Methods for Measuring T-Cell Memory to Vaccination: From Mouse to Man


Abstract
1. Introduction
2. Memory T-Cells
3. T-Cell-Inducing Vaccines
3.1. Immunogenicity Requirements for Vaccine Licensure
3.2. Factors to Consider in the Design of Vaccine Studies to Assess T-Cell Memory
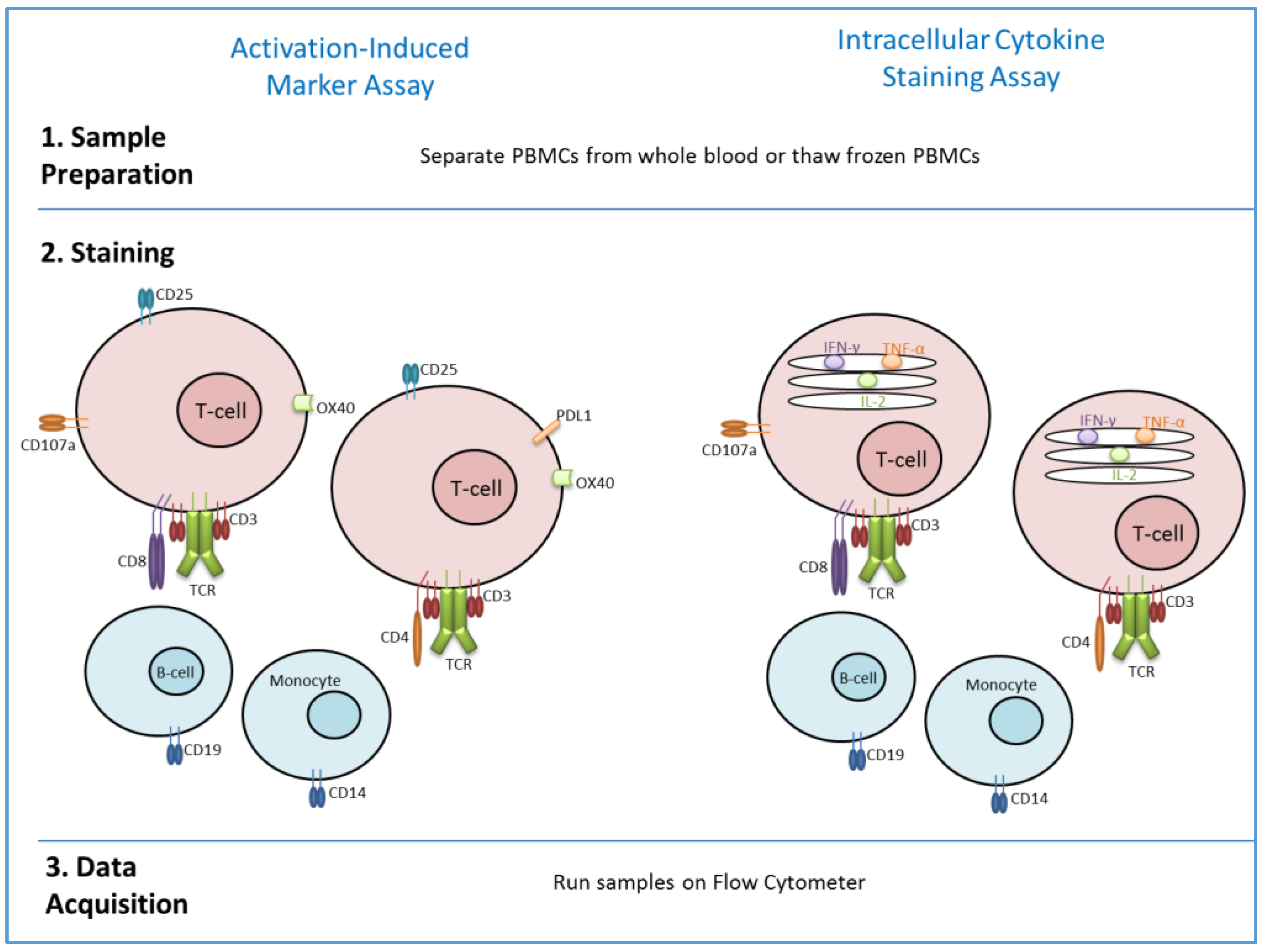
4. Methods to Assess T-Cell Memory in Vaccine Studies
4.1. Advantages and Disadvantages of Established Methods to Assess T-Cell Immunogenicity
4.2. Multiplex Cytokine Analysis
4.2.1. Luminex
4.2.2. LegendPlexTM
4.2.3. Meso Scale Discovery
4.2.4. CHIP Cytometry
4.2.5. CyTOF
4.3. Omics Approaches
5. Quantifying T-Cell Memory at Different Stages of Vaccine Development
5.1. Pre-Clinical Methods to Better Inform Clinical Trials
5.2. Limitations of Clinical Studies
5.2.1. Availability of Sample Types
5.2.2. Quantity of Sample Available
5.3. Technical Considerations in Clinical Trials
5.3.1. Fresh versus Frozen Samples
5.3.2. Performing Assays across Multiple Sites
5.3.3. Reagents and Equipment
5.4. Assay Validation
5.5. Efficacy Studies
5.6. Longevity Studies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Henao-Restrepo, A.M.; Longini, I.M.; Egger, M.; Dean, N.E.; Edmunds, W.J.; Camacho, A.; Carroll, M.W.; Doumbia, M.; Draguez, B.; Duraffour, S.; et al. Efficacy and effectiveness of an rvsv-vectored vaccine expressing ebola surface glycoprotein: Interim results from the guinea ring vaccination cluster-randomised trial. Lancet 2015, 386, 857–866. [Google Scholar] [CrossRef]
- Who Supports Ebola Vaccination of High Risk Populations in the Democratic Republic of the Congo. Available online: http://www.who.int/news-room/detail/21-05-2018-who-supports-ebola-vaccination-of-high-risk-populations-in-the-democratic-republic-of-the-congo (accessed on 6 June 2018).
- Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L.; Amara, R.R. T cell vaccines for microbial infections. Nat. Med. 2005, 11, S25–S32. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Saito, T. CD4 CTL, a cytotoxic subset of CD4+ T cells, their differentiation and function. Front. Immunol. 2017, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Ulvestad, E. Defending Life: The Nature of Host-Parasite Relations; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Borghans, J.; Ribeiro, R.M. The maths of memory. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Macallan, D.C.; Borghans, J.A.; Asquith, B. Human T cell memory: A dynamic view. Vaccines 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Omilusik, K.D.; Goldrath, A.W. The origins of memory T cells. Nature 2017, 552, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Graham, B.S.; Douek, D.C. The quest for a T cell-based immune correlate of protection against HIV: A story of trials and errors. Nat. Rev. Immunol. 2011, 11, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Verma, S.; Ellner, J.J.; Salgame, P. Quest for correlates of protection against tuberculosis. Clin. Vaccine Immunol. 2015, 22, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Complex correlates of protection after vaccination. Clin. Infect. Dis. 2013, 56, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.C.; Beura, L.K.; Masopust, D. Tissue resident memory T cells and viral immunity. Curr. Opin. Virol. 2017, 22, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.L.; Yudanin, N.A.; Restifo, N.P. Human memory T cells: Generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2014, 14, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Woodland, D.L.; Kohlmeier, J.E. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat. Rev. Immunol. 2009, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Ahmed, R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 2003, 4, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Hanekom, W.A. The immune response to BCG vaccination of newborns. Ann. N. Y. Acad. Sci. 2005, 1062, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.K.; Chodisetti, S.B.; Zeng, W.; Nadeem, S.; Maurya, S.K.; Pahari, S.; Janmeja, A.K.; Jackson, D.C.; Agrewala, J.N. A lipidated peptide of mycobacterium tuberculosis resuscitates the protective efficacy of BCG vaccine by evoking memory T cell immunity. J. Transl. Med. 2017, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Clemens, E.B.; van de Sandt, C.; Wong, S.S.; Wakim, L.M.; Valkenburg, S.A. Harnessing the power of T cells: The promising hope for a universal influenza vaccine. Vaccines 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Gans, H.A.; Yasukawa, L.L.; Alderson, A.; Rinki, M.; DeHovitz, R.; Beeler, J.; Audet, S.; Maldonado, Y.; Arvin, A.M. Humoral and cell-mediated immune responses to an early 2-dose measles vaccination regimen in the united states. J. Infect. Dis. 2004, 190, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Guideline on Clinical Evaluation of Vaccines. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2018/04/WC500248095.pdf (accessed on 12 June 2018).
