
Small Wonders—The Use of Nanoparticles for Delivering Antigen

Abstract
:1. Introduction

{kind=link}
| Disease | Number of Reported/Estimated Deaths | Number of Reported/Estimated Cases |
|---|---|---|
| AIDS (HIV) | 1,590,952/- | -/35,000,000 |
| Tuberculosis | -/1,072,678 | 5,725,317/11,468,559 |
| Malaria 1 | 107,225/624,568 | 48,231,939/207,400,000 |
| Hepatitis C | -/425,000 | -/140,000,000 |
| Leishmaniasis 2 | -/25,000 | 213,871/- |
| Schistosomaiasis | 23,313/- | -/300,000 |
| Trypanosomasis 3 | 19,026 4 | 6,314 newly reported/20,000 |
| Ebola virus (2014–2015 outbreak) # | 11,080 | 26,759 |
2. Choice of Material for Nanoparticle Vaccine
| Category | Nanoparticle Material | Size | Antigen (pathogen) | Ref. |
|---|---|---|---|---|
| Inorganic (Non-degradable) | Iron Silica | 20–300 nm | MSP1 (Plasmodium falciparum) BSA | [14,44] |
| Liposome (Non-viral lipids particle) | Cholesterol Lipid Lipid | 200 nm | Polysaccharides (Streptococcus pneumoniae serotype 14) VMP001 (Plasmodium vivax) RTS,S/AS01B (Plasmodium falciparum CSP + hepatitis B protein hybrid) | [45,46,47] |
| Virus-like particle | Viral capsid expressed in Bacurlovirus Bacteriophage expressed in E. coli C41 | 27–60 nm | Capsid protein L1 + L2 (HPV) Capsid protein L2 (HPV) | [19,20] |
