Improved Induction of Anti-Melanoma T Cells by Adenovirus-5/3 Fiber Modification to Target Human DCs

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Monocyte-Derived DC Generation
2.3. Flow Cytometric Phenotypic Analyses
2.4. Generation of Adenoviral Vectors
2.5. In Vitro Ad Transduction
2.6. In Vitro Induction of MART-1-Specific CD8+ Effector T Cell
2.7. Preparation, Transduction and Culture of Sentinel Lymph Node Single-Cell Suspensions
2.8. Intracellular Interferon γ (IFN-γ) Detection
2.9. Functional Avidity Analysis
2.10. Cytotoxicity Assay
2.11. Statistical Analysis
3. Results
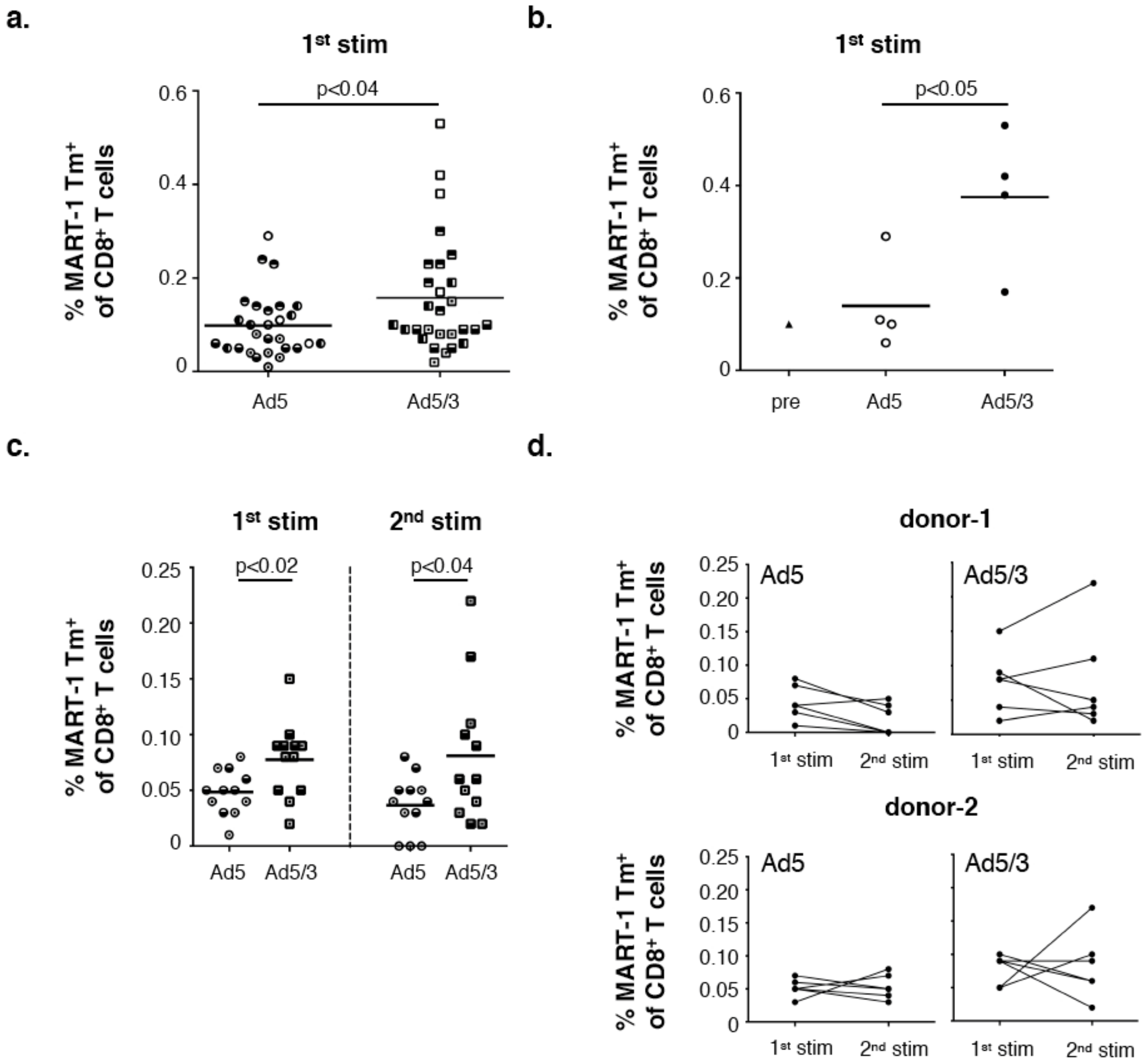
3.1. Superior Efficiency of MART-126–35 Specific CD8+ T Cell Priming by Ad5/3-MART-1 Transduced MoDCs as Compared to Ad5-MART-1 Transduced MoDCs
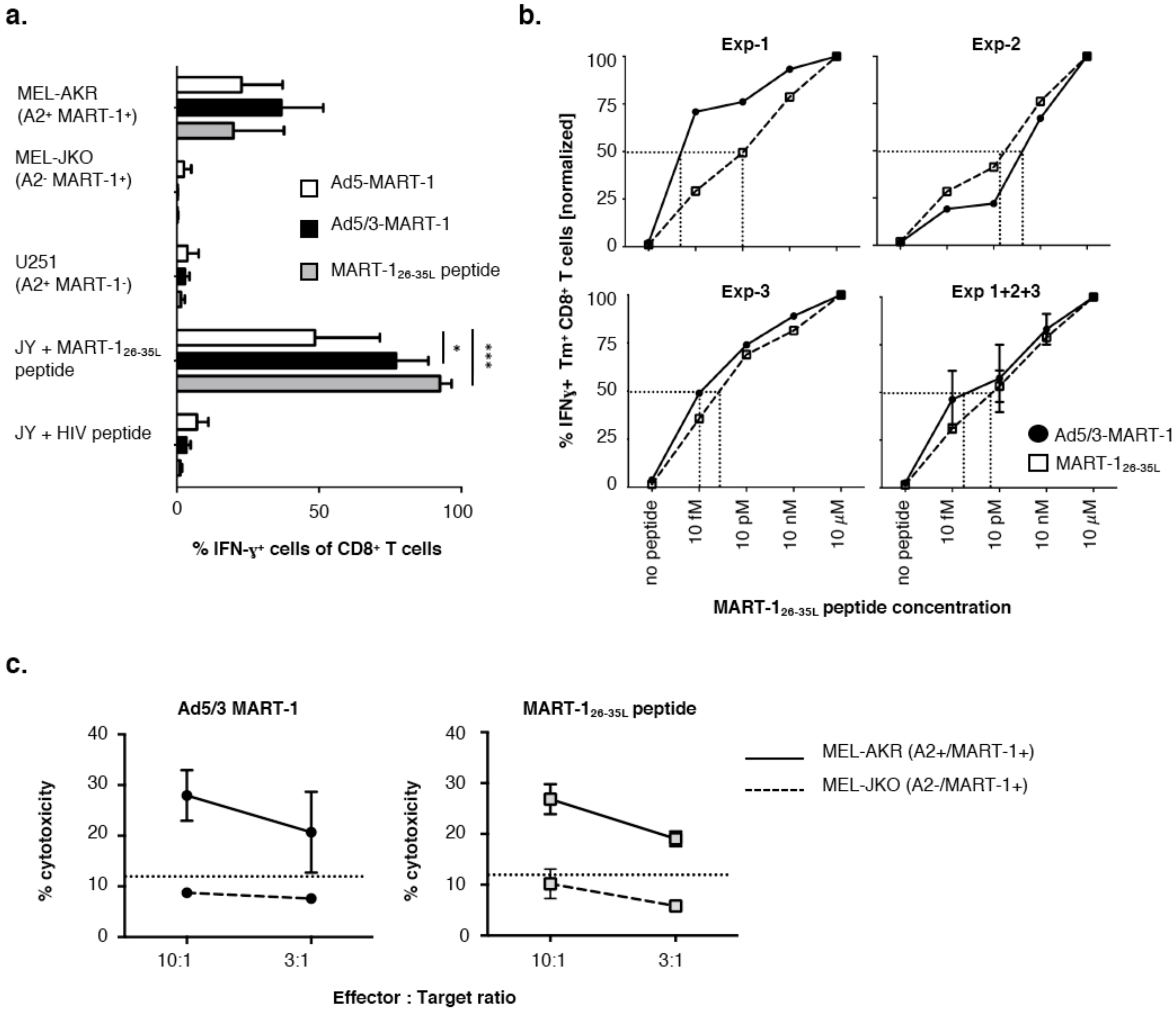
3.2. T Cells Primed with Ad5/3-MART-1 Transduced MoDCs Recognize Endogenously Processed MART-1
3.3. Functional Avidity of MART-126–35L-Specific CD8+ T Cells Primed by Ad5/3-MART-1 Targeted MoDCs
3.4. Melanoma Cytolysis by T Cells Primed with Ad5/3-MART-1 Transduced MoDCs
3.5. Superior Expansion of MART-1 Specific CD8+ T Cells Residing in Melanoma SLN Upon Ad5/3-MART-1 Infection as Compared to Ad5-MART-1
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Capone, M.; Urba, W.J.; Bifulco, C.B.; Botti, G.; Lugli, A.; Marincola, F.M.; Ciliberto, G.; Galon, J.; Fox, B.A. The additional facet of immunoscore: Immunoprofiling as a possible predictive tool for cancer treatment. J. Transl. Med. 2013, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H.; Comin-Anduix, B.; Vujanovic, L.; Lee, Y.; Dissette, V.B.; Yang, J.-Q.; Vu, H.T.; Seja, E.; Oseguera, D.K.; Potter, D.M.; et al. Adenovirus MART-1-engineered autologous dendritic cell vaccine for metastatic melanoma. J. Immunother. 2008, 31, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Redman, B.G.; Chang, A.E.; Whitfield, J.; Esper, P.; Jiang, G.; Braun, T.; Roessler, B.; Mulé, J.J. Phase Ib trial assessing autologous, tumor-pulsed dendritic cells as a vaccine administered with or without IL-2 in patients with metastatic melanoma. J. Immunother. 2008, 31, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, P.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Boullart, A.C.I.; Kok, E.; van Rossum, M.M.; Strijk, S.; Eijckeler, F.; Bonenkamp, J.J.; Jacobs, J.F.M.; et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin. Cancer Res. 2009, 15, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.W.; Palucka, A.K.; Paczesny, S.; Dhodapkar, M.; Johnston, D.A.; Burkeholder, S.; Ueno, H.; Banchereau, J. Long-term outcomes in patients with metastatic melanoma vaccinated with melanoma peptide-pulsed CD34(+) progenitor-derived dendritic cells. Cancer Immunol. Immunother. 2006, 55, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; de Vries, I.J.M.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Publ. Group 2007, 7, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Dendritic cells in vivo: A key target for a new vaccine science. Immunity 2008, 29, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Qian, Y.; Fondel, S.; Brueck, J.; Becker, C.; Enk, A.H. Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 2005, 65, 7007–7012. [Google Scholar] [CrossRef] [PubMed]