- Guidelines on clinical evaluation of vaccines: Regulatory expectations. Available online: http://www.who.int/biologicals/Clinical_guidelines_revised_IK_29_Oct_2015.pdf (accessed on 12 June 2018).
- Ogunjimi, B.; Beutels, P. Uk experience of herpes zoster vaccination can inform varicella zoster virus policies. Lancet Public Health 2018, 3, e57–e58. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Lal, H.; Kovac, M.; Chlibek, R.; Hwang, S.J.; Diez-Domingo, J.; Godeaux, O.; Levin, M.J.; McElhaney, J.E.; Puig-Barbera, J.; et al. Efficacy of the herpes zoster subunit vaccine in adults 70 years of age or older. N. Engl. J. Med. 2016, 375, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, I.; Leroux-Roels, G.; Clement, F.; Vandepapelière, P.; Vassilev, V.; Ledent, E.; Heineman, T.C. A phase 1/2 clinical trial evaluating safety and immunogenicity of a varicella zoster glycoprotein e subunit vaccine candidate in young and older adults. J. Infect. Dis. 2012, 206, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Chlibek, R.; Smetana, J.; Pauksens, K.; Rombo, L.; Van den Hoek, J.A.; Richardus, J.H.; Plassmann, G.; Schwarz, T.F.; Ledent, E.; Heineman, T.C. Safety and immunogenicity of three different formulations of an adjuvanted varicella-zoster virus subunit candidate vaccine in older adults: A phase II, randomized, controlled study. Vaccine 2014, 32, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Rts, S.C. Efficacy and safety of rts,s/as01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar]
- RTS,S. Available online: http://www.malariavaccine.org/malaria-and-vaccines/first-generation-vaccine/rtss (accessed on 6 February 2018).
- Kester, K.E.; Cummings, J.F.; Ofori-Anyinam, O.; Ockenhouse, C.F.; Krzych, U.; Moris, P.; Schwenk, R.; Nielsen, R.A.; Debebe, Z.; Pinelis, E.; et al. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: Safety, efficacy, and immunologic associates of protection. J. Infect. Dis. 2009, 200, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, I.R.; Sebastian, S. Novel viral vectors in infectious diseases. Immunology 2017, 153, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mohan, T.; Verma, P.; Rao, D.N. Novel adjuvants & delivery vehicles for vaccines development: A road ahead. Indian J. Med. Res. 2013, 138, 779–795. [Google Scholar] [PubMed]
- Charlton Hume, H.K.; Lua, L.H.L. Platform technologies for modern vaccine manufacturing. Vaccine 2017, 35, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.; Dorrell, L. Viral vectors as vaccine platforms: From immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Nolz, J.C.; Harty, J.T. Strategies and implications for prime-boost vaccination to generate memory CD8 T cells. Adv. Exp. Med. Biol. 2011, 780, 69–83. [Google Scholar] [PubMed]
- Schulze, K.; Ebensen, T.; Riese, P.; Prochnow, B.; Lehr, C.M.; Guzman, C.A. New horizons in the development of novel needle-free immunization strategies to increase vaccination efficacy. Curr. Top. Microbiol. Immunol. 2016, 398, 207–234. [Google Scholar] [PubMed]
- Scherliess, R. Delivery of antigens used for vaccination: Recent advances and challenges. Ther. Deliv. 2011, 2, 1351–1368. [Google Scholar] [CrossRef] [PubMed]
- Saade, F.; Gorski, S.A.; Petrovsky, N. Pushing the frontiers of T-cell vaccines: Accurate measurement of human T-cell responses. Expert Rev. Vaccines 2012, 11, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Lambe, T. Measuring cellular immunity to influenza: Methods of detection, applications and challenges. Vaccines 2015, 3, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, E.; Popescu, I.; Gigante, M. CTL elispot assay. Methods Mol. Biol. 2014, 1186, 75–86. [Google Scholar] [PubMed]
