
Can the Revolution in mRNA-Based Vaccine Technologies Solve the Intractable Health Issues of Current Ruminant Production Systems?


Abstract
1. Introduction
1.1. Sustainable Development Goals
1.2. Veterinary Vaccine Development
1.3. Emergence of mRNA Vaccination
1.4. Comparative Cost of mRNA Vaccines
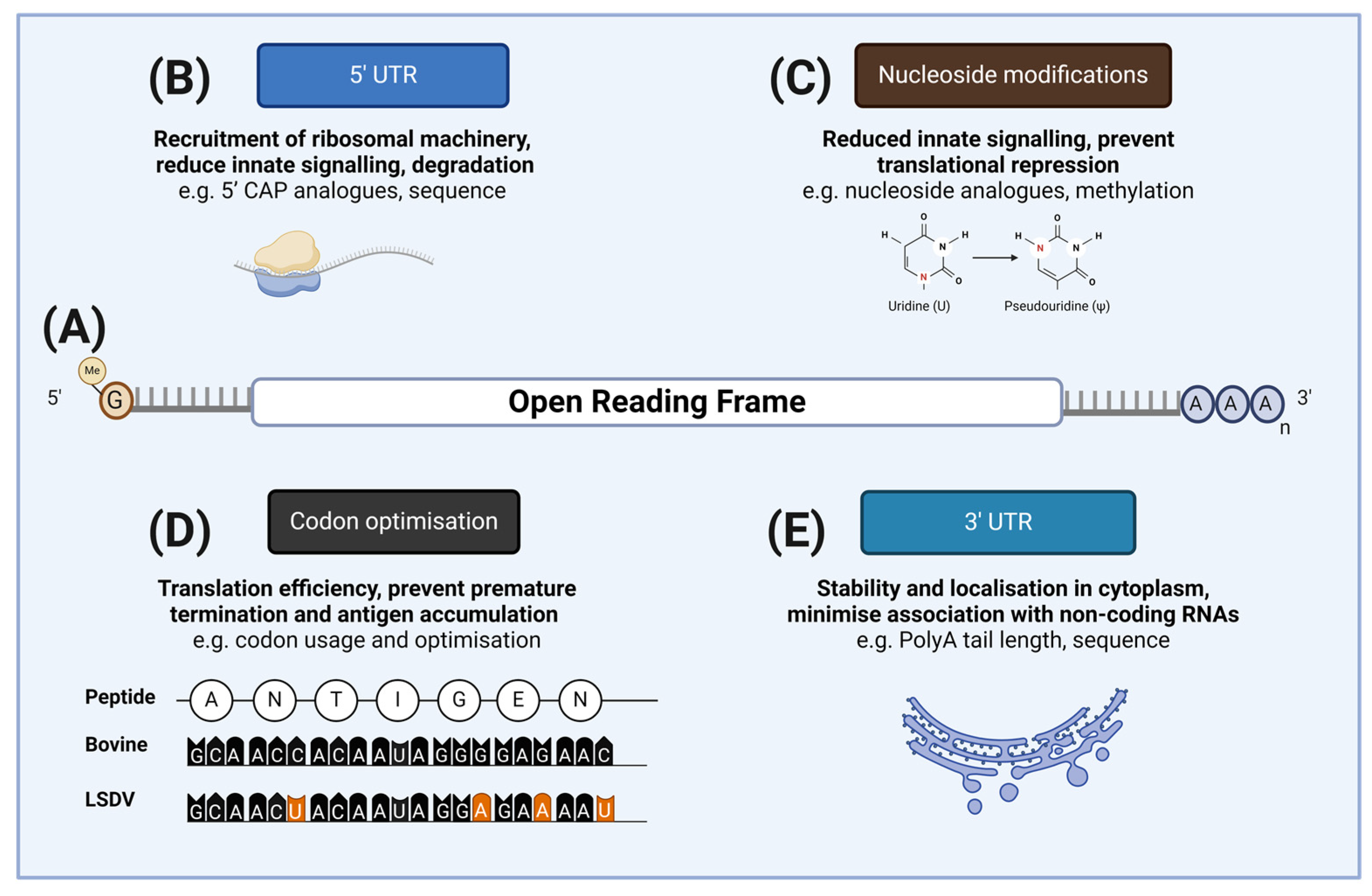
2. Design and Delivery of mRNA Vaccines
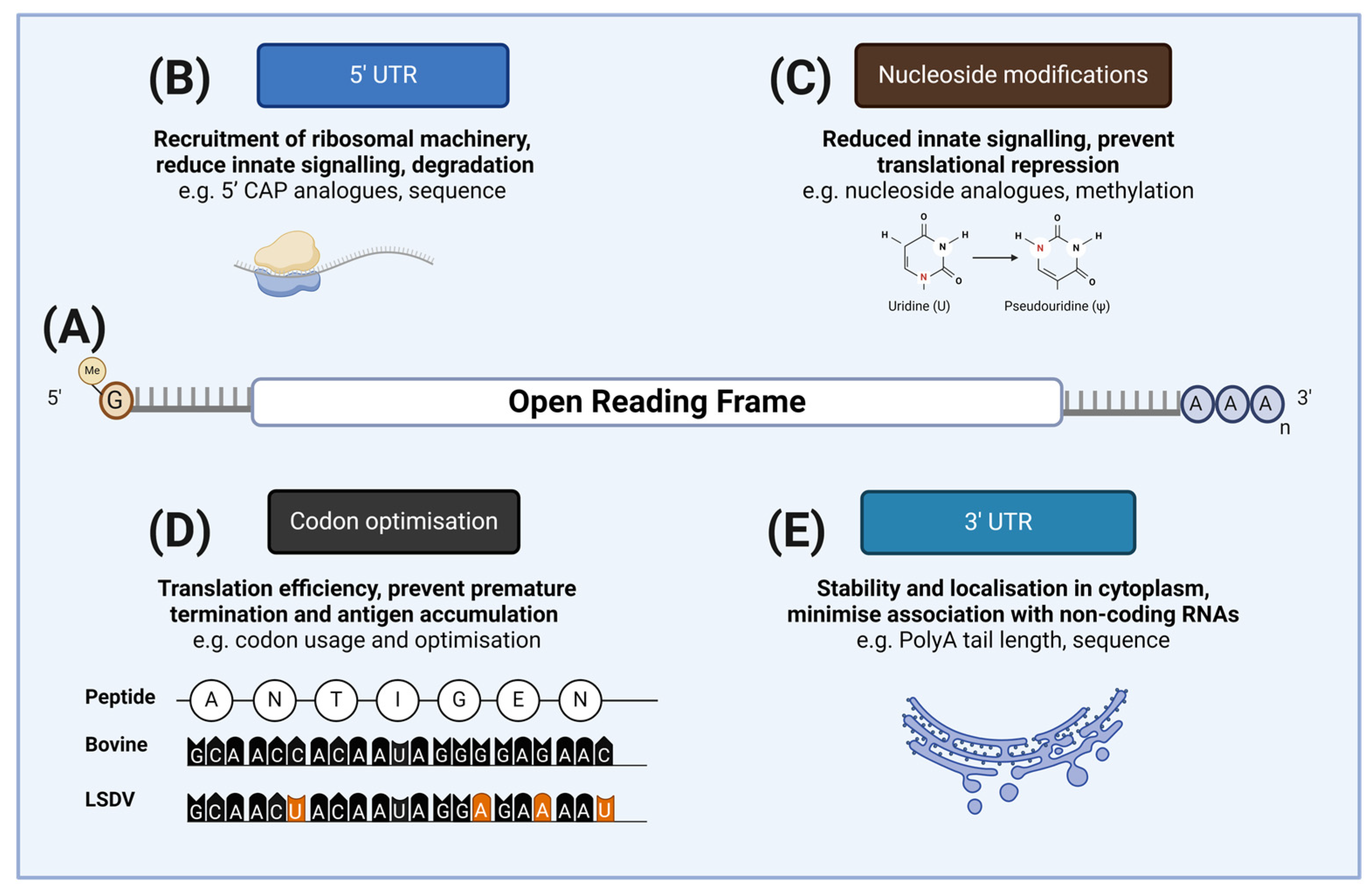
2.1. mRNA Vaccine Components
2.2. mRNA Vaccine Construction and Synthesis
2.3. mRNA Stabilization and Cellular Delivery
2.4. Mechanisms of mRNA Endosomal Escape
3. Immune Responses to mRNA Vaccination
3.1. Initiation of the Immune Response
3.2. Humoral Immune Responses
3.3. Cell-Mediated Immune Responses
3.4. Mucosal Immune Responses
4. Key Recent Advances in mRNA Vaccine Design and Delivery
4.1. Advances in mRNA Design
4.2. Dose-Optimization Strategies for mRNA Vaccines
4.3. Optimization of mRNA Delivery
4.4. Intranasal Vaccination with mRNA
4.5. Ribosomal Stalling and Ribosomal Frameshifting
5. Development of mRNA Vaccines for Important Livestock Diseases
5.1. Lumpy Skin Disease
5.2. Rift Valley Fever
5.3. Peste des Petits Ruminants
5.4. East Coast Fever
5.5. Bovine Respiratory Disease
5.6. Barber’s Pole Worm
6. Summary
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adam, D. How far will global population rise? Researchers can’t agree. Nature 2021, 597, 462–465. [Google Scholar] [CrossRef]
- Mottet, A.; Teillard, F.; Boettcher, P.; De’ Besi, G.; Besbes, B. Review: Domestic herbivores and food security: Current contribution, trends and challenges for a sustainable development. Animal 2018, 12, s188–s198. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, C.; Yang, S.; Stennett, R.; Miller, V.; Teo, K.; Anand, S.S.; Pare, G.; Yusuf, S.; Dehghan, M.; Mente, A. Changes in energy, macronutrient, and food consumption in 47 countries over the last 70 years (1950–2019): A systematic review and meta-analysis. Nutrition 2023, 108, 111941. [Google Scholar] [CrossRef] [PubMed]
- Laurance, W.F.; Sayer, J.; Cassman, K.G. Agricultural expansion and its impacts on tropical nature. Trends Ecol. Evol. 2014, 29, 107–116. [Google Scholar] [CrossRef] [PubMed]