- Breckpot, K.; Aerts, J.L.; Thielemans, K. Lentiviral vectors for cancer immunotherapy: Transforming infectious particles into therapeutics. Gene Ther. 2007, 14, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Altin, J.G.; Parish, C.R. Liposomal vaccines—Targeting the delivery of antigen. Methods 2006, 40, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Khare, R.; Chen, C.Y.; Weaver, E.A.; Barry, M.A. Advances and future challenges in adenoviral vector pharmacology and targeting. Curr. Gene Ther. 2011, 11, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Lotem, M.; Zhao, Y.; Riley, J.; Hwu, P.; Morgan, R.A.; Rosenberg, S.A.; Parkhurst, M.R. Presentation of tumor antigens by dendritic cells genetically modified with viral and nonviral vectors. J. Immunother. 2006, 29, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Tuettenberg, A.; Jonuleit, H.; Tüting, T.; Brück, J.; Knop, J.; Enk, A.H. Priming of T cells with Ad-transduced DC followed by expansion with peptide-pulsed DC significantly enhances the induction of tumor-specific CD8+ T cells: Implications for an efficient vaccination strategy. Gene Ther. 2003, 10, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Yang, J.C.; Kawakami, Y.; Spiess, P.; Wadsworth, S.C.; Cardoza, L.M.; Couture, L.A.; Smith, A.E.; Rosenberg, S.A. Antigen-specific tumor vaccines. Development and characterization of recombinant adenoviruses encoding MART1 or gp100 for cancer therapy. J. Immunol. 1996, 156, 700–710. [Google Scholar] [PubMed]
- Rea, D.; Havenga, M.J.; van Den Assem, M.; Sutmuller, R.P.; Lemckert, A.; Hoeben, R.C.; Bout, A.; Melief, C.J.; Offringa, R. Highly efficient transduction of human monocyte-derived dendritic cells with subgroup B fiber-modified adenovirus vectors enhances transgene-encoded antigen presentation to cytotoxic T cells. J. Immunol. 2001, 166, 5236–5244. [Google Scholar] [CrossRef] [PubMed]
- Noureddini, S.C.; Curiel, D.T. Genetic targeting strategies for adenovirus. Mol. Pharm. 2005, 2, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Tian, J.; Lozier, J.N.; Byrnes, A.P. Severe pulmonary pathology after intravenous administration of vectors in cirrhotic rats. Mol. Ther. 2004, 9, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Aichele, P.; Brduscha-Riem, K.; Zinkernagel, R.M.; Hengartner, H.; Pircher, H. T cell priming versus T cell tolerance induced by synthetic peptides. J. Exp. Med. 1995, 182, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Raki, M.; Sarkioja, M.; Escutenaire, S.; Kangasniemi, L.; Haavisto, E.; Kanerva, A.; Cerullo, V.; Joensuu, T.; Oksanen, M.; Pesonen, S.; et al. Switching the fiber knob of oncolytic adenoviruses to avoid neutralizing antibodies in human cancer patients. J. Gene Med. 2011, 13, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Nuzzo, M.; Urbanelli, L.; Monaci, P. General strategy for broadening adenovirus tropism. J. Virol. 2003, 77, 11094–11104. [Google Scholar] [CrossRef] [PubMed]
- Short, J.J.; Pereboev, A.V.; Kawakami, Y.; Vasu, C.; Holterman, M.J.; Curiel, D.T. Adenovirus serotype 3 utilizes CD80 (B7.1) and CD86 (B7.2) as cellular attachment receptors. Virology 2004, 322, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, R.; Lindenberg, J.J.; Oosterhoff, D.; van den Tol, M.P.; Rosalia, R.A.; Murakami, M.; Everts, M.; Scheffer, G.L.; Scheper, R.J.; de Gruijl, T.D.; et al. Selective transduction of mature DC in human skin and lymph nodes by CD80/CD86-targeted fiber-modified adenovirus-5/3. J. Immunother. 2009, 32, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Morton, D.L.; Stern, S.; Lana, A.M.; Essner, R.; Wen, D.R. Sentinel lymph nodes show profound downregulation of antigen-presenting cells of the paracortex: Implications for tumor biology and treatment. Mod. Pathol. 2001, 14, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Huang, R.-R.; Lee, J.; Itakura, E.; Leong, S.P.L.; Essner, R. Tumour-induced immune modulation of sentinel lymph nodes. Nat. Rev. Immunol. 2006, 6, 659–670. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, T.D.; van de Ven, R. Chapter six—Adenovirus-based immunotherapy of cancer: Promises to keep. Adv. Cancer Res. 2012, 115, 147–220. [Google Scholar] [PubMed]
- Radosević, K.; Rodriguez, A.; Lemckert, A.A.C.; van der Meer, M.; Gillissen, G.; Warnar, C.; von Eyben, R.; Pau, M.G.; Goudsmit, J. The Th1 immune response to Plasmodium falciparum circumsporozoite protein is boosted by adenovirus vectors 35 and 26 with a homologous insert. Clin. Vaccine Immunol. 2010, 17, 1687–1694. [Google Scholar]
- Rodriguez, A.; Mintardjo, R.; Tax, D.; Gillissen, G.; Custers, J.; Pau, M.G.; Klap, J.; Santra, S.; Balachandran, H.; Letvin, N.L.; et al. Evaluation of a prime-boost vaccine schedule with distinct adenovirus vectors against malaria in rhesus monkeys. Vaccine 2009, 27, 6226–6233. [Google Scholar] [CrossRef] [PubMed]
- Kahl, C.A.; Bonnell, J.; Hiriyanna, S.; Fultz, M.; Nyberg-Hoffman, C.; Chen, P.; King, C.R.; Gall, J.G.D. Potent immune responses and in vitro pro-inflammatory cytokine suppression by a novel adenovirus vaccine vector based on rare human serotype 28. Vaccine 2010, 28, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Molenkamp, B.G.; Sluijter, B.J.R.; van Leeuwen, P.A.M.; Santegoets, S.J.A.M.; Meijer, S.; Wijnands, P.G.J.T.B.; Haanen, J.B.A.G.; van den Eertwegh, A.J.M.; Scheper, R.J.; de Gruijl, T.D. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clin. Cancer Res. 2008, 14, 4532–4542. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, M.H.; Hooijberg, E.; Ruizendaal, J.J.; van der Weide, M.M.; Kueter, E.; Bakker, A.Q.; Schumacher, T.N.; Spits, H. Enrichment of an antigen-specific T cell response by retrovirally transduced human dendritic cells. Cell. Immunol. 1999, 195, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, M.W.J.; Scholten, K.B.J.; Kueter, E.W.M.; Ruizendaal, J.J.; Meijer, C.J.L.M.; Hooijberg, E. In vitro generation and life span extension of human papillomavirus type 16-specific, healthy donor-derived CTL clones. J. Immunol. 2003, 171, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Hangalapura, B.N.; Oosterhoff, D.; Aggarwal, S.; Wijnands, P.G.J.T.B.; van de Ven, R.; Santegoets, S.J.A.M.; van den Tol, M.P.; Hooijberg, E.; Pereboev, A.; van den Eertwegh, A.J.M.; et al. Selective transduction of dendritic cells in human lymph nodes and superior induction of high-avidity melanoma-reactive cytotoxic T cells by a CD40-targeted adenovirus. J. Immunother. 2010, 33, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Yssel, H.; De Vries, J.E.; Koken, M.; Van Blitterswijk, W.; Spits, H. Serum-free medium for generation and propagation of functional human cytotoxic and helper T cell clones. J. Immunol. Methods 1984, 72, 219–227. [Google Scholar] [CrossRef]
- Van de Ven, R.; van den Hout, M.F.C.M.; Lindenberg, J.J.; Sluijter, B.J.R.; van Leeuwen, P.A.M.; Lougheed, S.M.; Meijer, S.; van den Tol, M.P.; Scheper, R.J.; de Gruijl, T.D. Characterization of four conventional dendritic cell subsets in human skin-draining lymph nodes in relation to T-cell activation. Blood 2011, 118, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.A.M.; Bontkes, H.J.; Stam, A.G.M.; Bhoelan, F.; Ruizendaal, J.J.; van den Eertwegh, A.J.M.; Hooijberg, E.; Scheper, R.J.; de Gruijl, T.D. Inducing antitumor T cell immunity: Comparative functional analysis of interstitial versus Langerhans dendritic cells in a human cell line model. J. Immunol. 2008, 180, 4540–4549. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Hodi, F.S. Ipilimumab, vemurafenib, dabrafenib, and trametinib: Synergistic competitors in the clinical management of BRAF mutant malignant melanoma. Oncologist 2013, 18, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H. Correlates of immune and clinical activity of novel cancer vaccines. Semin. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Church, S.E.; Galon, J. Regulation of CTL Infiltration within the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 33–49. [Google Scholar] [PubMed]