- Lehmann, P.V.; Zhang, W. Unique strengths of elispot for T cell diagnostics. In Handbook of Elispot: Methods and Protocols; Kalyuzhny, A.E., Ed.; Humana Press: Totowa, NJ, USA, 2012. [Google Scholar]
- Calarota, S.A.; Baldanti, F. Enumeration and characterization of human memory T cells by enzyme-linked immunospot assays. Clin. Dev. Immunol. 2013, 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Sandoval, A.; Pearson, F.E.; Todryk, S.; Ewer, K. Potency assays for novel t-cell-inducing vaccines against malaria. Curr. Opin. Mol. Ther. 2009, 11, 72–80. [Google Scholar] [PubMed]
- Ahlborg, N.; Axelsson, B. Dual-and triple-color fluorospot. In Handbook of Elispot: Methods and Erotocols; Kalyuzhny, A.E., Ed.; Humana Press: Totowa, NJ, USA, 2012. [Google Scholar]
- T-spot. Available online: http://www.tspot.com/about-the-test/ (accessed on 13 July 2018).
- Lovelace, P.; Maecker, H.T. Multiparameter intracellular cytokine staining. In Flow Cytometry Protocols; Hawley, T.S., Hawley, R.G., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 165–178. [Google Scholar]
- Freer, G.; Rindi, L. Intracellular cytokine detection by fluorescence-activated flow cytometry: Basic principles and recent advances. Methods 2013, 61, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Foulds, K.E.; Chang-you, W.; Seder, R.A. Th1 memory: Implications for vaccine development. Immunol. Rev. 2006, 211, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory t cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.H.; Lerner, H.; et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Reiss, S.; Baxter, A.E.; Cirelli, K.M.; Dan, J.M.; Morou, A.; Daigneault, A.; Brassard, N.; Silvestri, G.; Routy, J.P.; Havenar-Daughton, C.; et al. Comparative analysis of activation induced marker (aim) assays for sensitive identification of antigen-specific CD4+ T cells. PloS ONE 2017, 12, e0186998. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Lindestam Arlehamn, C.S.; Weiskopf, D.; da Silva Antunes, R.; Havenar-Daughton, C.; Reiss, S.M.; Brigger, M.; Bothwell, M.; Sette, A.; Crotty, S. A cytokine-independent approach to identify antigen-specific human germinal center t follicular helper cells and rare antigen-specific CD4+ T cells in blood. J. Immunol. 2016, 197, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, G.; Rampling, T.; Powlson, J.; Morter, R.; Wright, D.; Hill, A.V.S.; Ewer, K.J. Activation-induced markers detect vaccine-specific CD4+ T cell responses not measured by assays conventionally used in clinical trials. Vaccines. (under review).
- Multiplex Immunoasssays. Available online: http://www.bio-rad.com/en-uk/applications-technologies/multiplex-immunoassays?ID=LUSM0E8UU (accessed on 14 June 2018).
- King, D.F.; McKay, P.F.; Mann, J.F.; Jones, C.B.; Shattock, R.J. Plasmid DNA vaccine co-immunisation modulates cellular and humoral immune responses induced by intranasal inoculation in mice. PloS ONE 2015, 10, e0141557. [Google Scholar] [CrossRef] [PubMed]
- Focke-Tejkl, M.; Weber, M.; Niespodziana, K.; Neubauer, A.; Huber, H.; Henning, R.; Stegfellner, G.; Maderegger, B.; Hauer, M.; Stolz, F.; et al. Development and characterization of a recombinant, hypoallergenic, peptide-based vaccine for grass pollen allergy. J. Allergy Clin. Immunol. 2015, 135. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, C.; Kasonta, R.; Lunemann, S.; Krahling, V.; Zinser, M.E.; Biedenkopf, N.; Fehling, S.K.; Ly, M.L.; Rechtien, A.; Stubbe, H.C.; et al. Dose-dependent T-cell dynamics and cytokine cascade following rvsv-zebov immunization. EBioMedicine 2017, 19, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Legendplex. Available online: https://www.biolegend.com/legendplex (accessed on 14 June 2018).