| Polymeric | Chitosan | 160–1000 nm | Hepatitis B | [30,48,49,50,51,52] |
| PLGA | Ovalbumin | |||
| PLGA | Tetanus toxoid | |||
| PVPONAlk | Ovalbumin | |||
| γ-PGA | gp120 (HIV-1) |
3. Nanoparticle as an Efficient Antigen Delivery System
3.1. Trafficking of Antigen to the Lymph Nodes
3.2. Nanoparticle-Induced Immunity
4. Disadvantages of Some Preparation Methods
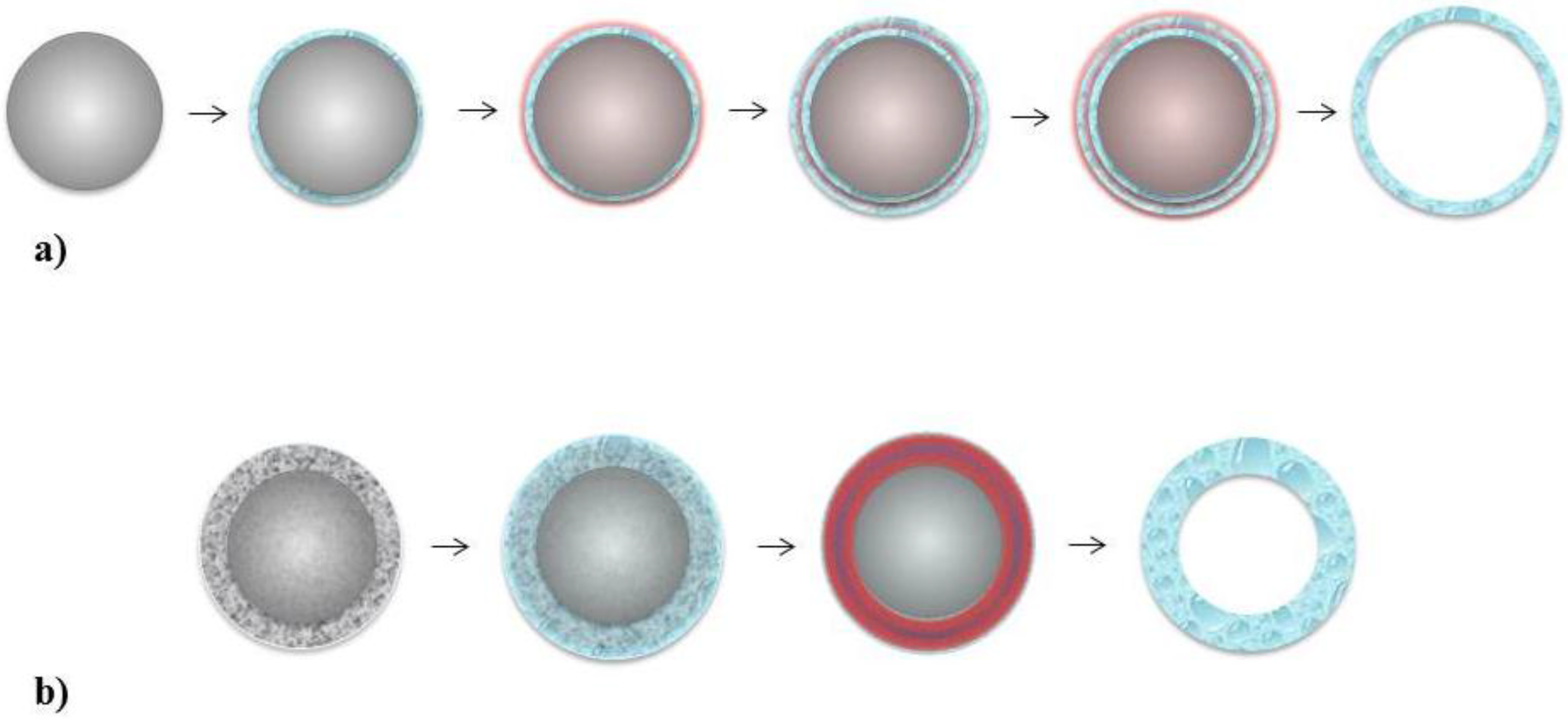
5. Novel Approach—Nanocapsule Assembly Using Templates
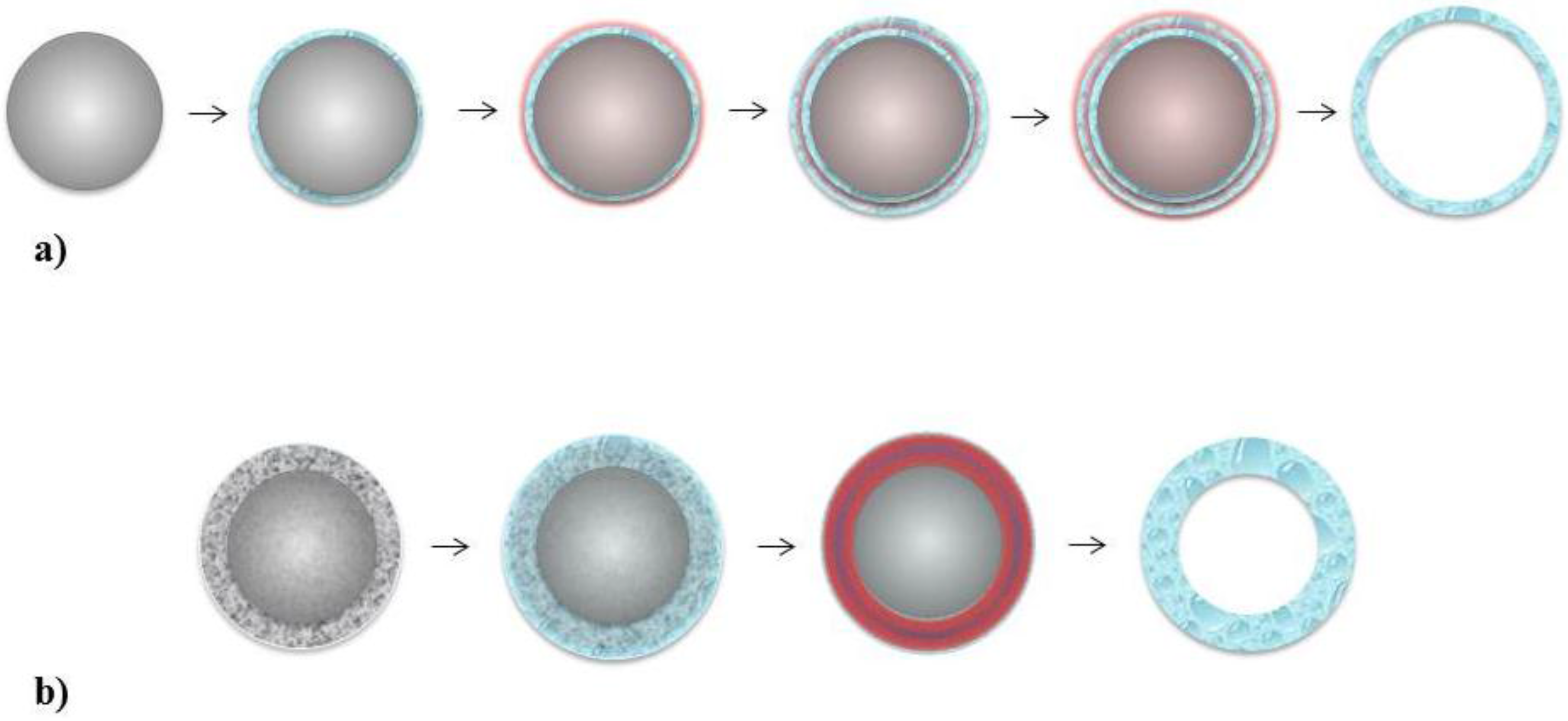
5.1. Layer-by-Layer
5.2. Single Step Assembly
6. Adjuvant
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nichol, K.L. The efficacy, effectiveness and cost-effectiveness of inactivated influenza virus vaccines. Vaccine 2003, 21, 1769–1775. [Google Scholar] [CrossRef]
- World Health Organization WHO. Ebola Situation Reports. Disease Outbreak News (DONs); WHO. Available online: http://apps.who.int/ebola/en/ebola-situation-reports (accessed on 14 May 2015).
- World Health Organization World Health Statistics: Selected infectious diseases. Available online: http://apps.who.int/gho/data/node.main.30?lang=en (accessed on 14 May 2015).