- Dutoit, V.; Rubio-Godoy, V.; Dietrich, P.Y.; Quiqueres, A.L.; Schnuriger, V.; Rimoldi, D.; Liénard, D.; Speiser, D.; Guillaume, P.; Batard, P.; et al. Heterogeneous T-cell response to MAGE-A10(254-262): High avidity-specific cytolytic T lymphocytes show superior antitumor activity. Cancer Res. 2001, 61, 5850–5856. [Google Scholar] [PubMed]
- Toes, R.E.; Offringa, R.; Blom, R.J.; Melief, C.J.; Kast, W.M. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc. Natl. Acad. Sci. USA 1996, 93, 7855–7860. [Google Scholar] [CrossRef] [PubMed]
- Slingluff, C.L. The present and future of peptide vaccines for cancer: Single or multiple, long or short, alone or in combination? Cancer J. 2011, 17, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Labow, D.; Lee, S.; Ginsberg, R.J.; Crystal, R.G.; Korst, R.J. Adenovirus vector-mediated gene transfer to regional lymph nodes. Hum. Gene Ther. 2000, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Molinier-Frenkel, V.; Gahery-Segard, M.; Le Boulaire, C.; Ribault, S.; Boulanger, P.; Tursz, T.; Guillet, J.G.; Farace, F. Immune response to recombinant adenovirus in humans: Capsid components from viral input are targets for vector-specific cytotoxic T lymphocytes. J. Virol. 2000, 74, 7678–7682. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.G.; Worgall, S.; Ely, S.; Leopold, P.L.; Crystal, R.G. Cellular immune responses of healthy individuals to intradermal administration of an E1-E3-adenovirus gene transfer vector. Hum. Gene Ther. 1999, 10, 2823–2837. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Nganga, D.; Brandariz, K.L.; Abbink, P.; et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [PubMed]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E.; et al. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Seshidhar Reddy, P.; Ganesh, S.; Limbach, M.P.; Brann, T.; Pinkstaff, A.; Kaloss, M.; Kaleko, M.; Connelly, S. Development of adenovirus serotype 35 as a gene transfer vector. Virology 2003, 311, 384–393. [Google Scholar] [CrossRef]
- Bru, T.; Salinas, S.; Kremer, E.J. An update on canine adenovirus type 2 and its vectors. Viruses 2010, 2, 2134–2153. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.; Tatsis, N.; Korioth-Schmitz, B.; Lasaro, M.O.; Hensley, S.E.; Lin, S.-W.; Li, Y.; Giles-Davis, W.; Cun, A.; Zhou, D.; et al. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J. Virol. 2007, 81, 6594–6604. [Google Scholar] [CrossRef] [PubMed]
- Stoff-Khalili, M.A.; Rivera, A.A.; Glasgow, J.N.; Le, L.P.; Stoff, A.; Everts, M.; Tsuruta, Y.; Kawakami, Y.; Bauerschmitz, G.J.; Mathis, J.M.; et al. A human adenoviral vector with a chimeric fiber from canine adenovirus type 1 results in novel expanded tropism for cancer gene therapy. Gene Ther. 2005, 12, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Lemckert, A.A.C.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; et al. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 2007, 81, 4654–4663. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, T.D.; Ophorst, O.J.A.E.; Goudsmit, J.; Verhaagh, S.; Lougheed, S.M.; Radosević, K.; Havenga, M.J.E.; Scheper, R.J. Intradermal delivery of adenoviral type-35 vectors leads to high efficiency transduction of mature, CD8+ T cell-stimulating skin-emigrated dendritic cells. J. Immunol. 2006, 177, 2208–2215. [Google Scholar] [CrossRef] [PubMed]