- Copland, A.; Diogo, G.R.; Hart, P.; Harris, S.; Tran, A.C.; Paul, M.J.; Singh, M.; Cutting, S.M.; Reljic, R. Mucosal delivery of fusion proteins with bacillus subtilis spores enhances protection against tuberculosis by bacillus calmette-guérin. Front. Immunol. 2018, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Our Technology. Available online: https://www.mesoscale.com/en/technical_resources/our_technology (accessed on 14 June 2018).
- Chowdhury, F.; Williams, A.; Johnson, P. Validation and comparison of two multiplex technologies, luminex® and mesoscale discovery, for human cytokine profiling. J. Immunol. Methods 2009, 340, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, S.E.; Reimer, J.M.; Karlsson, K.H.; Lilja, L.; Bengtsson, K.L.; Stertman, L. Immune enhancing properties of the novel matrix-m™ adjuvant leads to potentiated immune responses to an influenza vaccine in mice. Vaccine 2013, 31, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Hennig, C.; Adams, N.; Hansen, G. A versatile platform for comprehensive chip-based explorative cytometry. Cytometry A 2009, 75, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Technology. Available online: http://www.zellkraftwerk.com/technology/ (accessed on 14 June 2018).
- Hummert, M.W.; Alvermann, S.; Gingele, S.; Gross, C.C.; Wiendl, H.; Mirenska, A.; Hennig, C.; Stangel, M. Immunophenotyping of cerebrospinal fluid cells by chipcytometry. J. Neuroinflammation 2018, 15, 160. [Google Scholar] [CrossRef] [PubMed]
- Helios, a Cytof System. Available online: https://www.fluidigm.com/products/helios#overview (accessed on 14 June 2018).
- Swadling, L.; Capone, S.; Antrobus, R.D.; Brown, A.; Richardson, R.; Newell, E.W.; Halliday, J.; Kelly, C.; Bowen, D.; Fergusson, J.; et al. A human vaccine strategy based on chimpanzee adenoviral and mva vectors that primes, boosts, and sustains functional hcv-specific t cell memory. Sci. Transl. Med. 2014, 6, 261. [Google Scholar] [CrossRef] [PubMed]
- Mooney, M.; McWeeney, S.; Canderan, G.; Sékaly, R.-P. A systems framework for vaccine design. Curr. Opin. Immunol. 2013, 25, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Hagan, T.; Nakaya, H.I.; Subramaniam, S.; Pulendran, B. Systems vaccinology: Enabling rational vaccine design with systems biological approaches. Vaccine 2015, 33, 5294–5301. [Google Scholar] [CrossRef] [PubMed]
- Natesan, M.; Ulrich, R.G. Protein microarrays and biomarkers of infectious disease. Int. J. Mol. Sci. 2010, 11, 5165–5183. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Bi, L.; Zhou, L.; Guo, S.-J.; Fleming, J.; Jiang, H.-W.; Zhou, Y.; Gu, J.; Zhong, Q.; Wang, Z.-X.; et al. Mycobacterium tuberculosis proteome microarray for global studies of protein function and immunogenicity. Cell Rep. 2014, 9, 2317–2329. [Google Scholar] [CrossRef] [PubMed]
- Marsay, L.; Matsumiya, M.; Tanner, R.; Poyntz, H.; Griffiths, K.L.; Stylianou, E.; Marsh, P.D.; Williams, A.; Sharpe, S.; Fletcher, H.; et al. Mycobacterial growth inhibition in murine splenocytes as a surrogate for protection against mycobacterium tuberculosis (m. Tb). Tuberculosis 2013, 93, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.K.; Roederer, M. A mine is a terrible thing to waste: High content, single cell technologies for comprehensive immune analysis. Am. J. Transplant. 