- Atkins, H.S.; Morton, M.; Griffin, K.F.; Stokes, M.; Nataro, J.P.; Titball, R.W. Recombinant Salmonella vaccines for biodefence. Vaccine 2006, 24, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Beverley, P.C.L. Immunology of vaccination. Br. Med. Bull. 2002, 62, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Moron, G.; Dadaglio, G.; Leclerc, C. New tools for antigen delivery to the MHC class I pathway. Trends Immunol. 2004, 25, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, J.B.; Donnelly, J.J.; Parker, S.E.; Rhodes, G.H.; Felgner, P.L.; Dwarki, V.J.; Gromkowski, S.H.; Deck, R.R.; Dewitt, C.M.; Friedman, A.; et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 1993, 259, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Immunogenicity of DNA vaccines in humans: It takes two to tango. Hum. Vaccines 2014, 4, 449–452. [Google Scholar] [CrossRef]
- Taki, A.; Kikidopoulos, N.; Smooker, P. Improving the Immunogenicity of DNA Vaccines: A Nano-Sized Task? Nova Science Publisher: New York, NY, USA, 2011; pp. 37–65. [Google Scholar]
- Atkinson, W.; Wolfe, C.; Hamborsky, J. Epidemiology and Prevention of Vaccine-Preventable Diseases; Public Health Foundation Publications: Atlanta, GA, USA, 2011. [Google Scholar]
- Epidemiology and Prevention of Vaccine-Preventable Diseases; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2015.
- Mody, K.T.; Popat, A.; Mahony, D.; Cavallaro, A.S.; Yu, C.; Mitter, N. Mesoporous silica nanoparticles as antigen carriers and adjuvants for vaccine delivery. Nanoscale 2013, 5, 5167–5179. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef] [PubMed]
- Pusic, K.; Aguilar, Z.; McLoughlin, J.; Kobuch, S.; Xu, H.; Tsang, M.; Wang, A.; Hui, G. Iron oxide nanoparticles as a clinically acceptable delivery platform for a recombinant blood-stage human malaria vaccine. FASEB J. 2013, 27, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Lim, J.S. Induction of functional changes of dendritic cells by silica nanoparticles. Immune Netw. 2012, 12, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Vallhov, H.; Gabrielsson, S.; Strømme, M.; Scheynius, A.; Garcia-Bennett, A.E. Mesoporous silica particles induce size dependent effects on human dendritic cells. Nano Lett. 2007, 7, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Sanderson, B.J.S.; Wang, H. Cytotoxicity and genotoxicity of ultrafine crystalline SiO2 particulate in cultured human lymphoblastoid cells. Environ. Mol. Mutagen 2007, 48, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, L.; Teng, X.; Huang, X.; Liu, H.; Chen, D.; Ren, J.; He, J.; Tang, F. Single and repeated dose toxicity of mesoporous hollow silica nanoparticles in intravenously exposed mice. Biomaterials 2011, 32, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Tyler, M.; Tumban, E.; Peabody, D.S.; Chackerian, B. The use of hybrid virus-like particles to enhance the immunogenicity of a broadly protective HPV vaccine. Biotechnol. Bioeng. 2014, 111, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Slupetzky, K.; Gambhira, R.; Culp, T.D.; Shafti-Keramat, S.; Schellenbacher, C.; Christensen, N.D.; Roden, R.B.S.; Kirnbauer, R. A papillomavirus-like particle (VLP) vaccine displaying HPV16 L2 epitopes induces cross-neutralizing antibodies to HPV11. Vaccine 2007, 25, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Korsholm, K.S.; Andersen, P.; Perrie, Y.; Christensen, D. Liposomal vaccine delivery systems. Expert Opin. Drug Deliv. 2011, 8, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Heurtault, B.; Frisch, B.; Pons, F. Liposomes as delivery systems for nasal vaccination: Strategies and outcomes. Expert Opin. Drug Deliv. 2010, 7, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Mauser, T.; Dejugnat, C.; Sukhorukov, G.B. Reversible pH-dependent properties of multilayer microcapsules made of weak polyelectrolytes. Macromol. Rapid Commun. 2004, 25, 1781–1785. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, Y.; Cui, J.; Hosta-Rigau, L.; Heath, J.K.; Nice, E.C.; Caruso, F. Encapsulation of water-insoluble drugs in polymer capsules prepared using mesoporous silica templates for intracellular drug delivery. Adv. Mater. 2010, 22, 4293–4297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bansal, V.; Zelikin, A.N.; Caruso, F. Templated synthesis of single-component polymer capsules and their application in drug delivery. Nano Lett. 2008, 8, 1741–1745. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Gao, C.; Moehwald, H. Single polyelectrolyte microcapsules fabricated by glutaraldehyde-mediated covalent layer-by-layer assembly. Macromol. Rapid Commun. 2006, 27, 2078–2083. [Google Scholar] [CrossRef]
- Zelikin, A.N.; Li, Q.; Caruso, F. Disulfide-stabilized poly(methacrylic acid) capsules: Formation, cross-linking, and degradation behavior. Chem. Mater. 2008, 20, 2655–2661. [Google Scholar] [CrossRef]