- Hangalapura, B.N.; Oosterhoff, D.; de Groot, J.; Boon, L.; Tüting, T.; van den Eertwegh, A.J.; Gerritsen, W.R.; van Beusechem, V.W.; Pereboev, A.; Curiel, D.T.; et al. Potent antitumor immunity generated by a CD40-targeted adenoviral vaccine. Cancer Res. 2011, 71, 5827–5837. [Google Scholar] [CrossRef] [PubMed]
- Bagaev, A.V.; Pichugin, A.V.; Lebedeva, E.S.; Lysenko, A.A.; Shmarov, M.M.; Logunov, D.Y.; Naroditsky, B.S.; Ataullakhanov, R.I.; Khaitov, R.M.; Gintsburg, A.L. Regulation of the target protein (transgene) expression in the adenovirus vector using agonists of toll-like receptors. Acta Nat. 2014, 6, 27–39. [Google Scholar]
- Nielsen, K.N.; Steffensen, M.A.; Christensen, J.P.; Thomsen, A.R. Priming of CD8 T cells by adenoviral vectors is critically dependent on B7 and dendritic cells but only partially dependent on CD28 ligation on CD8 T cells. J. Immunol. 2014, 193, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Wu, Y.; Ding, Z.-Y.; Luo, X.-M.; Zhong, W.-N.; Liu, J.; Xia, X.-Y.; Deng, G.-H.; Deng, Y.-T.; et al. Mannan-modified adenovirus encoding VEGFR-2 as a vaccine to induce anti-tumor immunity. J. Cancer Res. Clin. Oncol. 2014, 140, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Liljenfeldt, L.; Yu, D.; Chen, L.; Essand, M.; Mangsbo, S.M. A hexon and fiber-modified adenovirus expressing CD40L improves the antigen presentation capacity of dendritic cells. J. Immunother. 2014, 37, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hangalapura, B.N.; Timares, L.; Oosterhoff, D.; Scheper, R.J.; Curiel, D.T.; de Gruijl, T.D. CD40-targeted adenoviral cancer vaccines: The long and winding road to the clinic. J. Gene Med. 2012, 14, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Ophorst, O.J.A.E.; Kostense, S.; Goudsmit, J.; De Swart, R.L.; Verhaagh, S.; Zakhartchouk, A.; Van Meijer, M.; Sprangers, M.; Van Amerongen, G.; Yüksel, S.; et al. An adenoviral type 5 vector carrying a type 35 fiber as a vaccine vehicle: DC targeting, cross neutralization, and immunogenicity. Vaccine 2004, 22, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Sumida, S.M.; Truitt, D.M.; Lemckert, A.A.C.; Vogels, R.; Custers, J.H.H.V.; Addo, M.M.; Lockman, S.; Peter, T.; Peyerl, F.W.; Kishko, M.G.; et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J. Immunol. 2005, 174, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.E.