Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Bacterial Artificial Chromosome (BAC)-Cloning, and Viruses
2.2. Production and Purification of Virions and DB
2.3. SDS-PAGE, Silver/Instant Blue Staining, and Immunoblotting
2.4. Immunofluorescence
2.5. Analysis of Infectious Virus by IE1-Staining
2.6. IFN-β Treatment and Analysis of Genome Replication Kinetics
2.7. Statistical Analyses
3. Background, Results, and Strategy for Development
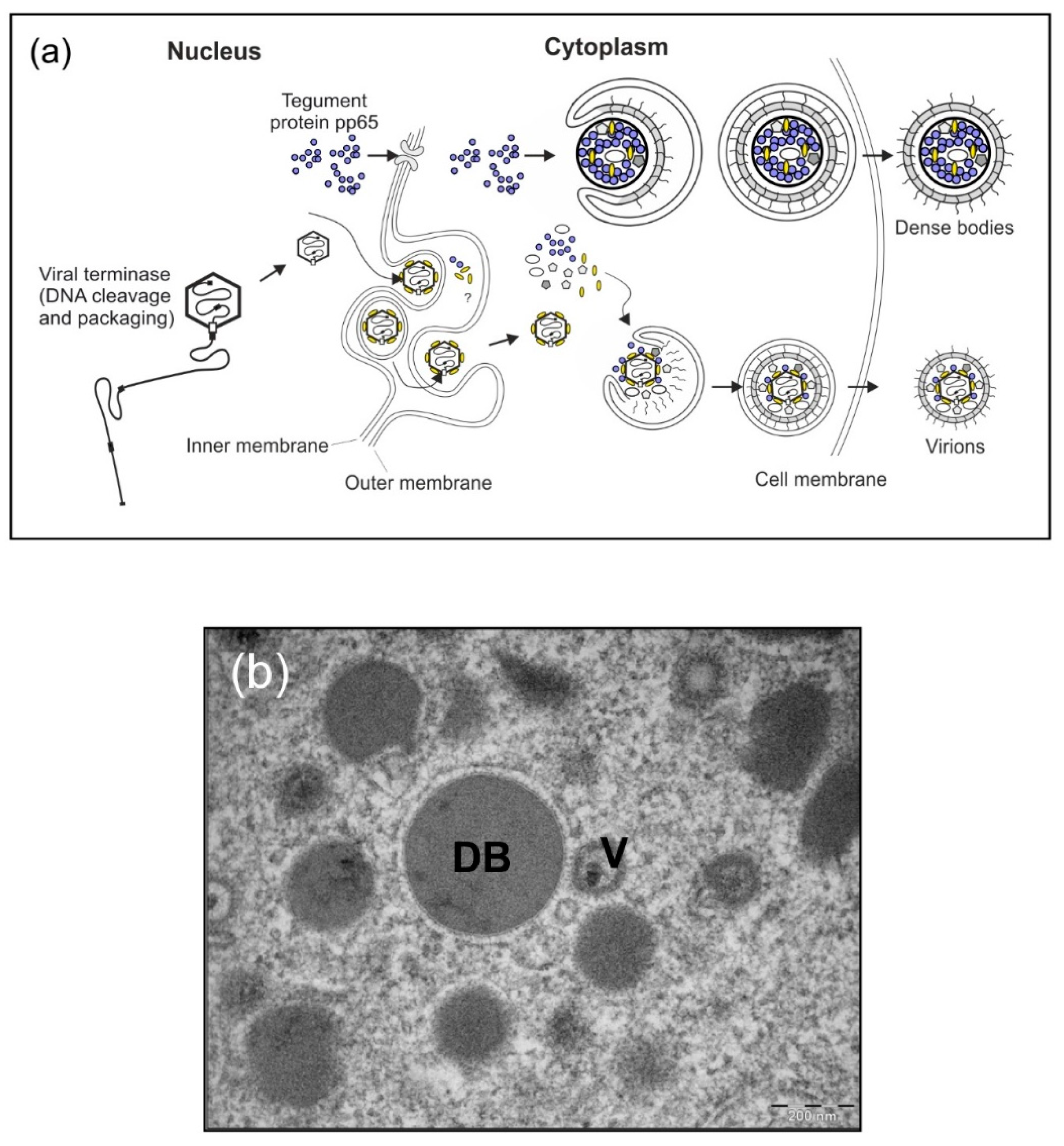
3.1. Background
3.2. Results and Strategy for Development
3.2.1. HCMV DB Material Can be Produced in a Large-Scale GMP-Compliant Manner
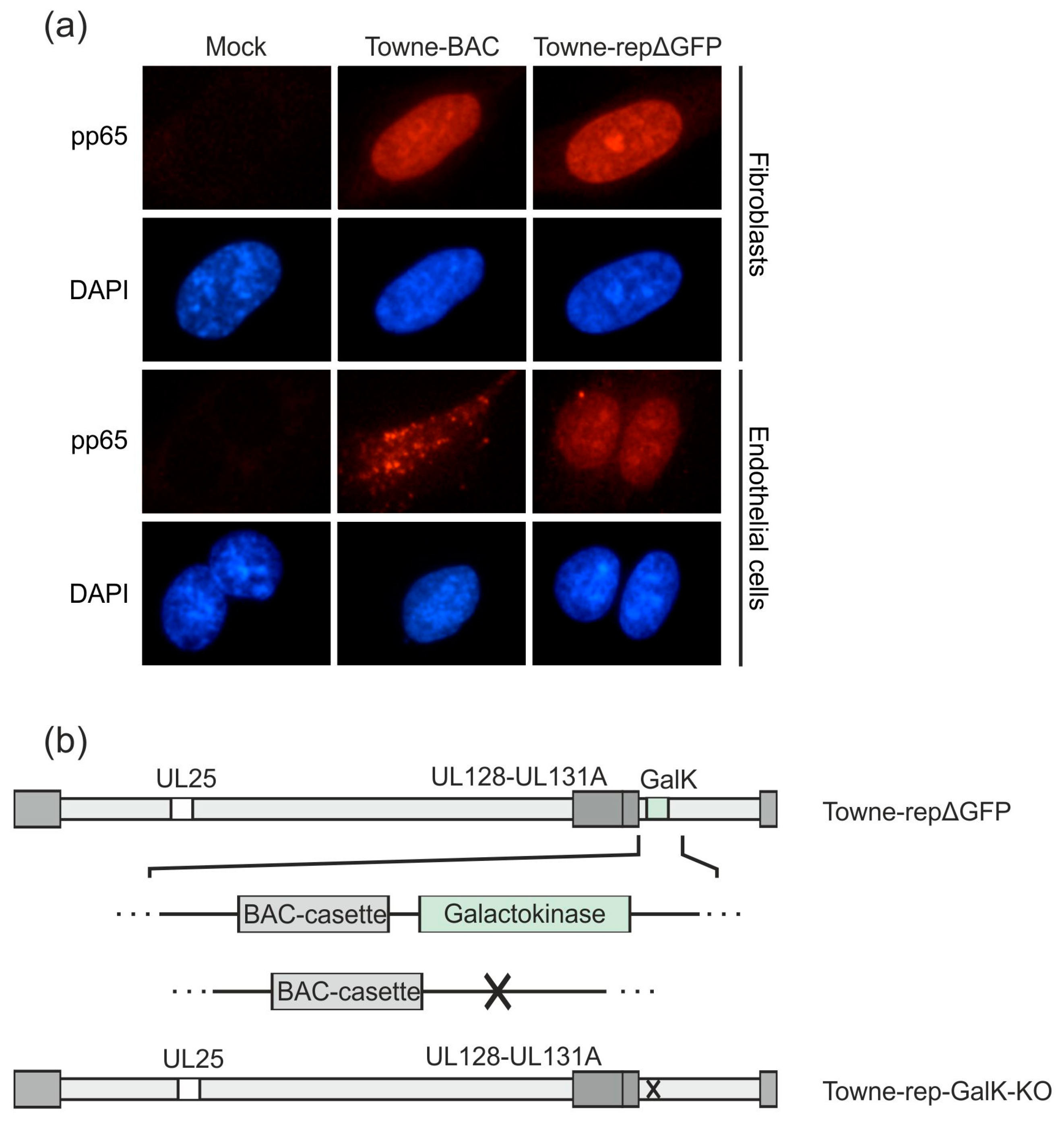
3.2.2. Further Development of Pentamer-Positive DB
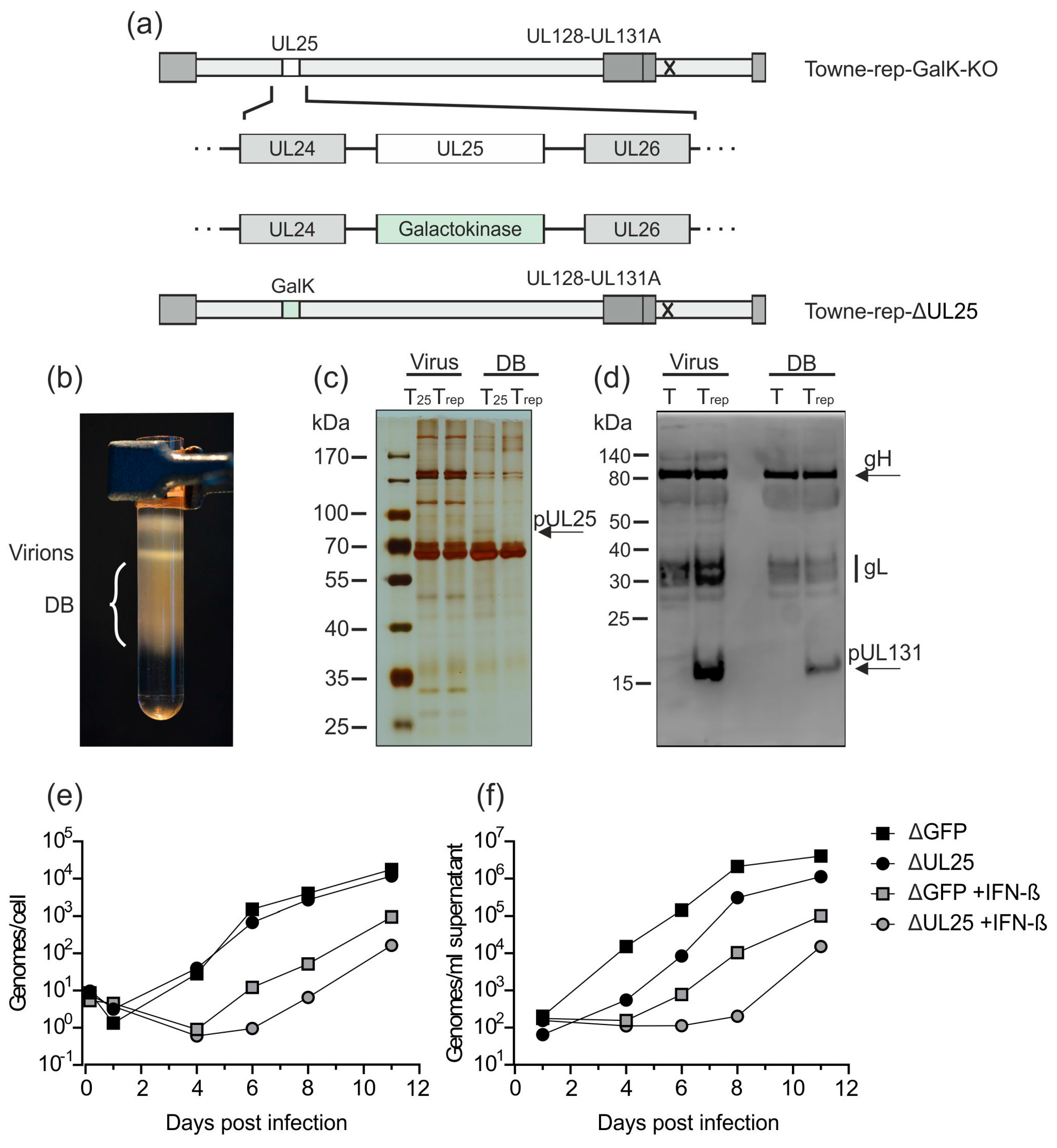
3.2.3. Establishment of a UL25-Deleted Virus Strain for DB Production
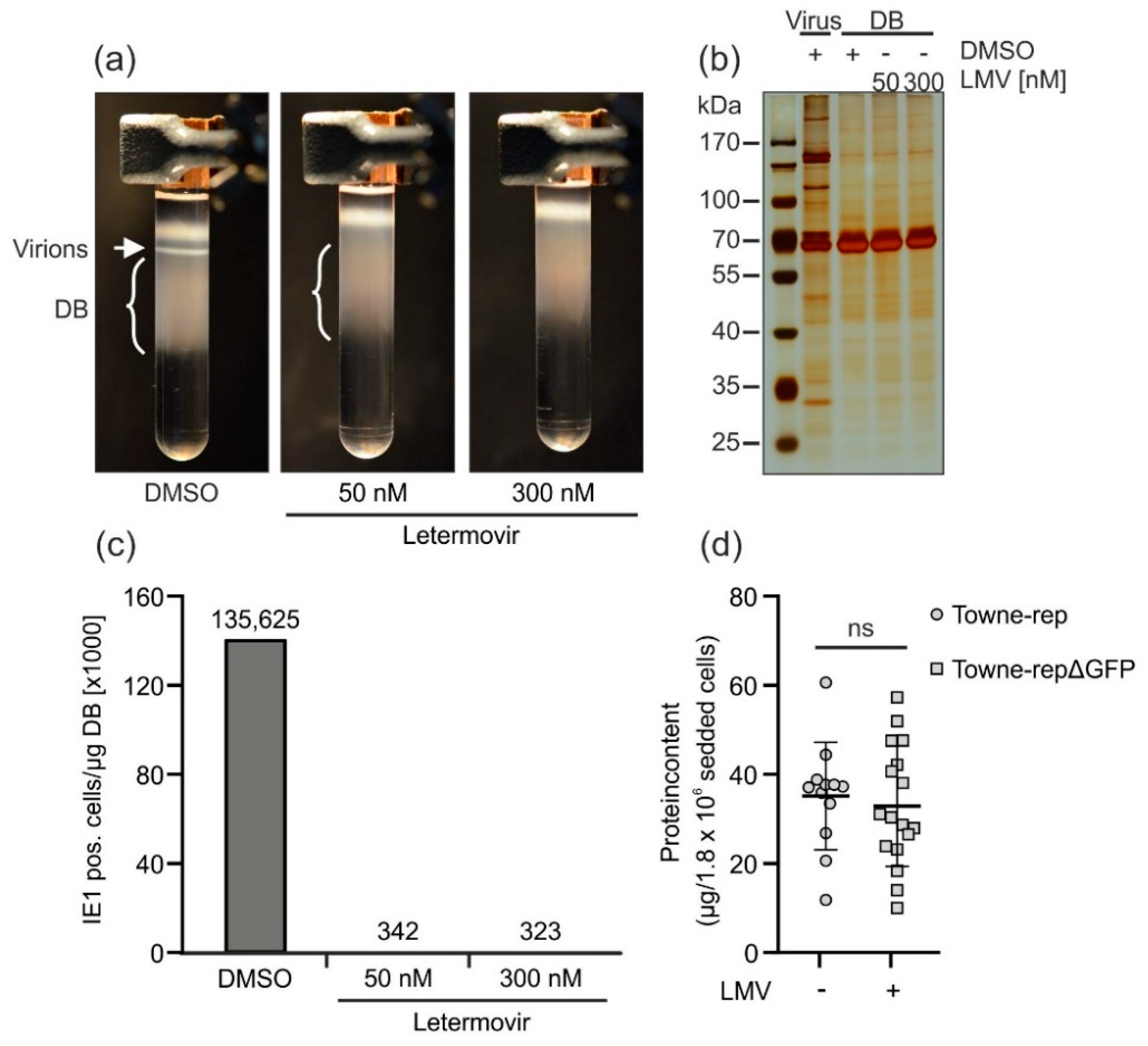
3.2.4. Application of the Terminase Inhibitor Letermovir in the DB Production Process
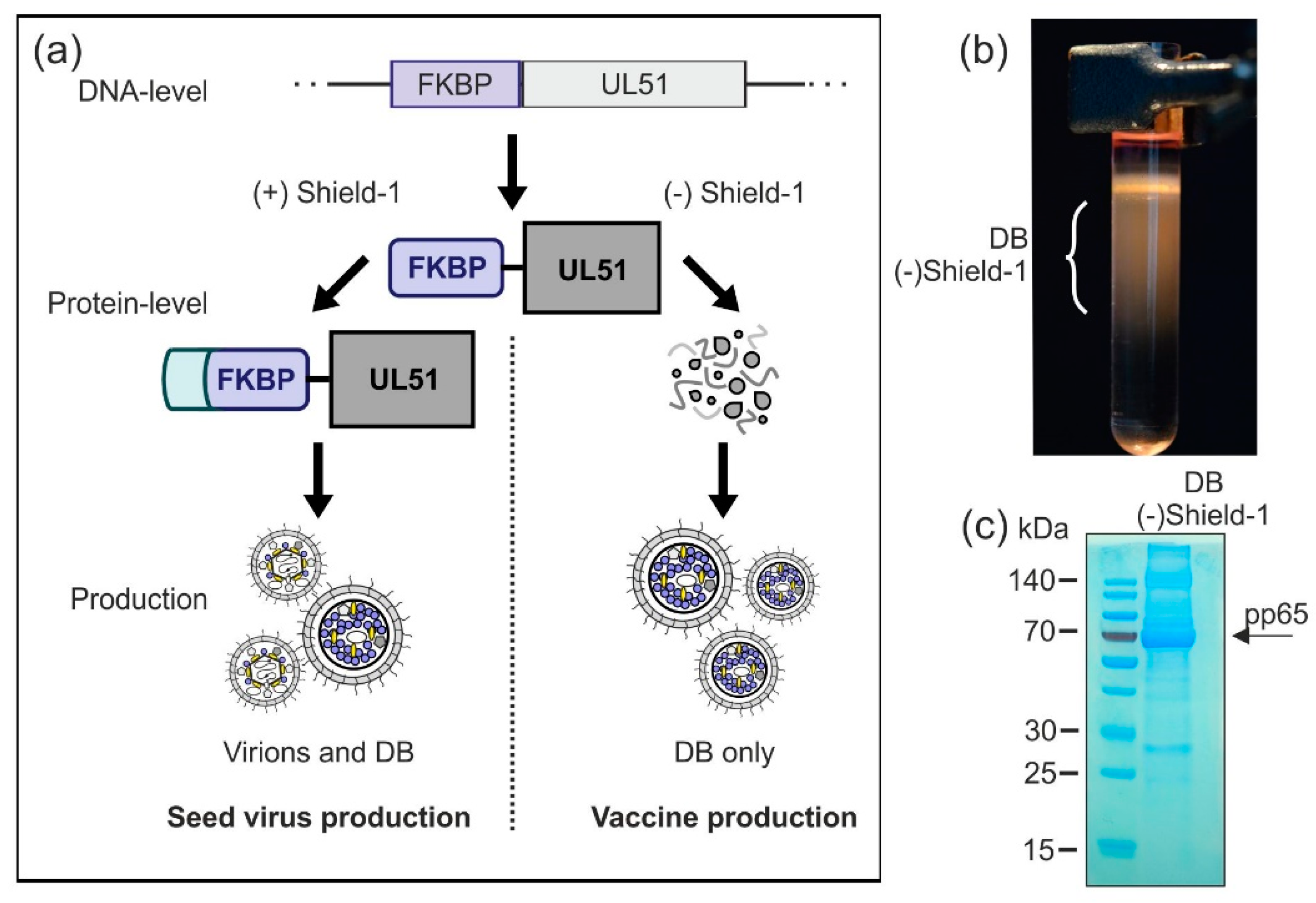
3.2.5. Establishment of a Shield-1-Dependent DB Production Process
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartlett, A.W.; McMullan, B.; Rawlinson, W.D.; Palasanthiran, P. Hearing and neurodevelopmental outcomes for children with asymptomatic congenital cytomegalovirus infection: A systematic review. Rev. Med. Virol. 2017, 27, e1938. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Dreher, A.M.; Arora, N.; Fowler, K.B.; Novak, Z.; Britt, W.J.; Boppana, S.B.; Ross, S.A. Spectrum of Disease and Outcome in Children with Symptomatic Congenital Cytomegalovirus Infection. J. Pediatr. 2014, 164, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.R.; Bialek, S.R.; Boppana, S.B.; Griffiths, P.D.; Laughlin, C.A.; Ljungman, P.; Mocarski, E.S.; Pass, R.F.; Read, J.S.; Schleiss, M.R.; et al. Priorities for CMV vaccine development. Vaccine 2013, 32, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Modlin, J.F.; Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R. Vaccine Development to Prevent Cytomegalovirus Disease: Report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid Organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Lee, D.-G.; Kim, H.-J. Cytomegalovirus Infections after Hematopoietic Stem Cell Transplantation: Current Status and Future Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2666. [Google Scholar] [CrossRef]
