Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress



Abstract
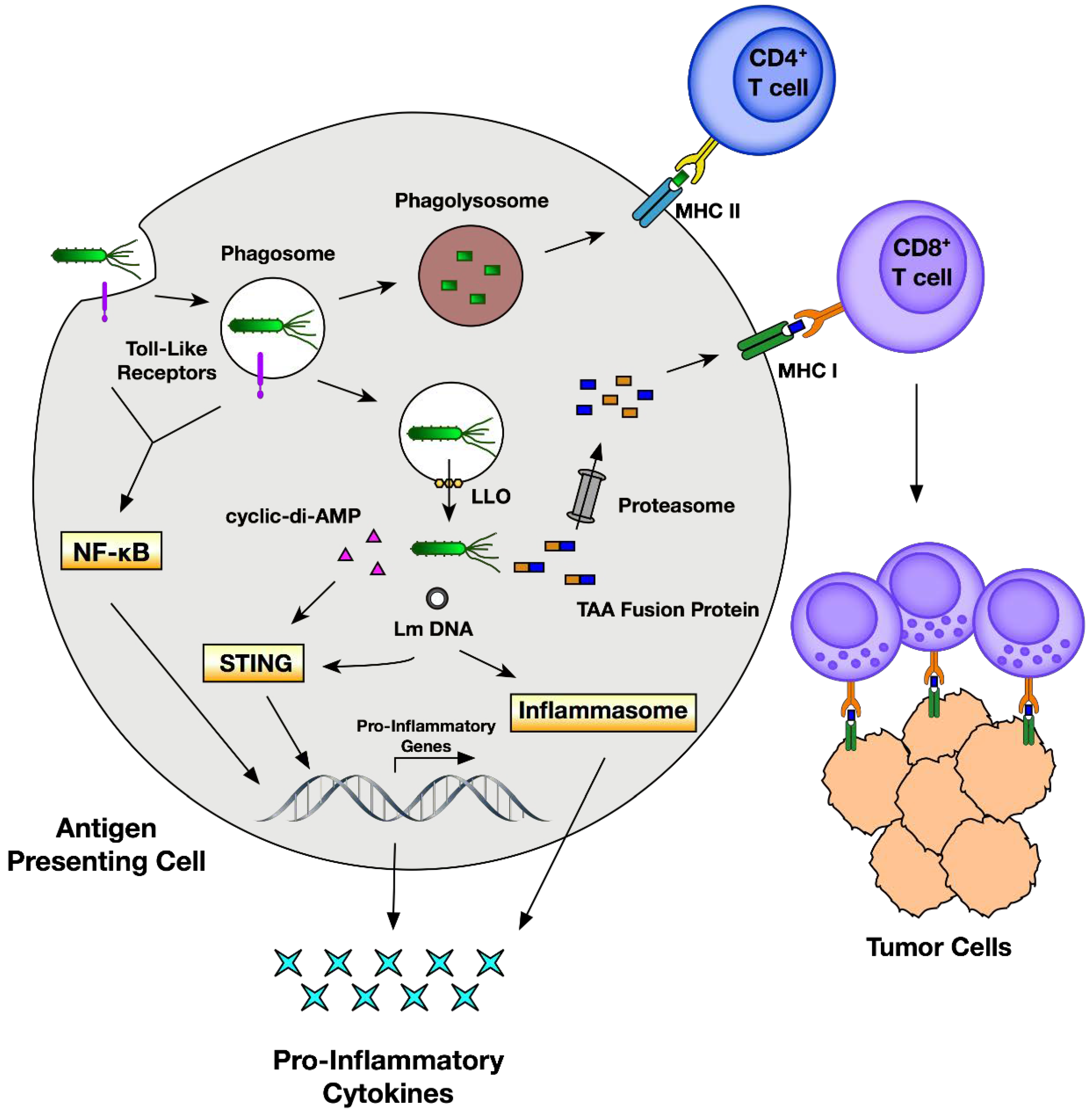
1. Introduction
2. Pathogenesis of Listeria Infection
3. Development of Attenuated Listeria Strains for Vaccination
3.1. Deletion of Virulence Genes
3.2. Episomal Replacement of Virulence or Metabolic Genes
3.3. Killed but Metabolically Active
4. Fusion with Listeria Antigens Enhances Antitumor Responses
4.1. Listeriolysin O
4.2. ActA
5. Listeria Vaccines in Clinical Trials
5.1. ADXS11-001 (Axalimogene Filolisbac [AXAL])
5.2. ADXS31-142
5.3. ADXS31-164
5.4. CRS-100 (ANZ-100)
5.5. CRS-207
5.6. ADU-623
5.7. JNJ-64041757 and JNJ-64041809
5.8. pLADD and ADXS-NEO
5.9. Adverse Effects of Listeria Vaccine Therapy
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Leong, M.L.; Hampl, J.; Liu, W.; Mathur, S.; Bahjat, K.S.; Luckett, W.; Dubensky, T.W.; Brockstedt, D.G. Impact of preexisting vector-specific immunity on vaccine potency: Characterization of Listeria monocytogenes-specific humoral and cellular immunity in humans and modeling studies using recombinant vaccines in mice. Infect. Immun. 2009, 77, 3958–3968. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.M.; Paterson, Y. Attenuated Listeria monocytogenes: A powerful and versatile vector for the future of tumor immunotherapy. Front. Cell. Infect. Microbiol. 2014, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas: With a report of ten original cases. Am. J. Med. Sci. 1893, 105, 487–511. [Google Scholar] [CrossRef]
- Mackaness, G.B. Cellular resistance to infection. J. Exp. Med. 1962, 116, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Schafer, R.; Portnoy, D.A.; Brassell, S.A.; Paterson, Y. Induction of a cellular immune response to a foreign antigen by a recombinant Listeria monocytogenes vaccine. J. Immunol. 1992, 149, 53–59. [Google Scholar] [PubMed]
- Pizarro-Cerdá, J.; Kühbacher, A.; Cossart, P. Entry of Listeria monocytogenes in mammalian epithelial cells: An updated view. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- De las Heras, A.; Cain, R.J.; Bielecka, M.K.; Vázquez-Boland, J.A. Regulation of Listeria virulence: PrfA master and commander. Curr. Opin. Microbiol. 2011, 14, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 2000, 103, 501–510. [Google Scholar] [CrossRef]
- Mengaud, J.; Ohayon, H.; Gounon, P.; Mege, R.-M.; Cossart, P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 1996, 84, 923–932. [Google Scholar] [CrossRef]
- Aubry, C.; Corr, S.C.; Wienerroither, S.; Goulard, C.; Jones, R.; Jamieson, A.M.; Decker, T.; O’Neill, L.A.J.; Dussurget, O.; Cossart, P. Both TLR2 and TRIF contribute to interferon-β production during Listeria infection. PLoS ONE 2012, 7, e33299. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, Y. Listeriolysin O as a strong immunogenic molecule for the development of new anti-tumor vaccines. Hum. Vaccines Immunother. 2013, 9, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Köster, S.; Van Pee, K.; Hudel, M.; Leustik, M.; Rhinow, D.; Kühlbrandt, W.; Chakraborty, T.; Yildiz, Ö. Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 2014, 5, 3690. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Sorbara, M.T.; Yang, C.; Tooze, S.A.; Philpott, D.J.; Girardin, S.E. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013, 32, 3066–3078. [Google Scholar] [CrossRef] [PubMed]
