [Google Scholar] [CrossRef]
- Kenyon, R.H.; McKee, K.T.; Zack, P.M.; Rippy, M.K.; Vogel, A.P.; York, C.; Meegan, J.; Crabbs, C.; Peters, C.J. Aerosol infection of rhesus macaques with junin virus. Intervirology 1992, 33, 23–31. [Google Scholar]
- Jahrling, P.B.; Smith, S.; Hesse, R.A.; Rhoderick, J.B. Pathogenesis of lassa virus infection in guinea pigs. Infect. Immun. 1982, 37, 771–778. [Google Scholar]
- Kenyon, R.H.; Green, D.E.; Peters, C.J. Effect of immunosuppression on experimental argentine hemorrhagic fever in guinea pigs. J. Virol. 1985, 53, 75–80. [Google Scholar]
- Bell, T.M.; Shaia, C.I.; Bunton, T.E.; Robinson, C.G.; Wilkinson, E.R.; Hensley, L.E.; Cashman, K.A. Pathology of experimental machupo virus infection, chicava strain, in cynomolgus macaques (macaca fascicularis) by intramuscular and aerosol exposure. Vet. Pathol. 2015, 52, 26–37. [Google Scholar] [CrossRef]
- Bell, T.M.; Bunton, T.E.; Shaia, C.I.; Raymond, J.W.; Honnold, S.P.; Donnelly, G.C.; Shamblin, J.D.; Wilkinson, E.R.; Cashman, K.A. Pathogenesis of bolivian hemorrhagic fever in guinea pigs. Vet. Pathol. 2016, 53, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Weissenbacher, M.C.; Avila, M.M.; Calello, M.A.; Merani, M.S.; McCormick, J.B.; Rodriguez, M. Effect of ribavirin and immune serum on junin virus-infected primates. Med. Microbiol. Immunol. 1986, 175, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Mateer, E.J.; Paessler, S.; Huang, C. Visualization of double-stranded rna colocalizing with pattern recognition receptors in arenavirus infected cells. Front. Cell Infect. Microbiol. 2018, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing rna virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U. Mechanisms of rig-i-like receptor activation and manipulation by viral pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, C. Regulation of mavs activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingström, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; et al. Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid rig-i-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Ronca, S.; Koma, T.; Paessler, S. Highly pathogenic new world and old world human arenaviruses induce distinct interferon responses in human cells. J. Virol. 2015, 89, 7079–7088. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Poussard, A.L.; Walker, A.G.; Brasier, A.R.; Zhao, Y.; Tian, B.; de la Torre, J.C.; et al. Junín virus infection activates the type i interferon pathway in a rig-i-dependent manner. PLoS Negl. Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef]
- Moreno, H.; Moller, R.; Fedeli, C.; Gerold, G.; Kunz, S. Comparison of the innate immune responses to pathogenic and nonpathogenic clade b new world arenaviruses. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Mateer, E.; Paessler, S.; Huang, C. Confocal imaging of double-stranded rna and pattern recognition receptors in negative-sense rna virus infection. J. Vis. Exp. 2019, 143. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Meurs, E.F.; Esteban, M. The dsrna protein kinase pkr: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Kolokoltsova, O.A.; Mateer, E.J.; Koma, T.; Paessler, S. Highly pathogenic new world arenavirus infection activates the pattern recognition receptor protein kinase r without attenuating virus replication in human cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- King, B.R.; Hershkowitz, D.; Eisenhauer, P.L.; Weir, M.E.; Ziegler, C.M.; Russo, J.; Bruce, E.A.; Ballif, B.A.; Botten, J. A map of the arenavirus nucleoprotein-host protein interactome reveals that junín virus selectively impairs the antiviral activity of double-stranded rna-activated protein kinase (pkr). J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the lassa virus nucleoprotein reveals a dsrna-specific 3’ to 5’ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. Structures of arenaviral nucleoproteins with triphosphate dsrna reveal a unique mechanism of immune suppression. J. Biol. Chem. 2013, 288, 16949–16959. [Google Scholar] [CrossRef]
- Huang, Q.; Shao, J.; Lan, S.; Zhou, Y.; Xing, J.; Dong, C.; Liang, Y.; Ly, H. In vitro and in vivo characterizations of pichinde viral nucleoprotein exoribonuclease functions. J Virol. 2015, 89, 6595–6607. [Google Scholar] [CrossRef]