- Herr, W.; Plachter, B. Cytomegalovirus and varicella–zoster virus vaccines in hematopoietic stem cell transplantation. Expert Rev. Vaccines 2009, 8, 999–1021. [Google Scholar] [CrossRef]
- Diamond, D.J.; La Rosa, C.; Chiuppesi, F.; Contreras, H.; Dadwal, S.; Wussow, F.; Bautista, S.; Nakamura, R.; Zaia, J.A. A fifty-year odyssey: Prospects for a cytomegalovirus vaccine in transplant and congenital infection. Expert Rev. Vaccines 2018, 17, 889–911. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Plachter, B. Cytomegalovirus Vaccine: On the Way to the Future. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 2nd ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 424–449. [Google Scholar]
- Luisi, K.; Sharma, M.; Yu, D. Development of a vaccine against cytomegalovirus infection and disease. Curr. Opin. Virol. 2017, 23, 23–29. [Google Scholar] [CrossRef]
- Schleiss, M.R. Cytomegalovirus vaccines under clinical development. J. Virus Erad. 2016, 2, 198–207. [Google Scholar] [PubMed]
- Wang, D.; Fu, T.-M. Progress on human cytomegalovirus vaccines for prevention of congenital infection and disease. Curr. Opin. Virol. 2014, 6, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Plachter, B. Prospects of a vaccine for the prevention of congenital cytomegalovirus disease. Med. Microbiol. Immunol. 2016, 205, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J.; Vugler, L.; Butfiloski, E.J.; Stephens, E.B. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): Use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J. Virol. 1990, 64, 1079–1085. [Google Scholar] [PubMed]
- Marshall, G.S.; Rabalais, G.P.; Stout, G.G.; Waldeyer, S.L. Antibodies to Recombinant-Derived Glycoprotein B after Natural Human Cytomegalovirus Infection Correlate with Neutralizing Activity. J. Infect. Dis. 1992, 165, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Klein, M.; Britt, W.J.; Hassfurther, E.; Mach, M. Glycoprotein H of human cytomegalovirus is a major antigen for the neutralizing humoral immune response. J. Gen. Virol. 