Neutralizing Anti-Hemagglutinin Monoclonal Antibodies Induced by Gene-Based Transfer Have Prophylactic and Therapeutic Effects on Influenza Virus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
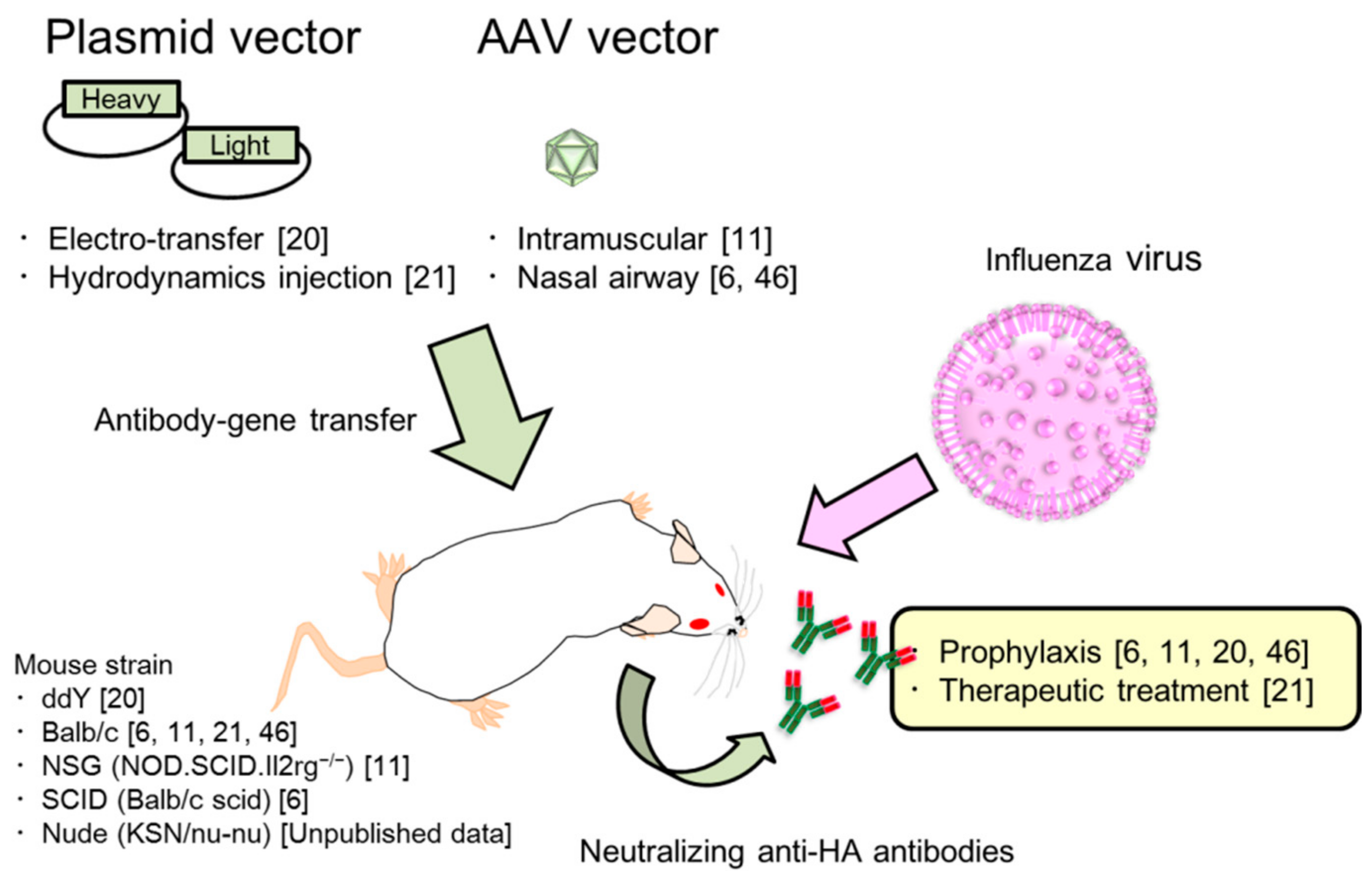
2. Passive Immunization for Influenza by the Antibody Gene Transfer Method
2.1. HA Is the Main Target to Protect Against Influenza Virus Infection
2.2. Long and Potent Expression of Neutralizing Antibodies by the Antibody Gene Transfer Method in Serum
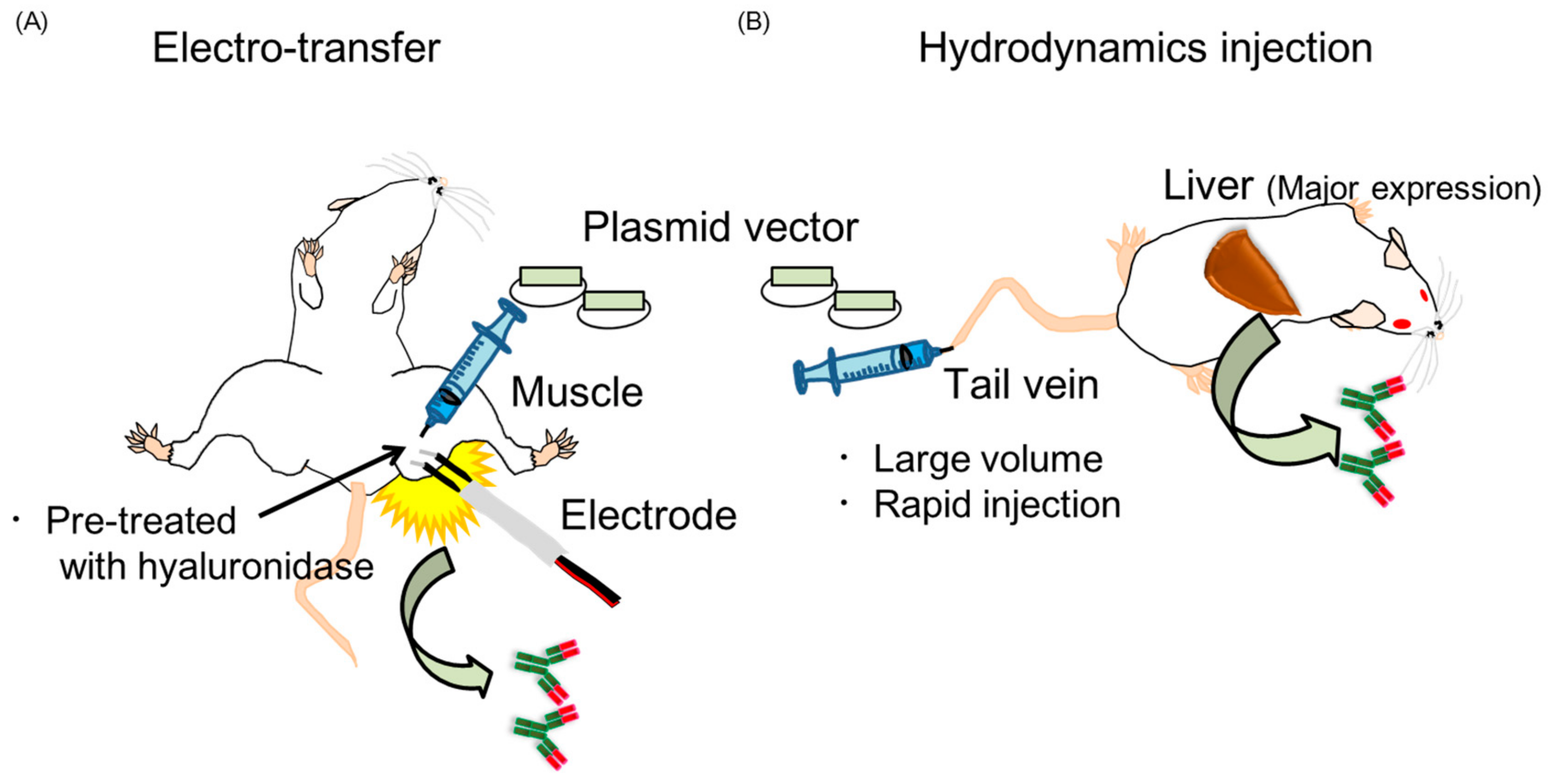
2.3. Two Kinds of Injection Methods, Electro-Transfer and Hydrodynamics Injection
2.4. Characterization of Antibody Gene Transfer with AAV Vector
2.5. Modification of the Expression Vector for the Increased Expression of Neutralizing Antibodies
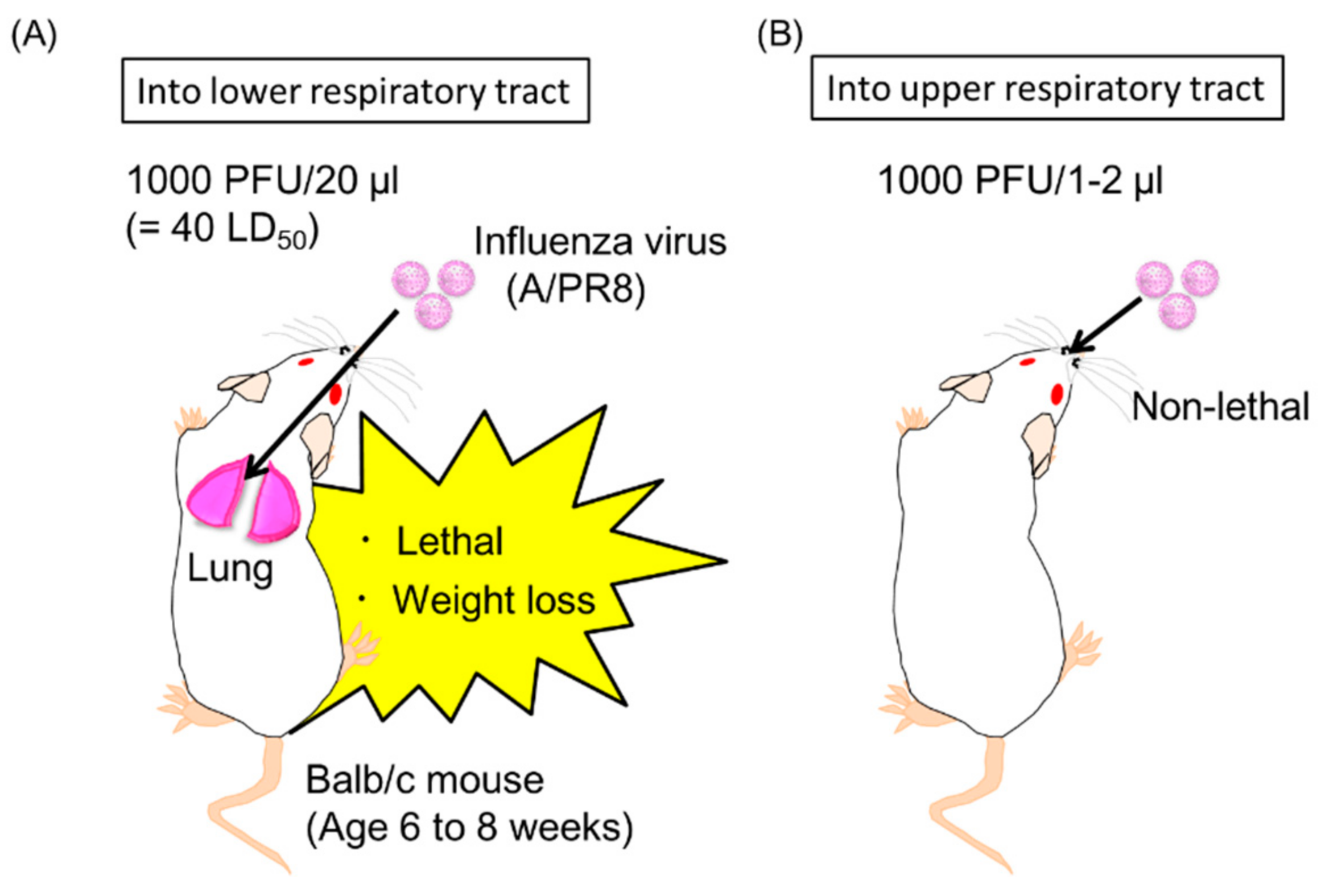
2.6. Induction of Neutralizing Anti-HA Antibodies in the Upper Respiratory Tract by Antibody Gene Transfer
2.7. Passive Prophylaxis for Immunocompetent and Immunocompromised Mice Using Antibody Gene Transfer
2.8. Passive Therapeutic Treatment Against Influenza Virus Infection by Antibody Gene Transfer
3. Perspectives of Passive Immunization with Antibody Genes for Clinical Trials
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kew, O.M.; Sutter, R.W.; de Gourville, E.M.; Dowdle, W.R.; Pallansch, M.A. Vaccine-Derived Polioviruses and the Endgame Strategy for Global Polio Eradication. Annu. Rev. Microbiol. 2005, 59, 587–635. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Li, C.; Van Domselaar, G.; Wang, J.; Farnsworth, A.; Cui, X.; Rode, H.; Cyr, T.D.; He, R.; Li, X. Universal antibodies and their applications to the quantitative determination of virtually all subtypes of the influenza A viral hemagglutinins. Vaccine 2008, 26, 6068–6076. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.M.; Jaentschke, B.; Van Domselaar, G.; Hashem, A.M.; Farnsworth, A.; Forbes, N.E.; Li, C.; Wang, J.; He, R.; Brown, E.G.; et al. The universal epitope of influenza a viral neuraminidase fundamentally contributes to enzyme activity and viral replication. J. Biol. Chem. 2013, 288, 18283–18289. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Ainai, A.; Suzuki, T.; Kurata, T.; Hasegawa, H. Intranasal Inactivated Influenza Vaccines: A Reasonable Approach to Improve the Efficacy of Influenza Vaccine? Jpn. J. Infect. Dis. 2016, 69, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Adam, V.S.; Crosariol, M.; Kumar, S.; Ge, M.Q.; Czack, S.E.; Roy, S.; Haczku, A.; Tretiakova, A.; Wilson, J.M.; Limberis, M.P. Adeno-associated virus 9-mediated airway expression of antibody protects old and immunodeficient mice against influenza virus. Clin. Vaccine Immunol. 