- Hanlon, D.; Saluja, S.; Sharp, F.; Hong, E.; Khalil, D.; Tigelaar, R.; Fahmy, T.; Edelson, R.; Robinson, E. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. Int. J. Nanomed. 2014, 9, 5231–5246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Liu, Y.; Chen, X.; Liu, Q.; Jia, J.; Yang, T.; Qiu, S.; Ma, G. Immune responses to vaccines involving a combined antigen-nanoparticle mixture and nanoparticle-encapsulated antigen formulation. Biomaterials 2014, 35, 6086–6097. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Wang, X.; Uto, T.; Baba, M.; Akashi, M. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials 2007, 28, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-W.; Hsu, P.Y.-J. The effect of poly(d,l-lactide-co-glycolide) microparticles with polyelectrolyte self-assembled multilayer surfaces on the cross-presentation of exogenous antigens. Biomaterials 2008, 29, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, K.; Ueminami, A.; Kurumada, S.; Nomura, Y. Self-association of cholesteryl-bearing poly(l-lysine) in water and control of its secondary structure by host-guest interaction with cyclodextrin. Macromolecules 2000, 33, 6752–6756. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, B. pH-Controlled drug loading and release from biodegradable microcapsules. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.Y.; Kobiasi, M.A.; Zeng, W.; Mainwaring, D.; Jackson, D.C. Chitosan-based particles as biocompatible delivery vehicles for peptide and protein-based vaccines. Procedia Vaccinol. 2012, 6, 74–79. [Google Scholar] [CrossRef]
- Goethals, E.C.; Elbaz, A.; Lopata, A.L.; Bhargava, S.K.; Bansal, V. Decoupling the effects of the size, wall thickness, and porosity of curcumin-loaded chitosan nanocapsules on their anticancer efficacy: Size is the winner. Langmuir 2013, 29, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Al-Qadi, S.; Grenha, A.; Carrión-Recio, D.; Seijo, B.; Remuñán-López, C. Microencapsulated chitosan nanoparticles for pulmonary protein delivery: In vivo evaluation of insulin-loaded formulations. J. Control. Release 2012, 157, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Langer, K.; Anhorn, M.G.; Steinhauser, I.; Dreis, S.; Celebi, D.; Schrickel, I.; Faust, S.; Vogel, V. Human serum albumin (HSA) nanoparticles: Reproducibility of preparation process and kinetics of enzymatic degradation. Int. J. Pharm. 2008, 347, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Langer, K.; Balthasar, S.; Vogel, V.; Dinauer, N.; von Briesen, H.; Schubert, D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2003, 257, 169–180. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Altintas, I.; Heukers, R.; van der Meel, R. Nanobody-albumin nanoparticles (NANAPs) for the delivery of a multikinase inhibitor 17864 to EGFR overexpressing tumor cells. J. Control. Release 2013, 165, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.S.; Endsley, A.N.; Huang, L. Design considerations for liposomal vaccines: Influence of formulation parameters on antibody and cell-mediated immune responses to liposome associated antigens. Vaccine 2012, 30, 2256–2272. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.V.; de Vos, P.; Niclou, S.P. Drug and cell encapsulation: Alternative delivery options for the treatment of malignant brain tumors. Adv. Drug Deliv. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle delivery of cancer drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Lee, K.; Choi, J.-N.; Hwang, Y.-K.; Yun, M.-Y.; Kim, H.-J.; Won, Y.S.; Kim, S.-J.; Kwon, H.; Huh, S. Intracellular protein delivery by hollow mesoporous silica capsules with a large surface hole. Nanotechnology 2012. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Bai, L.; Reboulet, R.; Matthew, R.; Engler, D.A.; Teyton, L.; Bendelac, A.; Savage, P.B. A peptide-free, liposome-based oligosaccharide vaccine, adjuvanted with a natural killer T cell antigen, generates robust antibody responses in vivo. Chem. Sci. 2014, 5, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Suh, H.; Li, A.V.; Ockenhouse, C.F.; Yadava, A.; Irvine, D.J. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. USA 2012, 109, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.L.; Rao, M.; Wassef, N.M.; Glenn, G.M.; Rothwell, S.W.; Alving, C.R. Liposomes containing lipid a serve as an adjuvant for induction of antibody and cytotoxic T-cell responses against RTS,S malaria antigen. Infect. Immun. 1998, 66, 2859–2865. [Google Scholar] [PubMed]
- Prego, C.; Paolicelli, P.; Diaz, B.; Vicente, S.; Sanchez, A.; Gonzalez-Fernandez, A.; Jose Alonso, M. Chitosan-based nanoparticles for improving immunization against hepatitis B infection. Vaccine 2010, 28, 2607–2614. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Ackerman, A.L.; Cody, V.; Giodini, A.; Hinson, E.R.; Cresswell, P.; Edelson, R.L.; Saltzman, W.M.; Hanlon, D.J. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 2006, 117, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Diwan, M.; Tafaghodi, M.; Samuel, J. Enhancement of immune responses by co-delivery of a CpG oligodeoxynucleotide and tetanus toxoid in biodegradable nanospheres. J. Control. Release 2002, 85, 247–262. [Google Scholar] [CrossRef]
- Mintern, J.D.; Percival, C.; Kamphuis, M.M. J.; Chin, W.J.; Caruso, F.; Johnston, A.P.R. Targeting dendritic cells: The role of specific receptors in the internalization of polymer capsules. Adv. Healthc. Mater. 2013, 2, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Uto, T.; Akagi, T.; Akashi, M.; Baba, M. Induction of potent CD8+ T-Cell responses by novel biodegradable nanoparticles carrying human immunodeficiency virus type 1 gp120. J. Virol. 2007, 81, 10009–10016. [Google Scholar] [CrossRef] [PubMed]