- Miles, B.A.; Monk, B.J.; Safran, H.P. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol. Oncol. Res. Pract. 2017, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Dubenksy, T.W.; Brockstedt, D.G. Clinical development of Listeria monocytogenes-based immunotherapies. Semin. Oncol. 2012, 39, 311–322. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.P.; Luo, S.; Ahmed-Qadri, F.; Zuck, M.; Thayer, E.F.; Goo, Y.A.; Hybiske, K.; Tong, L.; Woodward, J.J. Sensing of bacterial cyclic dinucleotides by the oxidoreductase RECON promotes NF-κB activation and shapes a proinflammatory antibacterial state. Immunity 2017, 46, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, A.; Gevaert, K.; Cossart, P.; Vandekerckhove, J.; Van Troys, M. Listeria comet tails: The actin-based motility machinery at work. Trends Cell Biol. 2008, 18, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, C.A.; Herzner, A.M.; Abdullah, Z.; Zillinger, T.; Jakobs, C.; Schuberth, C.; Coch, C.; Higgins, P.G.; Wisplinghoff, H.; Barchet, W.; et al. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS ONE 2013, 8, e62872. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, Z.; Schlee, M.; Roth, S.; Mraheil, M.A.; Barchet, W.; Böttcher, J.; Hain, T.; Geiger, S.; Hayakawa, Y.; Fritz, J.H.; et al. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J. 2012, 31, 4153–4164. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Prabakaran, T.; Laustsen, A.; Jørgensen, S.E.; Rahbæk, S.H.; Jensen, S.B.; Nielsen, R.; Leber, J.H.; Decker, T.; Horan, K.A.; et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014, 33, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Archer, K.A.; Durack, J.; Portnoy, D.A. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. 2014, 10, e1003861. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Bauernfeind, F.; Ablasser, A.; Hartmann, G.; Fitzgerald, K.A.; Latz, E.; Hornung, V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 2010, 40, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Brockstedt, D.G.; Giedlin, M.A.; Leong, M.L.; Bahjat, K.S.; Gao, Y.; Luckett, W.; Liu, W.; Cook, D.N.; Portnoy, D.A.; Dubensky, T.W. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 13832–13837. [Google Scholar] [CrossRef] [PubMed]
- Starks, H.; Bruhn, K.W.; Shen, H.; Barry, R.A.; Dubensky, T.W.; Brockstedt, D.; Hinrichs, D.J.; Higgins, D.E.; Miller, J.F.; Giedlin, M.; et al. Listeria monocytogenes as a vaccine vector: Virulence attenuation or existing antivector immunity does not diminish therapeutic efficacy. J. Immunol. 2004, 173, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Gunn, G.R.; Zubair, A.; Peters, C.; Pan, Z.K.; Wu, T.C.; Paterson, Y. Two Listeria monocytogenes vaccine vectors that express different molecular forms of human papilloma virus-16 (HPV-16) E7 induce qualitatively different T cell immunity that correlates with their ability to induce regression of established tumors immortalized by HPV-16. J. Immunol. 2001, 167, 6471–6479. [Google Scholar] [PubMed]
- Freitag, N.E.; Rong, L.; Portnoy, D.A. Regulation of the prfA transcriptional activator of Listeria monocytogenes: Multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect. Immun. 1993, 61, 2537–2544. [Google Scholar] [PubMed]