2014, 21, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P. Vaccination of immunocompromised patients. Clin. Microbiol. Infect. 2012, 18, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Guo, C.Y.; Tsai, M.S.; Cheng, Y.Y.; Lin, M.T.; Chen, C.H.; Shen, D.; Wang, J.R.; Sung, J.M. Poor immune response to a standard single dose non-adjuvanted vaccination against 2009 pandemic H1N1 influenza virus A in the adult and elder hemodialysis patients. Vaccine 2012, 30, 5009–5018. [Google Scholar] [CrossRef] [PubMed]
- Catania, J.; Que, L.G.; Govert, J.A.; Hollingsworth, J.W.; Wolfe, W. High intensive care unit admission rate for 2013-2014 influenza is associated with a low rate of vaccination. Am. J. Respir. Crit. Care Med. 2014, 189, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Stevens, N.E.; Hatjopolous, A.; Fraser, C.K.; Alsharifi, M.; Diener, K.R.; Hayball, J.D. Preserved antiviral adaptive immunity following polyclonal antibody immunotherapy for severe murine influenza infection. Sci. Rep. 2016, 6, 29154. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.B.; Bloom, J.D.; Hong, C.M.; Rao, D.S.; Baltimore, D. Broad protection against influenza infection by vectored immunoprophylaxis in mice. Nat. Biotechnol. 2013, 31, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Nunes-alves, C. Milestone 1: “Blood is a very unusual fluid”. Nat. Milest. Antib. 2016, S5. [Google Scholar] [CrossRef]
- Nachbagauer, R.; Palese, P. Development of next generation hemagglutinin-based broadly protective influenza virus vaccines. Curr. Opin. Immunol. 2018, 53, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Cameroni, E.; Guarino, B.; Nicole, L.; Zhu, Q.; Lanzavecchia, A. Tackling influenza with broadly neutralizing antibodies. Curr. Opin. Virol. 2017, 24, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Morell, A.; Terry, W.D.; Waldmann, T.A. Metabolic properties of IgG subclasses in man. J. Clin. Investig. 1970, 49, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, N.M.; Einarsdóttir, H.K.; Stemerding, A.M.; Vidarsson, G. The multiple facets of FcRn in immunity. Immunol. Rev. 2015, 268, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Hollevoet, K.; Declerck, P.J. State of play and clinical prospects of antibody gene transfer. J. Transl. Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Deshane, J.; Siegal, G.P.; Alvarez, R.D.; Wang, M.H.; Feng, M.; Cabrera, G.; Liu, T.; Kay, M.; Curiel, D.T. Targeted tumor killing via an intracellular antibody against erbB-2. J. Clin. Investig. 