1996, 77, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; Mcneal, M.M.; et al. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: A randomized clinical trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.-L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine Prevention of Maternal Cytomegalovirus Infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human Cytomegalovirus UL131-128 Genes Are Indispensable for Virus Growth in Endothelial Cells and Virus Transfer to Leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef]
- Gerna, G.; Percivalle, E.; Lilleri, D.; Lozza, L.; Fornara, C.; Hahn, G.; Baldanti, F.; Revello, M.G. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 2005, 86, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Fouts, A.E.; Chan, P.; Stephan, J.-P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 Complex Comprise the Majority of the Anti-Cytomegalovirus (Anti-CMV) Neutralizing Antibody Response in CMV Hyperimmune Globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [PubMed]
- Genini, E.; Percivalle, E.; Sarasini, A.; Revello, M.G.; Baldanti, F.; Gerna, G. Serum antibody response to the gH/gL/pUL128–131 five-protein complex of human cytomegalovirus (HCMV) in primary and reactivated HCMV infections. J. Clin. Virol. 2011, 52, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Büscher, N.; Krauter, S.; Krämer, N.; Wolfrum, U.; Sehn, E.; Tenzer, S.; Plachter, B. The Abundant Tegument Protein pUL25 of Human Cytomegalovirus Prevents Proteasomal Degradation of pUL26 and Supports Its Suppression of ISGylation. J. Virol. 2018, 92, e01180-18. [Google Scholar] [CrossRef] [PubMed]
- Krömmelbein, N.; Wiebusch, L.; Schiedner, G.; Büscher, N.; Sauer, C.; Florin, L.; Sehn, E.; Wolfrum, U.; Plachter, B. Adenovirus E1A/E1B Transformed Amniotic Fluid Cells Support Human Cytomegalovirus Replication. Viruses 2016, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Büscher, N.; Paulus, C.; Nevels, M.; Tenzer, S.; Plachter, B. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains. Med. Microbiol. Immunol. 2015, 204, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Sauer, C.; Klobuch, S.; Herr, W.; Thomas, S.; Plachter, B. Subviral Dense Bodies of Human Cytomegalovirus Stimulate Maturation and Activation of Monocyte-Derived Immature Dendritic Cells. J. Virol. 2013, 87, 11287–11291. [Google Scholar] [CrossRef]