- McElwain, T.F.; Thumbi, S.M. Animal pathogens and their impact on animal health, the economy, food security, food safety and public health. Rev. Sci. Tech. 2017, 36, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Cain, K. The many challenges of disease management in aquaculture. J. World Aquac. Soc. 2022, 53, 1080–1083. [Google Scholar] [CrossRef]
- Roth, J.A. Veterinary vaccines and their importance to animal health and public health. Procedia Vaccinol. 2011, 5, 127–136. [Google Scholar] [CrossRef]
- Vidovic, N.; Vidovic, S. Antimicrobial resistance and food animals: Influence of livestock environment on the emergence and dissemination of antimicrobial resistance. Antibiotics 2020, 9, 52. [Google Scholar] [CrossRef]
- Craig, J.; Sadoff, R.; Bennett, S.; Bahati, F.; Beauvais, W. Behavior-change interventions to improve antimicrobial stewardship in human health, animal health, and livestock agriculture: A systematic review. PLoS Glob. Public Health 2023, 3, e0001526. [Google Scholar] [CrossRef]
- Broderick, G.A. Review: Optimizing ruminant conversion of feed protein to human food protein. Animal 2018, 12, 1722–1734. [Google Scholar] [CrossRef]
- Li, Y.D.; Chi, W.Y.; Su, J.H.; Ferrall, L.; Hung, C.F.; Wu, T.C. Coronavirus vaccine development: From sars and mers to COVID-19. J. Biomed. Sci. 2020, 27, 104. [Google Scholar] [CrossRef]
- Jorge, S.; Dellagosti, O.A. The development of veterinary vaccines: A review of traditional methods and modern biotechnology approaches. Biotechnol. Res. Innov. 2017, 1, 6–13. [Google Scholar] [CrossRef]
- Meeusen, E.N.; Walker, J.; Peters, A.; Pastoret, P.P.; Jungersen, G. Current status of veterinary vaccines. Clin. Microbiol. Rev. 2007, 20, 489–510. [Google Scholar] [CrossRef]
- Choudhury, S.M.; Ma, X.; Dang, W.; Li, Y.; Zheng, H. Recent development of ruminant vaccine against viral diseases. Front. Vet. Sci. 2021, 8, 697194. [Google Scholar] [CrossRef] [PubMed]
- Lancet Commission on COVID-19 Vaccines and Therapeutics Task Force Members. Operation warp speed: Implications for global vaccine security. Lancet Glob. Health 2021, 9, e1017–e1021. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic t lymphocytes in vivo by liposome-entrapped mrna. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar] [PubMed]