- Singh, R.; Dominiecki, M.E.; Jaffee, E.M.; Paterson, Y. Fusion to Listeriolysin O and delivery by Listeria monocytogenes enhances the immunogenicity of HER-2/neu and reveals subdominant epitopes in the FVB/N mouse. J. Immunol. 2005, 175, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, V.; Reyes-Reyes, M.; Wallecha, A.; Rivera, S.; Paterson, Y.; Maciag, P. Development of a Listeria monocytogenes based vaccine against prostate cancer. Cancer Immunol. Immunother. 2008, 57, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, X.; Higgins, D.E.; Frankel, F.R. Conditional lethality yields a new vaccine strain of Listeria monocytogenes for the induction of cell-mediated immunity. Infect. Immun. 2005, 73, 5065–5073. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.J.; Bouwer, H.G.; Portnoy, D.A.; Frankel, F.R. Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires D-alanine for growth. Infect. Immun. 1998, 66, 3552–3561. [Google Scholar] [PubMed]
- Verch, T.; Pan, Z.-K.; Paterson, Y. Listeria monocytogenes-based antibiotic resistance gene-free antigen delivery system applicable to other bacterial vectors and DNA vaccines. Infect. Immun. 2004, 72, 6418–6425. [Google Scholar] [CrossRef] [PubMed]
- Rayevskaya, M.V.; Frankel, F.R. Systemic immunity and mucosal immunity are induced against human immunodeficiency virus Gag protein in mice by a new hyperattenuated strain of Listeria monocytogenes. J. Virol. 2001, 75, 2786–2791. [Google Scholar] [CrossRef] [PubMed]
- Wallecha, A.; Maciag, P.C.; Rivera, S.; Paterson, Y.; Shahabi, V. Construction and characterization of an attenuated Listeria monocytogenes strain for clinical use in cancer immunotherapy. Clin. Vaccine Immunol. 2009, 16, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, V.; Seavey, M.M.; Maciag, P.C.; Rivera, S.; Wallecha, A. Development of a live and highly attenuated Listeria monocytogenes-based vaccine for the treatment of HER2/neu-overexpressing cancers in human. Cancer Gene Ther. 2011, 18, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Brunt, L.M.; Portnoy, D.A.; Unanue, E.R. Presentation of Listeria monocytogenes to CD8+ T cells requires secretion of hemolysin and intracellular bacterial growth. J. Immunol. 1990, 145, 3540–3546. [Google Scholar] [PubMed]
- Bahjat, K.S.; Liu, W.; Lemmens, E.E.; Schoenberger, S.P.; Portnoy, D.A.; Dubensky, T.W.; Brockstedt, D.G. Cytosolic entry controls CD8+-T-cell potency during bacterial infection. Infect. Immun. 2006, 74, 6387–6397. [Google Scholar] [CrossRef] [PubMed]
- Sacco, J.J.; Evans, M.; Harrington, K.J.; Man, S.; Powell, N.; Shaw, R.J.; Jones, T.M. Systemic listeriosis following vaccination with the attenuated Listeria monocytogenes therapeutic vaccine, ADXS11-001. Hum. Vaccinces Immunother. 2016, 12, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Brockstedt, D.G.; Bahjat, K.S.; Giedlin, M.A.; Liu, W.; Leong, M.; Luckett, W.; Gao, Y.; Schnupf, P.; Kapadia, D.; Castro, G.; et al. Killed but metabolically active microbes: A new vaccine paradigm for eliciting effector T-cell responses and protective immunity. Nat. Med. 2005, 11, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Lauer, P.; Hanson, B.; Lemmens, E.E.; Liu, W.; Luckett, W.S.; Leong, M.L.; Allen, H.E.; Skoble, J.; Bahjat, K.S.; Freitag, N.E.; et al. Constitutive Activation of the PrfA regulon enhances the potency of vaccines based on live-attenuated and killed but metabolically active Listeria monocytogenes strains. Infect. Immun. 2008, 76, 3742–3753. [Google Scholar] [CrossRef] [PubMed]