- Becke, S.; Aue, S.; Thomas, D.; Schader, S.; Podlech, J.; Bopp, T.; Sedmak, T.; Wolfrum, U.; Plachter, B.; Reyda, S. Optimized recombinant dense bodies of human cytomegalovirus efficiently prime virus specific lymphocytes and neutralizing antibodies without the addition of adjuvant. Vaccine 2010, 28, 6191–6198. [Google Scholar] [CrossRef]
- Mersseman, V.; Besold, K.; Reddehase, M.J.; Wolfrum, U.; Strand, D.; Plachter, B.; Reyda, S. Exogenous introduction of an immunodominant peptide from the non-structural IE1 protein of human cytomegalovirus into the MHC class I presentation pathway by recombinant dense bodies. J. Gen. Virol. 2008, 89, 369–379. [Google Scholar] [CrossRef]
- Pepperl-Klindworth, S.; Frankenberg, N.; Riegler, S.; Plachter, B. Protein delivery by subviral particles of human cytomegalovirus. Gene Ther. 2003, 10, 278–284. [Google Scholar] [CrossRef][Green Version]
- Pepperl, S.; Mach, M.; Harris, J.R.; Plachter, B.; Münster, J. Dense Bodies of Human Cytomegalovirus Induce both Humoral and Cellular Immune Responses in the Absence of Viral Gene Expression. J. Virol. 2000, 74, 6132–6146. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Ohrum, K.; Cayatte, C.; Liu, Y.; Wang, Z.; Irrinki, A.; Cataniag, F.; Nguyen, N.; Lambert, S.; Liu, H.; Aslam, S.; et al. Production of Cytomegalovirus Dense Bodies by Scalable Bioprocess Methods Maintains Immunogenicity and Improves Neutralizing Antibody Titers. J. Virol. 2016, 90, 10133–10144. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cayatte, C.; Schneider-Ohrum, K.; Wang, Z.; Irrinki, A.; Nguyen, N.; Lu, J.; Nelson, C.; Servat, E.; Gemmell, L.; Citkowicz, A.; et al. Cytomegalovirus Vaccine Strain Towne-Derived Dense Bodies Induce Broad Cellular Immune Responses and Neutralizing Antibodies That Prevent Infection of Fibroblasts and Epithelial Cells. J. Virol. 2013, 87, 11107–11120. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Falk, J.J.; Büscher, N.; Penner, I.; Zimmermann, C.; Gogesch, P.; Sinzger, C.; Plachter, B. Dense bodies of a gH/gL/UL128-131 pentamer repaired Towne strain of human cytomegalovirus induce an enhanced neutralizing antibody response. J. Virol. 2019, 93, JYI-00391. [Google Scholar] [CrossRef] [PubMed]