1995, 96, 2980–2989. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.W.; Ponzio, T.A. Vectored immunoprophylaxis: An emerging adjunct to traditional vaccination. Trop. Dis. Travel Med. Vaccines 2017, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Nagashima, M.; Ninomiya, D.; Arai, Y.; Teshima, Y.; Fujimoto, A.; Ainai, A.; Hasegawa, H.; Chiba, J. Passive immune-prophylaxis against influenza virus infection by the expression of neutralizing anti-hemagglutinin monoclonal antibodies from plasmids. Jpn. J. Infect. Dis. 2011, 64, 40–49. [Google Scholar] [PubMed]
- Yamazaki, T.; Nagashima, M.; Ninomiya, D.; Ainai, A.; Fujimoto, A.; Ichimonji, I.; Takagi, H.; Morita, N.; Murotani, K.; Hasegawa, H.; et al. Neutralizing Antibodies Induced by Gene-Based Hydrodynamic Injection Have a Therapeutic Effect in Lethal Influenza Infection. Front. Immunol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.K.; Hanson, J.E.; Glick, G.D.; Brown, J.H.; Crowther, R.L.; Park, S.J.; Skehel, J.J.; Wiley, D.C. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: Analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry 1992, 31, 9609–9621. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426. [Google Scholar] [CrossRef] [PubMed]
- Knossow, M.; Skehel, J.J. Variation and infectivity neutralization in influenza. Immunology 2006, 119, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Agatic, G.; Bianchi, S.; Giacchetto-sasselli, I.; Calder, L.; Langedijk, J.P.M.; Skehel, J.J.; Lanzavecchia, A. A Neutralizing Antibody Selected from Plasma Cells That Binds to Group 1 and Group 2 Influenza A Hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef]
- Subbarao, K.; Joseph, T. Scientific barriers to developing vaccines against avian influenza viruses. Nat. Rev. Immunol. 2007, 7, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; García-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Sautto, G.A.; Kirchenbaum, G.A.; Ross, T.M. Towards a universal influenza vaccine: Different approaches for one goal. Virol. J. 2018, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, D.A.; Lee, F.E.H. Antibodies against conserved antigens provide opportunities for reform in influenza vaccine design. Front. Immunol. 2011, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.D.; Bajic, G.; Ferdman, J.; Suphaphiphat, P.; Settembre, E.C.; Moody, M.A.; Schmidt, A.G.; Harrison, S.C. Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc. Natl. Acad. Sci. USA 2017, 115, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.M.; Signori, E.; Wells, K.E.; Fazio, V.M.; Wells, D.J. Optimisation of electrotransfer of plasmid into skeletal muscle by pretreatment with hyaluronidase-increased expression with reduced muscle damage. Gene Ther. 2001, 8, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiu, S.; Zhang, X.; Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Kamensek, U.; Tesic, N.; Sersa, G.; Cemazar, M. Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B. Prospects for control of emerging infectious diseases with plasmid DNA vaccines. J. Immune Based Ther. Vaccines 2009, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Glenting, J.; Wessels, S. Ensuring safety of DNA vaccines. Microb. Cell Fact. 2005, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Aihara, H.; Miyazaki, J. Gene transfer into muscle by electroporation in vivo. Nat. Biotechnol. 1998, 16, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.M.; Ottensmeier, C.H.; Mulatero, C.; Lorigan, P.; Plummer, R.; Pandha, H.; Elsheikh, S.; Hadjimichael, E.; Villasanti, N.; Adams, S.E.; et al. Targeting gp100 and TRP-2 with a DNA vaccine: Incorporating T cell epitopes with a human IgG1 antibody induces potent T cell responses that are associated with favourable clinical outcome in a phase I/II trial. Oncoimmunology 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Van Drunen Littel-van den Hurk, S.; Hannaman, D. Electroporation for DNA immunization: Clinical application. Expert Rev. Vaccines 2010, 9, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Leen, E.; Picard, J.; Stebbing, J.; Abel, M.; Dhillon, T.; Wasan, H. Percutaneous irreversible electroporation with systemic treatment for locally advanced pancreatic adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ainai, A.; Ichinohe, T.; Tamura, S.; Kurata, T.; Sata, T.; Tashiro, M.; Hasegawa, H. Zymosan Enhances the Mucosal Adjuvant Activity of Poly(I:C) in a Nasal Influenza Vaccine. J. Med. Virol. 2010, 82, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Danko, I.; Wolff, J.A. Direct gene transfer into muscle. Vaccine 1994, 12, 1499–1502. [Google Scholar] [CrossRef]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Kitaguchi, K.; Toda, M.; Takekoshi, M.; Maeda, F.; Muramatsu, T.; Murai, A. Immune deficiency enhances expression of recombinant human antibody in mice after nonviral in vivo gene transfer. Int. J. Mol. Med. 2005, 16, 683–688. [Google Scholar] [PubMed]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Francoise, M.M.; Carole, M. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Limberis, M.P.; Adam, V.S.; Wong, G.; Gren, J.; Kobasa, D.; Ross, T.M.; Kobinger, G.P.; Tretiakova, A.; Wilson, J.M. Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci. Transl. Med. 2013, 5, 187ra72. [Google Scholar] [CrossRef] [PubMed]