- Dubensky, T.W.; Skoble, J.; Lauer, P.; Brockstedt, D.G. Killed but metabolically active vaccines. Curr. Opin. Biotechnol. 2012, 23, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, G.; Paterson, Y.; Kos, F.J.; Portnoy, D.A. Delivery of a viral antigen to the class I processing and presentation pathway by Listeria monocytogenes. J. Exp. Med. 1994, 180, 2209–2218. [Google Scholar] [CrossRef] [PubMed]
- Sewell, D.A.; Shahabi, V.; Gunn, G.R.; Pan, Z.-K.; Dominiecki, M.E.; Paterson, Y. Recombinant Listeria vaccines containing PEST sequences are potent immune adjuvants for the tumor-associated antigen human papillomavirus-16 E7. Cancer Res. 2004, 64, 8821–8825. [Google Scholar] [CrossRef] [PubMed]
- Decatur, A.L.; Portnoy, D.A. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 2000, 290, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Zhou, J.; Varshavsky, A.; Portnoy, D.A. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect. Immun. 2007, 75, 5135–5147. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Portnoy, D.A.; Decatur, A.L. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: Role of the PEST-like sequence. Cell. Microbiol. 2006, 8, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Wallecha, A.; Wood, L.; Pan, Z.-K.; Maciag, P.C.; Shahabi, V.; Paterson, Y. Listeria monocytogenes-derived listeriolysin O has pathogen-associated molecular pattern-like properties independent of its hemolytic ability. Clin. Vaccine Immunol. 2013, 20, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Paterson, Y. CD4+CD25+ regulatory T cells that secrete TGFβ and IL-10 are preferentially induced by a vaccine vector. J. Immunother. 2004, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ozbun, L.; Chong, N.; Wallecha, A.; Berzofsky, J.A.; Khleif, S.N. Episomal expression of truncated listeriolysin O in LmddA-LLO-E7 vaccine enhances antitumor efficacy by preferentially inducing expansions of CD4+FoxP3- and CD8+ T cells. Cancer Immunol. Res. 2014, 2, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Wallecha, A.; Singh, R.; Malinina, I. Listeria monocytogenes (Lm)-LLO immunotherapies reduce the immunosuppressive activity of myeloid-derived suppressor cells and regulatory T cells in the tumor microenvironment. J. Immunother. 2013, 36, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Souders, N.C.; Sewell, D.A.; Pan, Z.-K.; Hussain, S.F.; Rodriguez, A.; Wallecha, A.; Paterson, Y. Listeria-based vaccines can overcome tolerance by expanding low avidity CD8+ T cells capable of eradicating a solid tumor in a transgenic mouse model of cancer. Cancer Immun 2007, 7, 2. [Google Scholar] [PubMed]
- Jia, Q.; Dillon, B.J.; Masleša-galic, S.; Horwitz, M.A. Listeria-vectored vaccine expressing the Mycobacterium tuberculosis 30-kilodalton major secretory protein via the constitutively active prfA* regulon boosts Mycobacterium bovis BCG efficacy against tuberculosis. Infect. Immun. 2017, 85, 1–21. [Google Scholar]