- Craighead, J.E.; Kanich, R.E.; Almeida, J.D. Nonviral Microbodies with Viral Antigenicity Produced in Cytomegalovirus-Infected Cells. J. Virol. 1972, 10, 766–775. [Google Scholar] [PubMed]
- Roby, C.; Gibson, W. Characterization of phosphoproteins and protein kinase activity of virions, noninfectious enveloped particles, and dense bodies of human cytomegalovirus. J. Virol. 1986, 59, 714–727. [Google Scholar] [PubMed]
- Irmiere, A.; Gibson, W. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 1983, 130, 118–133. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Paša-Tolić, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S.; et al. Identification of Proteins in Human Cytomegalovirus (HCMV) Particles: The HCMV Proteome†. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Topilko, A.; Michelson, S. Hyperimmediate entry of human cytomegalovirus virions and dense bodies into human fibroblasts. Res. Virol. 1994, 145, 75–82. [Google Scholar] [CrossRef]
- Besold, K.; Wills, M.; Plachter, B. Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 2009, 391, 5–19. [Google Scholar] [CrossRef]
- Lieber, D.; Hochdorfer, D.; Stoehr, D.; Schubert, A.; Lotfi, R.; May, T.; Wirth, D.; Sinzger, C. A permanently growing human endothelial cell line supports productive infection with human cytomegalovirus under conditional cell growth arrest. Biotechniques 2015, 59, 127–136. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Butueva, M.; Bantner, S.; Markusic, D.; Seppen, J.; MacLeod, R.A.; Weich, H.; Hauser, H.; Wirth, D. Synthetic Gene Regulation Circuits for Control of Cell Expansion. Tissue Eng. Part A 2010, 16, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef] [PubMed]