2015, 15, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Afik, S.; Yates, K.B.; Bi, K.; Darko, S.; Godec, J.; Gerdemann, U.; Swadling, L.; Douek, D.C.; Klenerman, P.; Barnes, E.J.; et al. Targeted reconstruction of T cell receptor sequence from single cell rna-seq links cdr3 length to t cell differentiation state. Nucleic Acids. Res. 2017, 45, 16. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Hale, J.S.; Ahmed, R. T-cell memory differentiation: Insights from transcriptional signatures and epigenetics. Immunology 2013, 139, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.M.; Bett, A.J.; Cristescu, R.; Loboda, A.; ter Meulen, J. Transcriptional profiling of vaccine-induced immune responses in humans and non-human primates. Microb. Biotechnol. 2012, 5, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Faridi, P.; Wayne Purcell, A.; Croft, N.P. In immunopeptidomics we need a sniper instead of a shotgun. Proteomics 2018, 18, 1700464. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Wolint, P.; Schwarz, K.; Jager, P.; Oxenius, A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of il-7 receptor alpha and cd62l. J. Immunol. 2005, 175, 4686–4696. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.L. Novel vaccines and adjuvant systems: The utility of animal models for predicting immunogenicity in humans. Human Vaccines 2008, 4, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Rollier, C.S.; Hill, A.V.S.; Reyes-Sandoval, A. Influence of adenovirus and mva vaccines on the breadth and hierarchy of t cell responses. Vaccine 2016, 34, 4470–4474. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T.; Carey, J.B.; Li, Y.; Spencer, A.J.; van Laarhoven, A.; Mullarkey, C.E.; Vrdoljak, A.; Moore, A.C.; Gilbert, S.C. Immunity against heterosubtypic influenza virus induced by adenovirus and mva expressing nucleoprotein and matrix protein-1. Sci. Rep. 2013, 3, 1443. [Google Scholar] [CrossRef] [PubMed]
- Mullarkey, C.E.; Boyd, A.; van Laarhoven, A.; Lefevre, E.A.; Veronica Carr, B.; Baratelli, M.; Molesti, E.; Temperton, N.J.; Butter, C.; Charleston, B.; et al. Improved adjuvanting of seasonal influenza vaccines: Preclinical studies of mva-np+m1 coadministration with inactivated influenza vaccine. Eur. J. Immunol. 2013, 43, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, T.K.; Hamill, M.; Lillie, P.J.; Hwenda, L.; Collins, K.A.; Ewer, K.J.; Milicic, A.; Poyntz, H.C.; Lambe, T.; Fletcher, H.A.; et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza a vaccine, mva-np+m1. Clin. Infect. Dis. 2011, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, R.D.; Lillie, P.J.; Berthoud, T.K.; Spencer, A.J.; McLaren, J.E.; Ladell, K.; Lambe, T.; Milicic, A.; Price, D.A.; Hill, A.V.; et al. A t cell-inducing influenza vaccine for the elderly: Safety and immunogenicity of mva-np+m1 in adults aged over 50 years. PLoS ONE 2012, 7, e48322. [Google Scholar] [CrossRef] [PubMed]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a t-cell-based influenza vaccine, mva-np+m1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, R.D.; Berthoud, T.K.; Mullarkey, C.E.; Hoschler, K.; Coughlan, L.; Zambon, M.; Hill, A.V.; Gilbert, S.C. Coadministration of seasonal influenza vaccine and mva-np+m1 simultaneously achieves potent humoral and cell-mediated responses. Mol. Ther. 2014, 22, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Improved Novel Vaccine Combination Influenza Study (Invictus). Available online: https://clinicaltrials.gov/ct2/show/NCT03300362 (accessed on 5 July 2018).