- Sewell, D.A.; Douven, D.; Pan, Z.-K.; Rodriguez, A.; Paterson, Y. Regression of HPV-positive tumors treated with a new Listeria monocytogenes vaccine. Arch. Otolaryngol. Head Neck Surg. 2004, 130, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Sewell, D.A.; Pan, Z.K.; Paterson, Y. Listeria-based HPV-16 E7 vaccines limit autochthonous tumor growth in a transgenic mouse model for HPV-16 transformed tumors. Vaccine 2008, 26, 5315–5320. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.M.; Pan, Z.-K.; Shahabi, V.; Paterson, Y. Listeria-derived ActA is an effective adjuvant for primary and metastatic tumor immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Moors, M.A.; Auerbuch, V.; Portnoy, D.A. Stability of the Listeria monocytogenes ActA protein in mammalian cells is regulated by the N-end rule pathway. Cell. Microbiol. 1999, 1, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Lauer, P.; Chow, M.Y.N.; Loessner, M.J.; Portnoy, D.A.; Calendar, R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 2002, 184, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.S.; Stern, P.L. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat. Rev. Cancer 2018, 18, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; De Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Maciag, P.C.; Radulovic, S.; Rothman, J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: A Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine 2009, 27, 3975–3983. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Mehta, A.; Jain, M.; Gupta, S.; Nagarkar, R.V.; John, S.; Petit, R. A randomized phase 2 study of ADXS11-001 Listeria monocytogenes-listeriolysin O immunotherapy with or without cisplatin in treatment of advanced cervical cancer. Int. J. Gynecol. Cancer 2018, 28, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Safran, H.; Leonard, K.-L.; Perez, K.; Vrees, M.; Klipfel, A.; Schechter, S.; Oldenburg, N.; Roth, L.; Shah, N.; Rosati, K.; et al. Tolerability of ADXS11-001 Lm-LLO Listeria-based immunotherapy with mitomycin, fluorouracil, and radiation for anal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Petit, R.; Stein, M.; Tutrone, R.; Mega, A.; Agarwal, M.; Fong, L.; Haas, N. ADXS-PSA immunotherapy increases the magnitude and quality of prostate cancer-antigen-specific T cell responses in patients with metastatic castration-resistant prostate cancer. In Proceedings of the Society for Immunotherapy of Cancer’s (SITC) 32nd Annual Meeting 2017, National Harbor, MD, USA, 10–12 November 2017. [Google Scholar]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Ko, A.H.; Wainberg, Z.A.; Picozzi, V.J.; Kindler, H.L. Results from a phase 2b, randomized, multicenter study of GVAX pancreas and CRS-207 compared to chemotherapy in adults with previously-treated. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Jahan, T.; Hassan, R.; Alley, E.; Kindler, H.; Antonia, S.; Whiting, C.; Coussens, L.; Murphy, A.L.; Thomas, A.; Brockstedt, D.G. 208O_PR: CRS-207 with chemotherapy (chemo) in malignant pleural mesothelioma (MPM): Results from a phase 1b trial. J. Thorac. Oncol. 2016, 11, S156. [Google Scholar] [CrossRef]
- Crittenden, M.; Bahjat, K.S.; Li, R.; Gore, P.; Fountain, C.; Hanson, B.; Skoble, J.; Lauer, P.; Murphy, A.L.; Dubensky, T.; et al. Phase I study of safety and immunogenicity of ADU-623, a live-attenuated Listeria monocytogenes vaccine (ΔactA/ΔinlB) expressing EGFRVIII and NY-ESO-1, in patients with who grade III/IV astrocytomas. J. Immunother. Cancer 2015, 3, p162. [Google Scholar] [CrossRef]