- Borst, E.M.; Kleine-Albers, J.; Gabaev, I.; Babic, M.; Wagner, K.; Binz, A.; Degenhardt, I.; Kalesse, M.; Jonjic, S.; Bauerfeind, R.; et al. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J. Virol. 2013, 87, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Plachter, B.; Britt, W.; Vornhagen, R.; Stamminger, T.; Jahn, G. Analysis of Proteins Encoded by IE Regions 1 and 2 of Human Cytomegalovirus Using Monoclonal Antibodies Generated against Recombinant Antigens. Virology 1993, 193, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, M.; Faircloth, M.; Vugler, L.; Britt, W.J. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 1989, 23, 157–167. [Google Scholar] [CrossRef]
- Freed, D.C.; Tang, Q.; Tang, A.; Li, F.; He, X.; Huang, Z.; Meng, W.; Xia, L.; Finnefrock, A.C.; Durr, E.; et al. Pentameric complex of viral glycoprotein H is the primary target for potent neutralization by a human cytomegalovirus vaccine. Proc. Natl. Acad. Sci. USA 2013, 110, E4997–E5005. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Perez, L.; Lilleri, D.; Marcandalli, J.; Agatic, G.; Becattini, S.; Preite, S.; Fuschillo, D.; Percivalle, E.; Sallusto, F.; et al. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 17965–17970. [Google Scholar] [CrossRef] [PubMed]
- Wussow, F.; Chiuppesi, F.; Martinez, J.; Campo, J.; Johnson, E.; Flechsig, C.; Newell, M.; Tran, E.; Ortiz, J.; La Rosa, C.; et al. Human Cytomegalovirus Vaccine Based on the Envelope gH/gL Pentamer Complex. PLoS Pathog. 2014, 10, e1004524. [Google Scholar] [CrossRef]
- Reyda, S.; Tenzer, S.; Navarro, P.; Gebauer, W.; Saur, M.; Krauter, S.; Büscher, N.; Plachter, B. The Tegument Protein pp65 of Human Cytomegalovirus Acts as an Optional Scaffold Protein That Optimizes Protein Uploading into Viral Particles. J. Virol. 2014, 88, 9633–9646. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed]
- El Helou, G.; Razonable, R.R. Letermovir for the prevention of cytomegalovirus infection and disease in transplant recipients: An evidence-based review. Infect. Drug Resist. 2019, 12, 1481. [Google Scholar] [CrossRef] [PubMed]
- Borst, E.-M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the Human Cytomegalovirus (HCMV) Genome as an Infectious Bacterial Artificial Chromosome in Escherichia coli: A New Approach for Construction of HCMV Mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [PubMed]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP·Rapamycin·FRB Ternary Complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Freed, D.C.; He, X.; Li, F.; Tang, A.; Cox, K.S.; Dubey, S.A.; Cole, S.; Medi, M.B.; Liu, Y.; et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016, 8, 362ra145. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Boppana, S.B.; Novak, Z.; Wagatsuma, V.M.; Oliveira, P.D.F.; Duarte, G.; Britt, W.J. Human cytomegalovirus reinfection is associated with intrauterine transmission in a highly cytomegalovirus-immune maternal population. Am. J. Obstet. Gynecol. 2010, 202, 297.e1–297.e8. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J. Maternal Immunity and the Natural History of Congenital Human Cytomegalovirus Infection. Viruses 2018, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Noriega, V.; Redmann, V.; Gardner, T.; Tortorella, D. Diverse immune evasion strategies by human cytomegalovirus. Immunol. Res. 2012, 54, 140–151. [Google Scholar] [CrossRef]
- Patel, M.; Vlahava, V.-M.; Forbes, S.K.; Fielding, C.A.; Stanton, R.J.; Wang, E.C.Y. HCMV-Encoded NK Modulators: Lessons From in vitro and in vivo Genetic Variation. Front. Immunol. 2018, 9, 2214. [Google Scholar] [CrossRef]
- Marques, M.; Ferreira, A.R.; Ribeiro, D. The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses 2018, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Ciesla, J.H.; Munger, J. Who’s Driving? Human Cytomegalovirus, Interferon, and NFkappaB Signaling. Viruses 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [PubMed]
- Spindler, N.; Diestel, U.; Stump, J.D.; Wiegers, A.K.; Winkler, T.H.; Sticht, H.; Mach, M.; Muller, Y.A. Structural basis for the recognition of human cytomegalovirus glycoprotein B by a neutralizing human antibody. PLoS Pathog. 2014, 10, e1004377. [Google Scholar] [CrossRef] [PubMed]
- Lilleri, D.; Kabanova, A.; Lanzavecchia, A.; Gerna, G. Antibodies Against Neutralization Epitopes of Human Cytomegalovirus gH/gL/pUL128-130-131 Complex and Virus Spreading May Correlate with Virus Control In Vivo. J. Clin. Immunol. 2012, 32, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Buerger, I.; Reefschlaeger, J.; Bender, W.; Eckenberg, P.; Popp, A.; Weber, O.; Graeper, S.; Klenk, H.-D.; Ruebsamen-Waigmann, H.; Hallenberger, S. A Novel Nonnucleoside Inhibitor Specifically Targets Cytomegalovirus DNA Maturation via the UL89 and UL56 Gene Products. J. Virol. 2001, 75, 9077–9086. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Topic |