- Heath, W.R.; Carbone, F.R. The skin-resident and migratory immune system in steady state and memory: Innate lymphocytes, dendritic cells and T cells. Nat. Immunol. 2013, 14, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Villadangos, J.A. Antigen presentation by dendritic cells in vivo. Curr. Opin. Immunol. 2009, 21, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Albiston, A.L.; Wicks, I.P.; Chai, S.Y.; Villadangos, J.A. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20377–20381. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000, 151, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Ochando, J. Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 2008, 26, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.S.; Smith, C.M.; Belz, G.T.; van Lint, A.L.; Wakim, L.M.; Heath, W.R.; Carbone, F.R. Epidermal viral immunity induced by CD8 alpha+ dendritic cells but not by Langerhans cells. Science 2003, 301, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Lamsoul, I.; Metais, A.; Gouot, E.; Heuze, M.L.; Lennon-Dumenil, A.M.; Moog-Lutz, C.; Lutz, P.G. ASB2alpha regulates migration of immature dendritic cells. Blood 2013, 122, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, Y.; Yoshino, H. Lymphatic targeting with nanoparticulate system. Adv. Drug Deliv. Rev. 2001, 47, 55–64. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neill, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Pack, D.W. DNA delivery: Timing is everything. Nat. Mater. 2004, 3, 133–134. [Google Scholar] [CrossRef] [PubMed]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Norkin, L.C. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp. Cell Res. 1999, 246, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Mottram, P.L.; Leong, D.; Crimeen-Irwin, B.; Gloster, S.; Xiang, S.D.; Meanger, J.; Ghildyal, R.; Vardaxis, N.; Plebanski, M. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: Formulation of a model vaccine for respiratory syncytial virus. Mol. Pharm. 2007, 4, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Scheerlinck, J.; Gloster, S.; Gamvrellis, A.; Mottram, P.L. Systemic immune responses in sheep, induced by a novel nano-bead adjuvant. Vaccine 2006, 24, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Gamvrellis, A.; Gloster, S.; Jefferies, M.; Mottram, P.L.; Smooker, P.; Plebanski, M.; Scheerlinck, J.P.Y. Characterisation of local immune responses induced by a novel nano-particle based carrier-adjuvant in sheep. Vet. Immunol. Immunopathol. 2013, 155, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sneh-Edri, H.; Likhtenshtein, D.; Stepensky, D. Intracellular targeting of PLGA nanoparticles encapsulating antigenic peptide to the endoplasmic reticulum of dendritic cells and its effect on antigen cross-presentation in vitro. Mol. Pharm. 2011, 8, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.B.; Geary, S.M.; Salem, A.K. Biodegradable particles as vaccine delivery systems: Size matters. AAPS J. 2012, 15, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Reznikoff, G.; Dranoff, G.; Rock, K.L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 1997, 158, 2723–2730. [Google Scholar] [PubMed]
- Sexton, A.; Whitney, P.G.; Chong, S.-F.; Zelikin, A.N.; Johnston, A.P.R.; de Rose, R.; Brooks, A.G.; Caruso, F.; Kent, S.J. A protective vaccine delivery system for in vivo T cell stimulation using nanoengineered polymer hydrogel capsules. ACS Nano 2009, 3, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Kaba, S.A.; McCoy, M.E.; Doll, T.A.P.F.; Brando, C.; Guo, Q.; Dasgupta, D.; Yang, Y.; Mittelholzer, C.; Spaccapelo, R.; Crisanti, A.; et al. Protective antibody and CD8+ T-cell responses to the Plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS ONE 2012, 7, e48304. [Google Scholar] [CrossRef] [PubMed]
- Hirosue, S.; Kourtis, I.C.; van der Vlies, A.J.; Hubbell, J.A.; Swartz, M.A. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine 2010, 28, 7897–7906. [Google Scholar] [CrossRef] [PubMed]
- Plebanski, M.; Gilbert, S.C.; Schneider, J.; Hannan, C.M.; Layton, G.; Blanchard, T.; Becker, M.; Smith, G.; Butcher, G.; Sinden, R.E.; et al. Protection from Plasmodium berghei infection by priming and boosting T cells to a single class I-restricted epitope with recombinant carriers suitable for human use. Eur. J. Immunol. 1998, 28, 4345–4355. [Google Scholar] [CrossRef]
- Landsverk, O.J.B.; Bakke, O.; Gregers, T.F. MHC II and the endocytic pathway: Regulation by invariant chain. Scand. J. Immunol. 2009, 70, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Kautz, A.; Boehnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L. A new foreign policy: MHC class I molecules monitor the outside world. Immunol. Today 1996, 17, 131–137. [Google Scholar] [CrossRef]