- Spencer, A.J.; Longley, R.J.; Gola, A.; Ulaszewska, M.; Lambe, T.; Hill, A.V. The threshold of protection from liver-stage malaria relies on a fine balance between the number of infected hepatocytes and effector CD8+ T cells present in the liver. J. Immunol. 2017, 198, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Pembroke, T.; Gallimore, A.; Godkin, A. Tracking the kinetics of intrahepatic immune responses by repeated fine needle aspiration of the liver. J. Immunol. Methods 2015, 424, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Bliss, C.M.; Bowyer, G.; Anagnostou, N.A.; Havelock, T.; Snudden, C.M.; Davies, H.; de Cassan, S.C.; Grobbelaar, A.; Lawrie, A.M.; Venkatraman, N.; et al. Assessment of novel vaccination regimens using viral vectored liver stage malaria vaccines encoding me-trap. Sci. Rep. 2018, 8, 3390. [Google Scholar] [CrossRef] [PubMed]
- Hanekom, W.A.; Hughes, J.; Mavinkurve, M.; Mendillo, M.; Watkins, M.; Gamieldien, H.; Gelderbloem, S.J.; Sidibana, M.; Mansoor, N.; Davids, V.; et al. Novel application of a whole blood intracellular cytokine detection assay to quantitate specific t-cell frequency in field studies. J. Immunol. Methods 2004, 291, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Nemes, E.; Hesseling, A.C.; Tameris, M.; Mauff, K.; Downing, K.; Mulenga, H.; Rose, P.; van der Zalm, M.; Mbaba, S.; Van As, D.; et al. Safety and immunogenicity of newborn mva85a vaccination and selective, delayed bacille calmette-guerin for infants of human immunodeficiency virus-infected mothers: A phase 2 randomized, controlled trial. Clin. Infect. Dis. 2018, 66, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Ford, T.; Wenden, C.; Mbekeani, A.; Dally, L.; Cox, J.H.; Morin, M.; Winstone, N.; Hill, A.V.S.; Gilmour, J.; Ewer, K.J. Cryopreservation-related loss of antigen-specific ifngamma producing CD4+ T-cells can skew immunogenicity data in vaccine trials: Lessons from a malaria vaccine trial substudy. Vaccine 2017, 35, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Jaoko, W.; Karita, E.; Kayitenkore, K.; Omosa-Manyonyi, G.; Allen, S.; Than, S.; Adams, E.M.; Graham, B.S.; Koup, R.A.; Bailer, R.T.; et al. Safety and immunogenicity study of multiclade hiv-1 adenoviral vector vaccine alone or as boost following a multiclade hiv-1 DNA vaccine in Africa. PLoS ONE 2010, 5, e12873. [Google Scholar] [CrossRef] [PubMed]
- Breen, E.C.; Reynolds, S.M.; Cox, C.; Jacobson, L.P.; Magpantay, L.; Mulder, C.B.; Dibben, O.; Margolick, J.B.; Bream, J.H.; Sambrano, E.; et al. Multisite comparison of high-sensitivity multiplex cytokine assays. Clin. Vaccine Immunol. 2011, 18, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Kimani, D.; Jagne, Y.J.; Cox, M.; Kimani, E.; Bliss, C.M.; Gitau, E.; Ogwang, C.; Afolabi, M.O.; Bowyer, G.; Collins, K.A.; et al. Translating the immunogenicity of prime-boost immunization with chad63 and mva me-trap from malaria naive to malaria-endemic populations. Mol. Ther. 2014, 22, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, U.; Perret-Liaudet, A.; van Waalwijk van Doorn, L.J.; Blennow, K.; Chiasserini, D.; Engelborghs, S.; Fladby, T.; Genc, S.; Kruse, N.; Kuiperij, H.B.; et al. A practical guide to immunoassay method validation. Front. Neurol. 2015, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Flucop. Available online: http:www.flucop.eu (accessed on 15 June 2018).
- Transvac. Available online: http:www.transvac.org (accessed on 15 June 2018).