- Brahmer, J.; Johnson, M.; Awad, M.; Rajan, A.; Allred, A. P2. 07-058 first-in-human study of JNJ-64041757, a live attenuated Listeria monocytogenes immunotherapy, for non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, s2151. [Google Scholar] [CrossRef]
- Mkrtichyan, M.; Chong, N.; Eid, R.A.; Wallecha, A.; Singh, R.; Rothman, J.; Khleif, S.N. Anti-PD-1 antibody significantly increases therapeutic efficacy of Listeria monocytogenes (Lm)-LLO immunotherapy. J. Immunother. Cancer 2013, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Karan, D. Prostate immunotherapy: Should all guns be aimed at the prostate-specific antigen? Immunotherapy 2013, 5, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Hannan, R.; Zhang, H.; Wallecha, A.; Singh, R.; Liu, L.; Cohen, P.; Alfieri, A.; Rothman, J.; Guha, C. Combined immunotherapy with Listeria monocytogenes-based PSA vaccine and radiation therapy leads to a therapeutic response in a murine model of prostate cancer. Cancer Immunol. Immunother. 2012, 61, 2227–2238. [Google Scholar] [CrossRef] [PubMed]
- Bongiorno, E.K.; Baybutt, T.; Protocarrero, C.; Snook, A.; Dicker, A.; Hattier, G.; Addya, S.; Hayes, S.; Rodeck, U. Molecular signatures of combination immunotherapy of prostate cancer using a Listeria-based PSA vaccine and radiation. In Proceedings of the Society for Immunotherapy of Cancer’s (SITC) 32nd Annual Meeting 2017, National Harbor, MD, USA, 10–12 November 2017. [Google Scholar]
- Omar, N.; Yan, B.; Salto, M. HER2: An emerging biomarker in non-breast and non-gastric cancers. Pathogenesis 2015, 2, 1–9. [Google Scholar] [CrossRef]
- Scotlandi, K.; Manara, M.C.; Hattinger, C.M.; Benini, S.; Perdichizzi, S.; Pasello, M.; Bacci, G.; Zanella, L.; Bertoni, F.; Picci, P.; et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur. J. Cancer 2005, 41, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Fracol, M.; McMillan, M.T.; Berk, E.; Xu, S.; Goodman, N.; Lewis, D.A.; DeMichele, A.; Czerniecki, B.J. Association of depressed anti-HER2 T-helper type 1 response with recurrence in patients with completely treated HER2-positive breast cancer: Role for immune monitoring. JAMA Oncol. 2016, 2, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Nocera, N.F.; Lee, M.C.; De La Cruz, L.M.; Rosemblit, C.; Czerniecki, B.J. Restoring lost anti-HER-2 Th1 immunity in breast cancer: A crucial role for Th1 cytokines in therapy and prevention. Front. Pharmacol. 2016, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Seavey, M.M.; Pan, Z.-K.; Maciag, P.C.; Wallecha, A.; Rivera, S.; Paterson, Y.; Shahabi, V. A novel human HER-2/neu chimeric molecule expressed by Listeria monocytogenes can elicit potent HLA-A2 restricted CD8-positive T cell responses and impact the growth and spread of HER-2/neu-positive breast tumors. Clin. Cancer Res. 2009, 15, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Mason, N.J.; Gnanandarajah, J.S.; Engiles, J.B.; Gray, F.; Laughlin, D.; Gaurnier-Hausser, A.; Wallecha, A.; Huebner, M.; Paterson, Y. Immunotherapy with a HER2-targeting Listeria induces HER2-specific immunity and demonstrates potential therapeutic effects in a phase I trial in canine osteosarcoma. Clin. Cancer Res. 2016, 22, 4380–4390. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Paterson, Y. Vaccination strategy determines the emergence and dominance of CD8+ T-cell epitopes in a FVB/N rat HER-2/neu mouse model of breast cancer. Cancer Res. 2006, 66, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Pondé, N.F.; Lambertini, M.; De Azambuja, E. Twenty years of anti-HER2 therapy-associated cardiotoxicity. ESMO Open 2016, 1, e000073. [Google Scholar] [CrossRef] [PubMed]