- Weide, B.; Carralot, J.P.; Reese, A.; Scheel, B.; Eigentler, T.K.; Hoerr, I.; Rammensee, H.G.; Garbe, C.; Pascolo, S. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 2008, 31, 180–188. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nanotoday 2019, 28, 100766. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Borah, P.; Deb, P.K.; Al-Shar’i, N.A.; Dahabiyeh, L.A.; Venugopala, K.N.; Singh, V.; Shinu, P.; Hussain, S.; Deka, S.; Chandrasekaran, B.; et al. Perspectives on RNA vaccine candidates for COVID-19. Front. Mol. Biosci. 2021, 8, 635245. [Google Scholar] [CrossRef] [PubMed]
- Donadeu, M.; Nwankpa, N.; Abela-Ridder, B.; Dungu, B. Strategies to increase adoption of animal vaccines by smallholder farmers with focus on neglected diseases and marginalized populations. PLoS Negl. Trop. Dis. 2019, 13, e0006989. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, production scales and time required for producing rna vaccines for the global pandemic demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef]
- Munira, S.L.; Hendriks, J.T.; Atmosukarto, I.I.; Friede, M.H.; Carter, L.M.; Butler, J.R.G.; Clements, A.C.A. A cost analysis of producing vaccines in developing countries. Vaccine 2019, 37, 1245–1251. [Google Scholar] [CrossRef]
- Light, D.W.; Lexchin, J. The costs of coronavirus vaccines and their pricing. J. R. Soc. Med. 2021, 114, 502–504. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Gordon, N.F.; Stock, R.; Petrides, D. Adenoviral vector COVID-19 vaccines: Process and cost analysis. Processes 2021, 9, 1430. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.R.; Ahn, G.; Cho, B.K.; Ahn, J.Y.; Kim, Y.H. Modifications of mrna vaccine structural elements for improving mrna stability and translation efficiency. Mol. Cell. Toxicol. 2022, 18, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.S.; Kumari, M.; Chen, G.H.; Hong, M.H.; Yuan, J.P.; Tsai, J.L.; Wu, H.C. Mrna-based vaccines and therapeutics: An in-depth survey of current and upcoming clinical applications. J. Biomed. Sci. 2023, 30, 84. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Sacco, M.T.; Horner, S.M. How RNA modifications regulate the antiviral response. Immunol. Rev. 2021, 304, 169–180. [Google Scholar] [CrossRef]
- Brito Querido, J.; Diaz-Lopez, I.; Ramakrishnan, V. The molecular basis of translation initiation and its regulation in eukaryotes. Nat. Rev. Mol. Cell Biol. 2023, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Fox, D.M.; Branson, K.M.; Walker, R.C. mRNA codon optimization with quantum computers. PLoS ONE 2021, 16, e0259101. [Google Scholar] [CrossRef]