- Segura, E.; Villadangos, J.A. A modular and combinatorial view of the antigen cross-presentation pathway in dendritic cells. Traffic 2011, 12, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Akagi, T.; Akashi, M. Effect of hydrophobic side chains in the induction of immune responses by nanoparticle adjuvants consisting of amphiphilic poly(γ-glutamic acid). Bioconjugate Chem. 2015, 26, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.C.; Karulin, A.Y.; Tary-Lehmann, M.; Hesse, M.D.; Radeke, H.; Heeger, P.S.; Trezza, R.P.; Heinzel, F.P.; Forsthuber, T.; Lehmann, P.V. Adjuvant-guided type-1 and type-2 immunity: Infectious/noninfectious dichotomy defines the class of response. J. Immunol. 1999, 162, 3942–3949. [Google Scholar] [PubMed]
- Arkema, A.; Huckriede, A.; Schoen, P.; Wilschut, J.; Daemen, T. Induction of cytotoxic T lymphocyte activity by fusion-active peptide-containing virosomes. Vaccine 2000, 18, 1327–1333. [Google Scholar] [CrossRef]
- Shima, F.; Akagi, T.; Akashi, M. Synthesis and preparation of nanoparticles composed of amphiphilic poly(γ-glutamic acid) with different hydrophobic side chains and their potential of membrane disruptive activity. Colloid Polym. Sci. 2014, 292, 2663–2671. [Google Scholar] [CrossRef]
- Schwendeman, S.P. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit. Rev. Ther. Drug 2002, 19, 73–98. [Google Scholar] [CrossRef]
- Panyam, J.; Zhou, W.Z.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly(d,l-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Shima, F.; Akashi, M. Intracellular degradation and distribution of protein-encapsulated amphiphilic poly(amino acid) nanoparticles. Biomaterials 2011, 32, 4959–4967. [Google Scholar]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Toyama, M.; Nishi, Y.; Akagi, T.; Shima, F. Uptake of biodegradable poly (γ-glutamic acid) nanoparticles and antigen presentation by dendritic cells in vivo. Results Immunol. 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Akagi, T.; Hamasaki, T.; Akashi, M.; Baba, M. Modulation of innate and adaptive immunity by biodegradable nanoparticles. Immunol. Lett. 2009, 125, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Mohr, E.; Cunningham, A.F.; Toellner, K.M.; Bobat, S.; Coughlan, R.E.; Bird, R.A.; MacLennan, I.C.M.; Serre, K. IFN-gamma produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc. Natl. Acad. Sci. USA 2010, 107, 17292–17297. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Gamble, S.; Rothstein, L. Presentation of exogenous antigen with class I major histocompatibility complex molecules. Science 1990, 249, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.R.; Chiu, G.N.C. Liposomes as sterile preparations and limitations of sterilisation techniques in liposomal manufacturing. Asian J. Pharm. Sci. 2013, 8, 88–95. [Google Scholar] [CrossRef]
- Tamber, H.; Johansen, P.; Merkle, H.; Gander, B. Formulation aspects of biodegradable polymeric microspheres for antigen delivery. Adv. Drug Deliv. Rev. 2005, 57, 357–376. [Google Scholar] [PubMed]
- Borchert, U.; Lipprandt, U.; Bilang, M.; Kimpfler, A.; Rank, A.; Peschka-Suess, R.; Schubert, R.; Lindner, P.; Forster, S. pH-induced release from P2VP-PEO block copolymer vesicles. Langmuir 2006, 22, 5843–5847. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.V. Biopolymer albumin for diagnosis and in drug delivery. Drug Dev. Res. 2003, 58, 219–247. [Google Scholar] [CrossRef]
- Sundar, S.; Kundu, J.; Kundu, S.C. Biopolymeric nanoparticles. Sci. Technol. Adv. Mater. 2010. [Google Scholar] [CrossRef]
- Sah, H. Stabilization of proteins against methylene chloride water interface-induced denaturation and aggregation. J. Control. Release 1999, 58, 143–151. [Google Scholar] [CrossRef]
- Panyam, J.; Dali, M.M.; Sahoo, S.K.; Ma, W. Polymer degradation and in vitro release of a model protein from poly(d,l-lactide-co-glycolide) nano- and microparticles. J. Control Release 2003, 92, 173–187. [Google Scholar] [CrossRef]
- Akagi, T.; Baba, M.; Akashi, M. Biodegradable nanoparticles as vaccine adjuvants and delivery systems: Regulation of immune responses by nanoparticle-based vaccine. Polym. Nanomed. 2012, 247, 31–64. [Google Scholar]
- Kwon, Y.J.; Standley, S.M.; Goh, S.L.; Fréchet, J.M.J. Enhanced antigen presentation and immunostimulation of dendritic cells using acid-degradable cationic nanoparticles. J. Control. Release 2005, 105, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Kwon, S.M.; Chung, H.; Lee, S.-Y.; Kwon, S.-H.; Jeon, H.; Kim, Y.; Park, J.H.; Kim, J.; Her, S.; et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticle. J. Control Release 2009, 135, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Bieber, T.; Li, Y.; Elsässer, H.P.; Kissel, T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: Effect of molecular weight on transfection efficiency and cytotoxicity. Pharm. Res. 1999, 16, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.A.; Pillai, B.; Das, T.K.; Singh, Y.; Maiti, S. Molecular effects of uptake of gold nanoparticles in HeLa cells. ChemBioChem 2007, 8, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Okamoto, K.; Doi, K.; Harano, K.; Noiri, E.; Nakamura, E. siRNA Delivery targeting to the lung via agglutination-induced accumulation and clearance of cationic tetraamino fullerene. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Stellacci, F. Effect of surface properties on nanoparticle-cell interactions. Small 2010, 6, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Dokka, S.; Toledo, D.; Shi, X.G.; Castranova, V.; Rojanasakul, Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm. Res. 2000, 17, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.Z.; Mallery, S.R.; Schwendeman, S.P. Stabilization of proteins encapsulated in injectable poly (lactide-co-glycolide). Nat. Biotechnol. 2000, 18, 52–57. [Google Scholar] [PubMed]