- Bahjat, K.S.; Prell, R.A.; Allen, H.E.; Liu, W.; Lemmens, E.E.; Leong, M.L.; Portnoy, D.A.; Dubensky, T.W.; Brockstedt, D.G.; Giedlin, M.A. Activation of immature hepatic NK cells as immunotherapy for liver metastatic disease. J. Immunol. 2007, 179, 7376–7384. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin immunotherapy for cancer: Ready for prime time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: A pilot study of safety, feasibility, and immune activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 based immunotherapy of cancer: Current perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 569. [Google Scholar] [CrossRef] [PubMed]
- Coder, B.; Phee, H.; Mattershead, C.; Kelkar, D.; Filippova, E.; Ju, X.; Vander Lught, B.; Pryshchep, O.; Lesch, J.; Liu, X.; et al. Neoantigens that fail to elicit measurable T cell responses following peptide immunization can control tumor growth when delivered using a Listeria-based immunotherapy. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Coder, B.; Villarreal, D.; Armington, S.; Filippova, E.; L’Huillier, A.; Kelkar, D.; Ju, X.; Mattershead, C.; Balli, D.; Ramos, K.; et al. Targeting frameshift mutations with a Listeria monocytogenes immunotherapy drives neoantigens-specific anti-tumor immunity in the MC38 and CT26 mouse tumor models. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Villarreal, D.; Coder, B.; Armington, S.; L’Huillier, A.; Mattershead, C.; Filippova, E.; Thambi, N.; Ramos, K.; Balli, D.; Petit, R.; et al. Targeting shared hotspot cancer mutations with a Listeria monocytogenes immunotherapy induce potent anti-tumor immunity. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Denham, J.D.; Lee, D.H.; Castro, M.; Pandya, S.; Aslam, S.; Nanjappa, S.; Greene, J.N. Two cases of disseminated infection following live organism anti-cancer vaccine administration in cancer patients. Int. J. Infect. Dis. 2018, 72, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Seavey, M.M.; Paterson, Y. Antiangiogenesis immunotherapy induces epitope spreading to HER-2/neu resulting in breast tumor immunoediting. Breast Cancer (Dove Med. Press) 2009, 1, 19–30. [Google Scholar] [PubMed]
- Seavey, M.M.; Maciag, P.C.; Al-Rawi, N.; Sewell, D.; Paterson, Y. An anti-vascular endothelial growth factor receptor 2/fetal liver kinase-1 Listeria monocytogenes anti-angiogenesis cancer vaccine for the treatment of primary and metastatic HER-2/neu+ breast tumors in a mouse model. J. Immunol. 2009, 182, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.M.; Pan, Z.-K.; Guirnalda, P.; Tsai, P.; Seavey, M.; Paterson, Y. Targeting tumor vasculature with novel Listeria-based vaccines directed against CD105. Cancer Immunol. Immunother. 2011, 60, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Maciag, P.C.; Seavey, M.M.; Pan, Z.-K.; Ferrone, S.; Paterson, Y. Cancer immunotherapy targeting the high molecular weight melanoma-associated antigen protein results in a broad antitumor response and reduction of pericytes in the tumor vasculature. Cancer Res. 2008, 68, 8066–8075. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Vaccine | Antigen | Cancer Indication | Drug Combination | Phase | Completion | Studies | Identifier | Company |