- Stein, K.C.; Frydman, J. The stop-and-go traffic regulating protein biogenesis: How translation kinetics controls proteostasis. J. Biol. Chem. 2019, 294, 2076–2084. [Google Scholar] [CrossRef]
- Liu, Y. A code within the genetic code: Codon usage regulates co-translational protein folding. Cell Commun. Signal. 2020, 18, 145. [Google Scholar] [CrossRef]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117–E6125. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.A.; Coller, J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 2022, 23, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Mallet, L.; Blumel, J.; Cassart, J.P.; Knezevic, I.; Ng, S.H.S.; Wall, M.; Jakava-Viljanen, M.; Logvinoff, C.; Goios, A.; et al. Report of the third conference on next-generation sequencing for adventitious virus detection in biologics for humans and animals. Biologicals 2023, 83, 101696. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Tu, B.; Cui, L. Recombinant T7 RNA polymerase production using clearcoli BL21(DE3) and animal-free media for in vitro transcription. Appl. Microbiol. Biotechnol. 2024, 108, 41. [Google Scholar] [CrossRef] [PubMed]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Kariko, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Kariko, K. Nucleoside modifications in RNA limit activation of 2’-5’-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef]
- Kon, E.; Elia, U.; Peer, D. Principles for designing an optimal mRNA lipid nanoparticle vaccine. Curr. Opin. Biotechnol. 2022, 73, 329–336. [Google Scholar] [CrossRef]
- Verbeke, R.; Hogan, M.J.; Lore, K.; Pardi, N. Innate immune mechanisms of mRNA vaccines. Immunity 2022, 55, 1993–2005. [Google Scholar] [CrossRef]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic mRNA capping. Beilstein. J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef] [PubMed]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Satapathy, S.R.; Dutta, T. Delivery strategies for mRNA vaccines. Pharm. Med. 2022, 36, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Wei, J.; Hui, A.M. The delivery of mRNA vaccines for therapeutics. Life 2022, 12, 1254. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the messenger: Advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef]