|---|---|
| Pepperl et al. 2000 [32] | DB induce both cellular and humoral immune responses |
| Pepperl-Klindworth et al. 2003 [31] | DB-mediated protein delivery |
| Mersseman et al. 2008 [30] | DB-mediated delivery of heterologous peptides into MHC-class I presentation |
| Becke et al. 2010 [29] | Induction of CD8 T cell responses against an IE1-peptide, delivered by recombinant DB |
| Sauer et al. 2013 [28] | Maturation and activation of monocyte-derived iDCs upon stimulation with DB (Figure 2) |
| Cayatte et al. 2013 [34] | Induction of broad humoral and cellular immune responses by HCMV strain Towne DB |
| Krömmelbein et al. 2015 [26] | Limited production of DB in CAP cells |
| Büscher et al. 2015 [27] | Conserved protein composition of DB of various HCMV strains |
| Schneider-Ohrum et al. 2016 [33] | Development of a scalable bioprocess for DB production |
| Lehmann et al. 2019 [35] | Pentamer-positive DB are superior to pentamer-negative DB for the induction of neutralizing antibodies |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogesch, P.; Penner, I.; Krauter, S.; Büscher, N.; Grode, L.; Aydin, I.; Plachter, B. Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine. Vaccines 2019, 7, 104. https://doi.org/10.3390/vaccines7030104
Gogesch P, Penner I, Krauter S, Büscher N, Grode L, Aydin I, Plachter B. Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine. Vaccines. 2019; 7(3):104. https://doi.org/10.3390/vaccines7030104
Chicago/Turabian StyleGogesch, Patricia, Inessa Penner, Steffi Krauter, Nicole Büscher, Leander Grode, Inci Aydin, and Bodo Plachter. 2019. "Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine" Vaccines 7, no. 3: 104. https://doi.org/10.3390/vaccines7030104
APA StyleGogesch, P., Penner, I., Krauter, S., Büscher, N., Grode, L., Aydin, I., & Plachter, B. (2019). Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine. Vaccines, 7(3), 104. https://doi.org/10.3390/vaccines7030104