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; Walsh, S.R.; Seaman, M.S.; Johnson, J.A.; Tucker, R.P.; Kleinjan, J.A.; Gothing, J.A.; Engelson, B.A.; Carey, B.R.; Oza, A.; et al. First-in-human evaluation of a hexon chimeric adenovirus vector expressing HIV-1 Env (IPCAVD 002). J. Infect. Dis. 2014, 210, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Icyuz, M.; Krendelchtchikova, V.; Krendelchtchikov, A.; Johnston, A.E.; Matthews, Q.L. Development of an Ad5H3 Chimera Using the “Antigen Capsid-Incorporation” Strategy for an Alternative Vaccination Approach. Open Virol. J. 2016, 10, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Kawabata, K.; Mizuguchi, H. Adenovirus vectors composed of subgroup B adenoviruses. Curr. Gene Ther. 2007, 7, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. Immunologic basis of vaccine vectors. Immunity 2010, 33, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.-Y.; Liu, Y.; Persson, J.; Beyer, I.; ller, T.M.O.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2010, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Cook, M.G.; John, R.J.; Powell, B.W.E.M.; Pandha, H.; Dalgleish, A.G. Successful live cell harvest from bisected sentinel lymph nodes research report. J. Immunol. Methods 2004, 291, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Parviainen, S.; Siurala, M.; Hemminki, O.; Havunen, R.; Tähtinen, S.; Bramante, S.; Vassilev, L.; Wang, H.; Lieber, A.; et al. Intravenously usable fully serotype 3 oncolytic adenovirus coding for CD40L as an enabler of dendritic cell therapy. Oncoimmunology 2017, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chondronasiou, D.; Eisden, T.-J.T.H.D.; Stam, A.G.M.; Matthews, Q.L.; Icyuz, M.; Hooijberg, E.; Dmitriev, I.; Curiel, D.T.; De Gruijl, T.D.; Van de Ven, R. Improved Induction of Anti-Melanoma T Cells by Adenovirus-5/3 Fiber Modification to Target Human DCs. Vaccines 2018, 6, 42. https://doi.org/10.3390/vaccines6030042
Chondronasiou D, Eisden T-JTHD, Stam AGM, Matthews QL, Icyuz M, Hooijberg E, Dmitriev I, Curiel DT, De Gruijl TD, Van de Ven R. Improved Induction of Anti-Melanoma T Cells by Adenovirus-5/3 Fiber Modification to Target Human DCs. Vaccines. 2018; 6(3):42. https://doi.org/10.3390/vaccines6030042
Chicago/Turabian StyleChondronasiou, Dafni, Tracy-Jane T. H. D. Eisden, Anita G. M. Stam, Qiana L. Matthews, Mert Icyuz, Erik Hooijberg, Igor Dmitriev, David T. Curiel, Tanja D. De Gruijl, and Rieneke Van de Ven. 2018. "Improved Induction of Anti-Melanoma T Cells by Adenovirus-5/3 Fiber Modification to Target Human DCs" Vaccines 6, no. 3: 42. https://doi.org/10.3390/vaccines6030042
APA StyleChondronasiou, D., Eisden, T.-J. T. H. D., Stam, A. G. M., Matthews, Q. L., Icyuz, M., Hooijberg, E., Dmitriev, I., Curiel, D. T., De Gruijl, T. D., & Van de Ven, R. (2018). Improved Induction of Anti-Melanoma T Cells by Adenovirus-5/3 Fiber Modification to Target Human DCs. Vaccines, 6(3), 42. https://doi.org/10.3390/vaccines6030042