- Balhorn, R. The protamine family of sperm nuclear proteins. Genome Biol. 2007, 8, 227. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkic-Zrna, S.; Probst, J.; Kallen, K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Scheel, B.; Teufel, R.; Probst, J.; Carralot, J.P.; Geginat, J.; Radsak, M.; Jarrossay, D.; Wagner, H.; Jung, G.; Rammensee, H.G.; et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. [Google Scholar] [CrossRef]
- Li, M.; Zhao, M.; Fu, Y.; Li, Y.; Gong, T.; Zhang, Z.; Sun, X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J. Control. Release 2016, 228, 9–19. [Google Scholar] [CrossRef]
- Li, D.; Song, H.; Li, J.; Liu, X.; Gao, X.; Wu, T.; Zhang, Z.; Li, Y. Expression and evaluation of a novel PPRV nanoparticle antigen based on ferritin self-assembling technology. Pharmaceutics 2022, 14, 1902. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; DuRoss, A.; Sun, C.; Murphy-Benenato, K.E.; Mihai, C.; Almarsson, O.; Sahay, G. Boosting intracellular delivery of lipid nanoparticle-encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef]
- Paramasivam, P.; Franke, C.; Stoter, M.; Hoijer, A.; Bartesaghi, S.; Sabirsh, A.; Lindfors, L.; Arteta, M.Y.; Dahlen, A.; Bak, A.; et al. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 2022, 221, e202110137. [Google Scholar] [CrossRef] [PubMed]
- Richeson, J.T.; Hughes, H.D.; Broadway, P.R.; Carroll, J.A. Vaccination management of beef cattle: Delayed vaccination and endotoxin stacking. Vet. Clin. N. Am. Food Anim. Pract. 2019, 35, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, M.F.; Palomares, R.A. Bovine respiratory disease vaccination against viral pathogens: Modified-live versus inactivated antigen vaccines, intranasal versus parenteral, what is the evidence? Vet. Clin. N. Am. Food Anim. Pract. 2020, 36, 461–472. [Google Scholar] [CrossRef]
- Roeder, P.; Mariner, J.; Kock, R. Rinderpest: The veterinary perspective on eradication. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120139. [Google Scholar] [CrossRef] [PubMed]
- Berche, P. Life and death of smallpox. Presse Med. 2022, 51, 104117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ge, X.; Yang, H. Porcine reproductive and respiratory syndrome modified live virus vaccine: A “leaky” vaccine with debatable efficacy and safety. Vaccines 2021, 9, 362. [Google Scholar] [CrossRef]