- Carnec, X.; Mateo, M.; Page, A.; Reynard, S.; Hortion, J.; Picard, C.; Yekwa, E.; Barrot, L.; Barron, S.; Vallve, A.; et al. A vaccine platform against arenaviruses based on a recombinant hyperattenuated mopeia virus expressing heterologous glycoproteins. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase ikkε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, W.W.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa b. J. Virol. 2012, 86, 8185–8197. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Huang, Q.; Liu, X.; Di, D.; Liang, Y.; Ly, H. Arenaviral nucleoproteins suppress pact-induced augmentation of rig-i function to inhibit type i interferon production. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.H.; Lui, P.Y.; Ng, M.H.; Siu, K.L.; Au, S.W.; Jin, D.Y. The double-stranded RNA-binding protein pact functions as a cellular activator of rig-i to facilitate innate antiviral response. Cell Host Microbe. 2011, 9, 299–309. [Google Scholar] [CrossRef]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the ebola virus vp35 protein and the rig-i activator pact determines infection outcome. Cell Host Microbe. 2013, 14, 74–84. [Google Scholar] [CrossRef]
- Tawaratsumida, K.; Phan, V.; Hrincius, E.R.; High, A.A.; Webby, R.; Redecke, V.; Häcker, H. Quantitative proteomic analysis of the influenza a virus nonstructural proteins ns1 and ns2 during natural cell infection identifies pact as an ns1 target protein and antiviral host factor. J. Virol. 2014, 88, 9038–9048. [Google Scholar] [CrossRef]
- Siu, K.L.; Yeung, M.L.; Kok, K.H.; Yuen, K.S.; Kew, C.; Lui, P.Y.; Chan, C.P.; Tse, H.; Woo, P.C.; Yuen, K.Y.; et al. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded rna-binding protein that suppresses pact-induced activation of rig-i and mda5 in the innate antiviral response. J. Virol. 2014, 88, 4866–4876. [Google Scholar] [CrossRef]
- Kew, C.; Lui, P.Y.; Chan, C.P.; Liu, X.; Au, S.W.; Mohr, I.; Jin, D.Y.; Kok, K.H. Suppression of pact-induced type i interferon production by herpes simplex virus 1 us11 protein. J. Virol. 2013, 87, 13141–13149. [Google Scholar] [CrossRef]
- Patel, R.C.; Sen, G.C. Pact, a protein activator of the interferon-induced protein kinase, pkr. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef]
- Loureiro, M.E.; Zorzetto-Fernandes, A.L.; Radoshitzky, S.; Chi, X.; Dallari, S.; Marooki, N.; Lèger, P.; Foscaldi, S.; Harjono, V.; Sharma, S.; et al. Ddx3 suppresses type i interferons and favors viral replication during arenavirus infection. PLoS Pathog. 2018, 14, e1007125. [Google Scholar] [CrossRef]
- Xing, J.; Ly, H.; Liang, Y. The z proteins of pathogenic but not nonpathogenic arenaviruses inhibit rig-i-like receptor-dependent interferon production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of new world arenaviruses bind rig-i and interfere with type i interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type i interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Roldán, J.S.; Candurra, N.A.; Colombo, M.I.; Delgui, L.R. Junín virus promotes autophagy to facilitate the virus life cycle. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Perez Vidakovics, M.L.A.; Ure, A.E.; Arrías, P.N.; Romanowski, V.; Gómez, R.M. Junín virus induces autophagy in human a549 cells. PLoS ONE 2019, 14, e0218730. [Google Scholar] [CrossRef]
- Baillet, N.; Krieger, S.; Journeaux, A.; Caro, V.; Tangy, F.; Vidalain, P.O.; Baize, S. Autophagy promotes infectious particle production of mopeia and lassa viruses. Viruses 2019, 11, 293. [Google Scholar] [CrossRef]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The atg5 atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef]
- Takeshita, F.; Kobiyama, K.; Miyawaki, A.; Jounai, N.; Okuda, K. The non-canonical role of atg family members as suppressors of innate antiviral immune signaling. Autophagy 2008, 4, 67–69. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Russier, M.; Reynard, S.; Tordo, N.; Baize, S. Nk cells are strongly activated by lassa and mopeia virus-infected human macrophages in vitro but do not mediate virus suppression. Eur. J. Immunol. 2012, 42, 1822–1832. [Google Scholar] [CrossRef]
- Russier, M.; Reynard, S.; Carnec, X.; Baize, S. The exonuclease domain of lassa virus nucleoprotein is involved in antigen-presenting-cell-mediated nk cell responses. J. Virol. 2014, 88, 13811–13820. [Google Scholar] [CrossRef]