- Rose, B.Y.J.A.; Berns, K.I.; Hoggan, M.D.; Koczot, F.J. Evidence for a single-stranded adenovirus-associated virus genome: Formation of a DNA density hybrid on release of viral DNA. Proc. Natl. Acad. Sci. USA 1969, 64, 863–869. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Qian, J.-J.; Yi, S.; Harding, T.C.; Tu, G.H.; VanRoey, M.; Jooss, K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotechnol. 2005, 23, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.B.; Chen, J.; Hong, C.M.; Rao, D.S.; Yang, L.; Baltimore, D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 2011, 481, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, H.; Xu, Z.; Ishii-Watabe, A.; Uchida, E.; Hayakawa, T. IRES-Dependent Second Gene Expression Is Significantly Lower Than Cap-Dependent First Gene Expression in a Bicistronic Vector. Mol. Ther. 2000, 1, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Kurata, T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn. J. Infect. Dis. 2004, 57, 236–247. [Google Scholar] [PubMed]
- Tamura, S.; Tanimoto, T.; Kurata, T. Mechanisms of broad cross-protection provided by influenza virus infection and their application to vaccines. Jpn. J. Infect. Dis. 2005, 58, 195–207. [Google Scholar] [PubMed]
- Asahi, Y.; Yoshikawa, T.; Watanabe, I.; Iwasaki, T.; Hasegawa, H.; Sato, Y.; Shimada, S.; Nanno, M.; Matsuoka, Y.; Ohwaki, M.; et al. Protection Against Influenza Virus Infection in Polymeric Ig Receptor Knockout Mice Immunized Intranasally with Adjuvant-Combined Vaccines. J. Immunol. 2002, 168, 2930–2938. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kawaguchi, A.; Ainai, A.; Tamura, S.; Ito, R.; Multihartina, P.; Setiawaty, V.; Pangesti, K.N.A.; Odagiri, T.; Tashiro, M.; et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc. Natl. Acad. Sci. USA 2015, 112, 7809–7814. [Google Scholar] [CrossRef] [PubMed]
- Hohenadl, C.; Wodal, W.; Kerschbaum, A.; Fritz, R.; Howard, M.K.; Farcet, M.R.; Portsmouth, D.; McVey, J.K.; Baker, D.A.; Ehrlich, H.J.; et al. Hyperimmune intravenous immunoglobulin containing high titers of pandemic H1N1 hemagglutinin and neuraminidase antibodies provides dose-dependent protection against lethal virus challenge in SCID mice. Virol. J. 2014, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-scid IL2R null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.L.; Mayner, R.E.; Barry, D.W.; Ennis, F.A. Influenza virus infection in nude mice. J. Infect. Dis. 1976, 133, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Kiso, M.; Lopes, T.J.S.; Yamayoshi, S.; Ito, M.; Yamashita, M.; Nakajima, N.; Hasegawa, H.; Neumann, G.; Kawaoka, Y. Combination Therapy With Neuraminidase and Polymerase Inhibitors in Nude Mice Infected With Influenza Virus. J. Infect. Dis. 2018, 217, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Kiso, M.; Shinya, K.; Shimojima, M.; Takano, R.; Takahashi, K.; Katsura, H.; Kakugawa, S.; Le, M.T.Q.; Yamashita, M.; Furuta, Y.; et al. Characterization of Oseltamivir-Resistant 2009 H1N1 Pandemic Influenza A Viruses. PLoS Pathog. 2010, 6, e1001079. [Google Scholar] [CrossRef] [PubMed]
- Yetter, R.A.; Lehrer, S.; Ramphal, R.; Small, P.A. Outcome of influenza infection: Effect of site of initial infection and heterotypic immunity. Infect. Immun. 1980, 29, 654–662. [Google Scholar] [PubMed]
- Renegar, K.B.; Small, P.A., Jr. Passive transfer of local immunity to influenza virus infection by IgA antibody. J. Immunol. 1991, 146, 1972–1978. [Google Scholar] [PubMed]
- Renegar, K.B.; Small, P.A., Jr.; Boykins, L.G.; Wright, P.F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 2004, 173, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Seibert, C.W.; Rahmat, S.; Krause, J.C.; Eggink, D.; Albrecht, R.A.; Goff, P.H.; Krammer, F.; Duty, J.A.; Bouvier, N.M.; Garcia-Sastre, A.; et al. Recombinant IgA Is Sufficient to Prevent Influenza Virus Transmission in Guinea Pigs. J. Virol. 2013, 87, 7793–7804. [Google Scholar] [CrossRef] [PubMed]
- Johansen, F.E.; Braathen, R.; Brandtzaeg, P. Role of J chain in secretory immunoglobulin formation. Scand. J. Immunol. 2000, 52, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and mast cells in host defense against parasites and venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef] [PubMed]
- Platzer, B.; Elpek, K.G.; Cremasco, V.; Baker, K.; Stout, M.M.; Schultz, C.; Dehlink, E.; Shade, K.T.C.; Anthony, R.M.; Blumberg, R.S.; et al. IgE/FcεRI-Mediated Antigen Cross-Presentation by Dendritic Cells Enhances Anti-Tumor Immune Responses. Cell Rep. 2015, 10, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xu, W.; Wilson, M.; He, B.; Miller, N.W.; Bengtén, E.; Edholm, E.S.; Santini, P.A.; Rath, P.; Chiu, A.; et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat. Immunol. 2009, 10, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Fabre, J.W.; Grehan, A.; Whitehorne, M.; Sawyer, G.J.; Dong, X.; Salehi, S.; Eckley, L.; Zhang, X.; Seddon, M.; Shah, A.; et al. Hydrodynamic gene delivery to the pig liver via an isolated segment of the inferior vena cava. Gene Ther. 2008, 15, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.D.; Barnes, M.N.; Gomez-navarro, J.; Wang, M.; Strong, T.V.; Arafat, W.; Arani, R.B.; Johnson, M.R.; Roberts, B.L.; Siegal, G.P.; et al. A Cancer Gene Therapy Approach Utilizing an Anti-erbB-2 Single- Chain Antibody-encoding Adenovirus (AD21): A Phase I Trial. Clin. Cancer Res. 2000, 6, 3081–3087. [Google Scholar] [PubMed]