|---|---|---|---|---|---|---|---|---|
| ADXS11-001 | HPV 16 E7 | Cervical | Vaccine alone | I | 2009 | [65] | N/A | Advaxis |
| I/II | 12/2018 1 | NCT02164461 | ||||||
| II | 04/2016 | NCT01116245 | ||||||
| II | 10/2018 1 | NCT01266460 | ||||||
| III | 06/2021 | NCT02853604 | ||||||
| Vaccine + chemotherapy | II | 2017 | [66] | CTRI/2010/091/001232 | ||||
| Cervical and Oropharyngeal | Vaccine + αPD-1 | I/II | 12/2019 | NCT02291055 | ||||
| Oropharyngeal | Vaccine alone | I | 11/2014 | NCT01598792 | ||||
| II | 08/2019 | NCT02002182 | ||||||
| Anal | Vaccine + chemoradiation | I/II | 02/2018 | [67] | NCT01671488 | |||
| Vaccine alone | II | 03/2022 | NCT02399813 | |||||
| Lung | Vaccine + chemotherapy | II | 03/2019 1 | NCT02531854 | ||||
| ADXS31-142 | PSA | Prostate | Vaccine + αPD-1 | I/II | 12/2019 | [68] | NCT02325557 | |
| ADXS31-164 | HER2 | HER2 + Solid Tumors | Vaccine alone | I/II | 12/2018 | NCT02386501 | ||
| ADXS-NEO | Personal Neo-antigens | Colon, Lung, Head and Neck | Vaccine alone | I | 09/2020 | NCT03265080 | ||
| CRS-100 (ANZ-100) | None | Hepatic metastases | Vaccine alone | I | 02/2008 | [69] | NCT00327652 | Aduro |
| CRS-207 | Mesothelin | Pancreatic, Lung, Ovarian and Mesothelioma | Vaccine alone | I | 02/2009 | [69] | NCT00585845 | |
| Pancreatic | Vaccine + Cy/GVAX | II | 08/2016 | [70] | NCT02004262 | |||
| Vaccine + Cy/GVAX | II | 02/2017 | [71] | NCT01417000 | ||||
| Vaccine + αPD-1 + Cy/GVAX | II | 01/2019 | NCT02243371 | |||||
| Vaccine + αPD-1 + αCTLA-4 + Cy/GVAX | II | 10/2019 | NCT03190265 | |||||
| Vaccine + αPD-1 + IDO1 inhibitor + Cy/GVAX | II | 06/2023 | NCT03006302 | |||||
| Ovarian, Fallopian and Peritoneal | Vaccine + αPD-1 + IDO1 inhibitor | I/II | 12/2018 | NCT02575807 | ||||
| Gastroesophageal | Vaccine + αPD-1 | II | 05/2019 | NCT03122548 | ||||
| Mesothelioma | Vaccine + chemotherapy | I | 12/2018 | [72] | NCT01675765 | |||
| Vaccine + αPD-1 | II | 03/2019 1 | NCT03175172 | |||||
| ADU-623 | EGFRvIII and NY-ESO-1 | Brain | Vaccine alone | I | 12/2018 | [73] | NCT01967758 | |
| pLADD | Personal Neo-antigens | Colorectal | Vaccine alone | I | 12/2020 | NCT03189030 | ||
| JNJ-64041757(ADU-214) | EGFRvIII and mesothelin | Lung | Vaccine alone | I | 03/2020 | [74] | NCT02592967 | Janssen 2 |
| Vaccine + αPD-1 | I/II | 03/2022 | NCT03371381 | |||||
| JNJ-64041809(ADU-741) | Multiple prostate antigens | Prostate | Vaccine alone | I | 06/2018 | NCT02625857 | ||
| Vaccine + anti-androgen | II | 09/2018 | NCT02906605 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flickinger, J.C., Jr.; Rodeck, U.; Snook, A.E. Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress. Vaccines 2018, 6, 48. https://doi.org/10.3390/vaccines6030048
Flickinger JC Jr., Rodeck U, Snook AE. Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress. Vaccines. 2018; 6(3):48. https://doi.org/10.3390/vaccines6030048
Chicago/Turabian StyleFlickinger, John C., Jr., Ulrich Rodeck, and Adam E. Snook. 2018. "Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress" Vaccines 6, no. 3: 48. https://doi.org/10.3390/vaccines6030048
APA StyleFlickinger, J. C., Jr., Rodeck, U., & Snook, A. E. (2018). Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress. Vaccines, 6(3), 48. https://doi.org/10.3390/vaccines6030048