- Wauquier, N.; Petitdemange, C.; Tarantino, N.; Maucourant, C.; Coomber, M.; Lungay, V.; Bangura, J.; Debré, P.; Vieillard, V. Hla-c-restricted viral epitopes are associated with an escape mechanism from kir2dl2. EBioMedicine 2019, 40, 605–613. [Google Scholar] [CrossRef]
- Ambrosio, A.; Saavedra, M.; Mariani, M.; Gamboa, G.; Maiza, A. Argentine hemorrhagic fever vaccines. Hum. Vaccin. 2011, 7, 694–700. [Google Scholar] [CrossRef]
- Koma, T.; Patterson, M.; Huang, C.; Seregin, A.V.; Maharaj, P.D.; Miller, M.; Smith, J.N.; Walker, A.G.; Hallam, S.; Paessler, S. Machupo virus expressing gpc of the candid#1 vaccine strain of junin virus is highly attenuated and immunogenic. J. Virol. 2016, 90, 1290–1297. [Google Scholar]
- Lukashevich, I.S.; Carrion, R., Jr.; Salvato, M.S.; Mansfield, K.; Brasky, K.; Zapata, J.; Cairo, C.; Goicochea, M.; Hoosien, G.E.; Ticer, A.; et al. Safety, immunogenicity, and efficacy of the ml29 reassortant vaccine for lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Patterson, J.; Carrion, R.; Moshkoff, D.; Ticer, A.; Zapata, J.; Brasky, K.; Geiger, R.; Hubbard, G.B.; Bryant, J.; et al. A live attenuated vaccine for lassa fever made by reassortment of lassa and mopeia viruses. J. Virol. 2005, 79, 13934–13942. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jones, S.; Fritz, E.A.; Shurtleff, A.C.; Geisbert, J.B.; Liebscher, R.; Grolla, A.; Stroher, U.; Fernando, L.; Daddario, K.M.; et al. Development of a new vaccine for the prevention of lassa fever. PLoS Med. 2005, 2, e183. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Hutwagner, L.; Brown, B.; McCormick, J.B. Effective vaccine for lassa fever. J. Virol. 2000, 74, 6777–6783. [Google Scholar] [CrossRef]
- Salazar, M.; Yun, N.E.; Poussard, A.L.; Smith, J.N.; Smith, J.K.; Kolokoltsova, O.A.; Patterson, M.J.; Linde, J.; Paessler, S. Effect of ribavirin on junin virus infection in guinea pigs. Zoonoses Public Health 2012, 59, 278–285. [Google Scholar] [CrossRef]
- Eddy, G.A.; Wagner, F.S.; Scott, S.K.; Mahlandt, B.J. Protection of monkeys against machupo virus by the passive administration of bolivian haemorrhagic fever immunoglobulin (human origin). Bull. World Health Organ. 1975, 52, 723–727. [Google Scholar]
- Cashman, K.A.; Wilkinson, E.R.; Zeng, X.; Cardile, A.P.; Facemire, P.R.; Bell, T.M.; Bearss, J.J.; Shaia, C.I.; Schmaljohn, C.S. Immune-mediated systemic vasculitis as the proposed cause of sudden-onset sensorineural hearing loss following lassa virus exposure in cynomolgus macaques. MBio 2018, 9. [Google Scholar] [CrossRef]
- Yun, N.E.; Ronca, S.; Tamura, A.; Koma, T.; Seregin, A.V.; Dineley, K.T.; Miller, M.; Cook, R.; Shimizu, N.; Walker, A.G.; et al. Animal model of sensorineural hearing loss associated with lassa virus infection. J. Virol. 2015, 90, 2920–2927. [Google Scholar] [CrossRef]
- Gary, J.M.; Welch, S.R.; Ritter, J.M.; Coleman-McCray, J.; Huynh, T.; Kainulainen, M.H.; Bollweg, B.C.; Parihar, V.; Nichol, S.T.; Zaki, S.R.; et al. Lassa virus targeting of anterior uvea and endothelium of cornea and conjunctiva in eye of guinea pig model. Emerg. Infect. Dis. 2019, 25, 865–874. [Google Scholar] [CrossRef]
- Liu, D.X.; Perry, D.L.; DeWald, L.E.; Cai, Y.; Hagen, K.R.; Cooper, T.K.; Huzella, L.M.; Hart, R.; Bonilla, A.; Bernbaum, J.G.; et al. Persistence of lassa virus associated with severe systemic arteritis in convalescing guinea pigs (cavia porcellus). J. Infect. Dis. 2019, 219, 1818–1822. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantlo, E.; Paessler, S.; Huang, C. Differential Immune Responses to Hemorrhagic Fever-Causing Arenaviruses. Vaccines 2019, 7, 138. https://doi.org/10.3390/vaccines7040138
Mantlo E, Paessler S, Huang C. Differential Immune Responses to Hemorrhagic Fever-Causing Arenaviruses. Vaccines. 2019; 7(4):138. https://doi.org/10.3390/vaccines7040138
Chicago/Turabian StyleMantlo, Emily, Slobodan Paessler, and Cheng Huang. 2019. "Differential Immune Responses to Hemorrhagic Fever-Causing Arenaviruses" Vaccines 7, no. 4: 138. https://doi.org/10.3390/vaccines7040138
APA StyleMantlo, E., Paessler, S., & Huang, C. (2019). Differential Immune Responses to Hemorrhagic Fever-Causing Arenaviruses. Vaccines, 7(4), 138. https://doi.org/10.3390/vaccines7040138