- Jiang, W.L.; Schwendeman, S.P. Stabilization and controlled release of bovine serum albumin encapsulated in poly(d,l-lactide) and poly(ethylene glycol) microsphere blends. Pharm. Res. 2001, 18, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Harashima, H. The polyethyleneglycol dilemma: Advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Fisher, M.; Juliano, R.L. Targeted albumin-based nanoparticles for delivery of amphipathic drugs. Bioconjug. Chem. 2011, 22, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Ryan, A.J. Bilayers and interdigitation in block copolymer vesicles. J. Am. Chem. Soc. 2005, 127, 8757–8764. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Yang, S.R.; An, E.J.; Kim, J.-D. Biodegradable polymersomes from poly(2-hydroxyethyl aspartamide) grafted with lactic acid oligomers in aqueous solution. Macromolecules 2006, 39, 4938–4940. [Google Scholar] [CrossRef]
- Hauschild, S.; Lipprandt, U.; Rumplecker, A.; Borchert, U.; Rank, A.; Schubert, R.; Forster, S. Direct preparation and loading of lipid and polymer vesicles using inkjets. Small 2005, 1, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Rahimnejad, M.; Mokhtarian, N.; Ghasemi, M. Production of protein nanoparticles for food and drug delivery system. Afr. J. Biotechol. 2009, 8, 4738–4743. [Google Scholar]
- Weber, C.; Kreuter, J.; Langer, K. Desolvation process and surface characteristics of HSA-nanoparticles. Int. J. Pharm. 2000, 196, 197–200. [Google Scholar] [CrossRef]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal uptake of biodegradable microparticles: Effect of particle size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Marrano, P.; Kumar, S.; Leadley, M.; Elias, E.; Thorner, P.; Baruchel, S. Nab-Paclitaxel is an active drug in preclinical model of pediatric solid tumors. Clin. Cancer Res. 2013, 19, 5972–5983. [Google Scholar] [CrossRef] [PubMed]
- Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davis, S.A.; Mohwald, H. Novel hollow polymer shells by colloid-templated assembly of polyelectrolytes. Angew. Chem. Int. Ed. 1998, 37, 2202–2205. [Google Scholar] [CrossRef]
- Caruso, F.; Caruso, R.A.; Mohwald, H. Nanoengineering of inorganic and hybrid hollow spheres by colloidal templating. Science 1998, 282, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; van Koeverden, M.P.; Müllner, M.; Kempe, K.; Caruso, F. Emerging methods for the fabrication of polymer capsules. Adv. Colloid Interface Sci. 2014, 207, 14–31. [Google Scholar] [CrossRef] [PubMed]
- De Rose, R.; Zelikin, A.N.; Johnston, A.P.R.; Sexton, A.; Chong, S.F.; Cortez, C.; Mulholland, W.; Caruso, F.; Kent, S.J. Binding, internalization, and antigen presentation of vaccine-loaded nanoengineered capsules in blood. Adv. Mater. 2008, 20, 4698–4703. [Google Scholar] [CrossRef]
- Decher, G.; Hong, J.D. Buildup of ultrathin multilayer films by a self-assembly process, 1 consecutive adsorption of anionic and cationic bipolar amphiphiles on charged surfaces. Makromol. Chem. 2011, 46, 321–327. [Google Scholar] [CrossRef]
- Chong, S.-F.; Sexton, A.; de Rose, R.; Kent, S.J.; Zelikin, A.N.; Caruso, F. A paradigm for peptide vaccine delivery using viral epitopes encapsulated in degradable polymer hydrogel capsules. Biomaterials 2009, 30, 5178–5186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Caruso, F. Nanoporous protein particles through templating mesoporous silica spheres. Adv. Mater. 2006, 18, 795–800. [Google Scholar] [CrossRef]
- Wang, Y.J.; Caruso, F. Mesoporous silica spheres as supports for enzyme immobilization and encapsulation. Chem. Mater. 2005, 17, 953–961. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Büchel, G.; Unger, K.K.; Matsumoto, A.; Tsutsumi, K. A novel pathway for synthesis of submicrometer-size solid core/mesoporous shell silica spheres. Adv. Mater. 1998, 10, 1036–1038. [Google Scholar] [CrossRef]
- Romero, E.L.; Morilla, M.J.; Regts, J.; Koning, G.A.; Scherphof, G.L. On the mechanism of hepatic transendothelial passage of large liposomes. FEBS Lett. 1999, 448, 193–196. [Google Scholar] [CrossRef]
- Reed, S.G.; Bertholet, S.; Coler, R.N.; Friede, M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009, 30, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; O’Hagan, D.T.; Amiji, M.M.; Brito, L.A. The impact of size on particulate vaccine adjuvants. Nanomedicine 2014, 9, 2671–2681. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques, 3rd ed.; Academic Press: Oxford, UK, 2013. [Google Scholar]
- Di Marco, M.; Shamsuddin, S.; Razak, K.A.; Aziz, A.A.; Devaux, C.; Borghi, E.; Levy, L.; Sadun, C. Overview of the main methods used to combine proteins with nanosystems: Absorption, bioconjugation, and encapsulation. Int. J. Nanomed. 2010, 5, 37. [Google Scholar] [CrossRef]
- Demento, S.L.; Siefert, A.L.; Bandyopadhyay, A. Pathogen-associated molecular patterns on biomaterials: A paradigm for engineering new vaccines. Trends Biotechnol. 2011, 29, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [PubMed]