- Epstein, J.E.; Tewari, K.; Lyke, K.E.; Sim, B.K.; Billingsley, P.F.; Laurens, M.B.; Gunasekera, A.; Chakravarty, S.; James, E.R.; Sedegah, M.; et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science 2011, 334, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.J.; O’Hara, G.A.; Duncan, C.J.; Collins, K.A.; Sheehy, S.H.; Reyes-Sandoval, A.; Goodman, A.L.; Edwards, N.J.; Elias, S.C.; Halstead, F.D.; et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-mva immunisation. Nat. Commun. 2013, 4, 2836. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362–367. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Factor | Example | References |
|---|---|---|
| Vaccine Platform | Protein, VLP, DNA vaccine, viral vector, use of adjuvant | [32,33,34,35] |
| Vaccine Regimen | Prime only, Homologous or Heterologous Prime-Boost | [36] |
| Route of administration | Intramuscular, subcutaneous, intranasal, sublingual, aerosol | [37,38] |
| Timing of sampling for immunogenicity assessment | Hours, days or weeks post immunisation | [36] |
| Technical Aspect | Luminex | LegendPlexTM | Meso Scale Discovery |
|---|---|---|---|
| Principle of assay | Fluorescent beads | Fluorescent beads | Electrochemiluniscence |
| Bead based? | Yes | Yes | No |
| Plate based? | Yes | No | Yes |
| Species available | Human, mouse, rat, NHP, canine and others | Human, mouse, rat, NHP | Human, mouse, rat, NHP |
| Maximum Number of Analytes | 50–80 | 13 | 10 per plate |
| Minimum Sample volume required | 12.5 µL or 25 µL, depending on kit manufacturer | Supernatant—25 µL Serum/plasma—12.5 µL | 12.5 µL |
| Number of tests/samples | 39 per plate (performed in duplicate) | 100 tests per kit | 40 per plate (performed in duplicate) |
| Estimated sample preparation time including incubations | 4–5 h | 4–5 h | 4.5–5 h |
| Estimated cost per multiplex plate/kit | £1000–5000 | ~£1000 | £1000–1500 |
| Data Acquisition time | ~1 h per plate | As for flow cytometry: 2–3 min per sample plus set up time | <5 min per plate |
| Dedicated data Acquisition instrument? | Yes, e.g., Magpix, Luminex 200, FLEXMAP 3D | No, Flow cytometer that can detect PE and APC | Yes, SECTOR range of instruments or Quickplex |
| Data analysis software | xPONENT, usually purchased with instrument | LEGENDplex™ data analysis software—Free Download | MSD Discovery workbench—Free Download |
| Technical Aspect | CHIP Cytometry | CyTOF |
|---|---|---|
| Principle of assay | Flow Cytometry | Mass spectrometry |
| Species available | Human | Human. Mouse |
| Number of Analytes | 90+ | 40+ |
| Assay volume/cell number | 100 µL per chip | Human PBMC assay: 3 × 106 |
| Dedicated data Acquisition instrument? | Yes, ZellScanner ONE™ (manual) or CYTOBOT (automated) | Yes, Helios or CyTOF® 2 Instrument |
| Data analysis software | ZellExplorer | Cytobank |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flaxman, A.; Ewer, K.J. Methods for Measuring T-Cell Memory to Vaccination: From Mouse to Man. Vaccines 2018, 6, 43. https://doi.org/10.3390/vaccines6030043
Flaxman A, Ewer KJ. Methods for Measuring T-Cell Memory to Vaccination: From Mouse to Man. Vaccines. 2018; 6(3):43. https://doi.org/10.3390/vaccines6030043
Chicago/Turabian StyleFlaxman, Amy, and Katie J. Ewer. 2018. "Methods for Measuring T-Cell Memory to Vaccination: From Mouse to Man" Vaccines 6, no. 3: 43. https://doi.org/10.3390/vaccines6030043
APA StyleFlaxman, A., & Ewer, K. J. (2018). Methods for Measuring T-Cell Memory to Vaccination: From Mouse to Man. Vaccines, 6(3), 43. https://doi.org/10.3390/vaccines6030043