- Khorsandi, S.E.; Bachellier, P.; Weber, J.C.; Greget, M.; Jaeck, D.; Zacharoulis, D.; Rountas, C.; Helmy, S.; Helmy, A.; Salama, H.; et al. Minimally invasive and selective hydrodynamic gene therapy of liver segments in the pig and human. Cancer Gene Ther. 2008, 15, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Palli, S.R.; Kapitskaya, M.Z.; Kumar, M.B.; Cress, D.E. Improved ecdysone receptor-based inducible gene regulation system. Eur. J. Biochem. 2003, 270, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Puthumana, J.; Lee, M.C.; Han, J.; Kim, H.S.; Hwang, D.S.; Lee, J.S. Ecdysone receptor (EcR) and ultraspiracle (USP) genes from the cyclopoid copepod Paracyclopina nana: Identification and expression in response to water accommodated fractions (WAFs). Comp. Biochem. Physiol. Part C 2017, 192, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Cakouros, D.; Daish, T.J.; Kumar, S. Ecdysone receptor directly binds the promoter of the Drosophila caspase dronc, regulating its expression in specific tissues. J. Cell Biol. 2004, 165, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Sun, L.; Miao, J.; Krishman, S.; Lebel, F.; Barrett, J.A. Plasma Pharmacokinetics of Veledimex, a Small-Molecule Activator Ligand for a Proprietary Gene Therapy Promoter System, in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2017, 6, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Boisgérault, F.; Mingozzi, F. The skeletal muscle environment and its role in immunity and tolerance to AAV vector-mediated gene transfer. Curr. Gene Ther. 2015, 15, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.P.; Desrosiers, R.C. Promise and problems associated with the use of recombinant AAV for the delivery of anti-HIV antibodies. Mol. Ther. Methods Clin. Dev. 2016, 16, 16068. [Google Scholar] [CrossRef] [PubMed]
- Cao, O.; Hoffman, B.E.; Moghimi, B.; Nayak, S.; Cooper, M.; Zhou, S.; Ertl, H.C.J.; High, K.A.; Herzog, R.W. Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Mol. Ther. 2009, 17, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Somanathan, S.; Qin, Q.; Betts, M.R.; Rech, A.J.; Vonderheide, R.H.; Mueller, C.; Flotte, T.R.; Wilson, J.M. Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for α-1-antitrypsin deficiency. Proc. Natl. Acad. Sci. USA 2017, 114, 1655–1659. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 1, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.J. Systemic AAV Gene Therapy Close to Clinical Trials for Several Neuromuscular Diseases. Mol. Ther. 2017, 25, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Navio, J.M.; Fuchs, S.P.; Pedreño-López, S.; Rakasz, E.G.; Gao, G.; Desrosiers, R.C. Host anti-antibody responses following adeno-associated virus-mediated delivery of antibodies against HIV and SIV in rhesus monkeys. Mol. Ther. 2016, 24, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Meunier, L.; Hendershot, L.M. pERp1 is significantly up-regulated during plasma cell differentiation and contributes to the oxidative folding of immunoglobulin. Proc. Natl. Acad. Sci. USA 2009, 106, 17013–17018. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.R.; Schnepp, B.C.; Zhang, J.; Connell, M.J.; Greene, S.M.; Yuste, E.; Desrosiers, R.C.; Reed Clark, K. Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys. Nat. Med. 2009, 15, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; Du Bridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Deal, C.; Balazs, A.B.; Espinosa, D.A.; Zavala, F.; Baltimore, D.; Ketner, G. Vectored antibody gene delivery protects against Plasmodium falciparum sporozoite challenge in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 12528–12532. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazaki, T.; Chiba, J.; Akashi-Takamura, S. Neutralizing Anti-Hemagglutinin Monoclonal Antibodies Induced by Gene-Based Transfer Have Prophylactic and Therapeutic Effects on Influenza Virus Infection. Vaccines 2018, 6, 35. https://doi.org/10.3390/vaccines6030035
Yamazaki T, Chiba J, Akashi-Takamura S. Neutralizing Anti-Hemagglutinin Monoclonal Antibodies Induced by Gene-Based Transfer Have Prophylactic and Therapeutic Effects on Influenza Virus Infection. Vaccines. 2018; 6(3):35. https://doi.org/10.3390/vaccines6030035
Chicago/Turabian StyleYamazaki, Tatsuya, Joe Chiba, and Sachiko Akashi-Takamura. 2018. "Neutralizing Anti-Hemagglutinin Monoclonal Antibodies Induced by Gene-Based Transfer Have Prophylactic and Therapeutic Effects on Influenza Virus Infection" Vaccines 6, no. 3: 35. https://doi.org/10.3390/vaccines6030035
APA StyleYamazaki, T., Chiba, J., & Akashi-Takamura, S. (2018). Neutralizing Anti-Hemagglutinin Monoclonal Antibodies Induced by Gene-Based Transfer Have Prophylactic and Therapeutic Effects on Influenza Virus Infection. Vaccines, 6(3), 35. https://doi.org/10.3390/vaccines6030035