- Demento, S.L.; Bonafe, N.; Cui, W.; Kaech, S.M.; Caplan, M.J.; Fikrig, E.; Ledizet, M.; Fahmy, T.M. TLR9-targeted biodegradable nanoparticles as immunization vectors protect against West Nile encephalitis. J. Immunol. 2010, 185, 2989–2997. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Van der Aar, A.M. G.; Sylva-Steenland, R.M.R.; Bos, J.D.; Kapsenberg, M.L.; de Jong, E.C.; Teunissen, M.B.M. Cutting edge: Loss of TLR2, TLR4, and TLR5 on Langerhans cells abolishes bacterial recognition. J. Immunol. 2007, 178, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Means, T.K.; Hayashi, F.; Smith, K.D.; Aderem, A.; Luster, A.D. The Toll-like receptor 5 stimulus bacterial flagellin induces maturation and chemokine production in human dendritic cells. J. Immunol. 2003, 170, 5165–5175. [Google Scholar] [CrossRef] [PubMed]
- McDermott, P.F.; Ciacci-Woolwine, F.; Snipes, J.A.; Mizel, S.B. High-affinity interaction between gram-negative flagellin and a cell surface polypeptide results in human monocyte activation. Infec. Immun. 2000, 68, 5525–5529. [Google Scholar] [CrossRef]
- Eaves-Pyles, T.; Murthy, K.; Liaudet, L.; Virág, L.; Ross, G.; Soriano, F.G.; Szabó, C.; Salzman, A.L. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J. Immunol. 2001, 166, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Tang, N.; Jan, J.T.; Huang, M.H.; Lu, C.Y.; Chiang, B.L.; Huang, L.M.; Wu, S.C. Use of recombinant flagellin in oil-in-water emulsions enhances hemagglutinin-specific mucosal IgA production and IL-17 secreting T cells against H5N1 avian influenza virus infection. Vaccine 2015. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Duluc, D.; Fremaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct Stimulation of human T cells via TLR5 and TLR7/8: Flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, D.; Sokolova, V.; Zhong, M.; Zhang, E.; Yang, J.; Li, W.; Yang, Y.; Buer, J.; Westendorf, A.M.; Epple, M.; Yan, H. Calcium phosphate nanoparticles show an effective activation of the innate immune response in vitro and in vivo after functionalization with flagellin. Virol. Sin. 2013, 29, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Knuschke, T.; Sokolova, V.; Rotan, O.; Wadwa, M.; Tenbusch, M.; Hansen, W.; Staeheli, P.; Epple, M.; Buer, J.; Westendorf, A.M. Immunization with biodegradable nanoparticles efficiently induces cellular immunity and protects against influenza virus infection. J. Immunol. 2013, 190, 6221–6229. [Google Scholar] [CrossRef] [PubMed]
- Waithman, J.; Zanker, D.; Xiao, K.; Oveissi, S.; Wylie, B.; Ng, R.; Toegel, L.; Chen, W. Resident CD8+ and migratory CD103+ dendritic cells control CD8 T cell immunity during acute influenza infection. PLoS ONE 2013, 8, e66136. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Willart, M.A.M.; van Rijt, L.S.; Muskens, F.; Kool, M.; Baas, C.; Thielemans, K.; Bennett, C.; Clausen, B.E.; Hoogsteden, H.C.; et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J. Exp. Med. 2008, 205, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Desch, A.N.; Randolph, G.J.; Murphy, K.; Gautier, E.L.; Kedl, R.M.; Lahoud, M.H.; Caminschi, I.; Shortman, K.; Henson, P.M.; Jakubzick, C.V. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J. Exp. Med. 2011, 208, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Pooley, J.L.; Heath, W.R.; Shortman, K. Cutting edge: Intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 2001, 166, 5327–5330. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, A.M.; Brown, G.D. C-type lectins and phagocytosis. Immunobiology 2009, 214, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Engering, A.J.; Cella, M.; Fluitsma, D.; Brockhaus, M.; Hoefsmit, E.C.; Lanzavecchia, A.; Pieters, J. The mannose receptor functions as a high capacity and broad specificity antigen receptor in human dendritic cells. Eur. J. Immunol. 1997, 27, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Conde, B.; Song, E.-H.; Chavez-Santoscoy, A.; Phanse, Y.; Ramer-Tait, A.E.; Pohl, N.L.B.; Wannemuehler, M.J.; Bellaire, B.H.; Narasimhan, B. Mannose-functionalized “pathogen-like” polyanhydride nanoparticles target C-type lectin receptors on dendritic cells. Mol. Pharm. 2011, 8, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Le Cabec, V.; Emorine, L.J.; Toesca, I.; Cougoule, C.; Maridonneau-Parini, I. The human macrophage mannose receptor is not a professional phagocytic receptor. J. Leukoc. Biol. 2005, 77, 934–943. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taki, A.; Smooker, P. Small Wonders—The Use of Nanoparticles for Delivering Antigen. Vaccines 2015, 3, 638-661. https://doi.org/10.3390/vaccines3030638
Taki A, Smooker P. Small Wonders—The Use of Nanoparticles for Delivering Antigen. Vaccines. 2015; 3(3):638-661. https://doi.org/10.3390/vaccines3030638
Chicago/Turabian StyleTaki, Aya, and Peter Smooker. 2015. "Small Wonders—The Use of Nanoparticles for Delivering Antigen" Vaccines 3, no. 3: 638-661. https://doi.org/10.3390/vaccines3030638
APA StyleTaki, A., & Smooker, P. (2015). Small Wonders—The Use of Nanoparticles for Delivering Antigen. Vaccines, 3(3), 638-661. https://doi.org/10.3390/vaccines3030638