- Clemente, B.; Denis, M.; Silveira, C.P.; Schiavetti, F.; Brazzoli, M.; Stranges, D. Straight to the point: Targeted mRNA-delivery to immune cells for improved vaccine design. Front. Immunol. 2023, 14, 1294929. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, H.; Meng, L.; Li, F.; Yu, C. Comparison of immune responses elicited by SARS-CoV-2 mRNA and recombinant protein vaccine candidates. Front. Immunol. 2022, 13, 906457. [Google Scholar] [CrossRef]
- Lim, J.M.E.; Hang, S.K.; Hariharaputran, S.; Chia, A.; Tan, N.; Lee, E.S.; Chng, E.; Lim, P.L.; Young, B.E.; Lye, D.C.; et al. A comparative characterization of SARS-CoV-2-specific t cells induced by mRNA or inactive virus COVID-19 vaccines. Cell Rep. Med. 2022, 3, 100793. [Google Scholar] [CrossRef]
- Azzi, L.; Dalla Gasperina, D.; Veronesi, G.; Shallak, M.; Ietto, G.; Iovino, D.; Baj, A.; Gianfagna, F.; Maurino, V.; Focosi, D.; et al. Mucosal immune response in BNT162B2 COVID-19 vaccine recipients. EBioMedicine 2022, 75, 103788. [Google Scholar] [CrossRef]
- Nickel, O.; Rockstroh, A.; Wolf, J.; Landgraf, S.; Kalbitz, S.; Kellner, N.; Borte, M.; Pietsch, C.; Fertey, J.; Lubbert, C.; et al. Evaluation of the systemic and mucosal immune response induced by COVID-19 and the BNT162B2 mRNA vaccine for SARS-CoV-2. PLoS ONE 2022, 17, e0263861. [Google Scholar] [CrossRef] [PubMed]
- van Splunter, M.; van Hoffen, E.; Floris-Vollenbroek, E.G.; Timmerman, H.; de Bos, E.L.; Meijer, B.; Ulfman, L.H.; Witteman, B.; Wells, J.M.; Brugman, S.; et al. Oral cholera vaccination promotes homing of iga(+) memory b cells to the large intestine and the respiratory tract. Mucosal Immunol. 2018, 11, 1254–1264. [Google Scholar] [CrossRef]
- Cuburu, N.; Kweon, M.N.; Song, J.H.; Hervouet, C.; Luci, C.; Sun, J.B.; Hofman, P.; Holmgren, J.; Anjuere, F.; Czerkinsky, C. Sublingual immunization induces broad-based systemic and mucosal immune responses in mice. Vaccine 2007, 25, 8598–8610. [Google Scholar] [CrossRef]
- Hanson, S.M.; Singh, S.; Tabet, A.; Sastry, K.J.; Barry, M.; Wang, C. Mucoadhesive wafers composed of binary polymer blends for sublingual delivery and preservation of protein vaccines. J. Control. Release 2021, 330, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.A.; Chamorro, M.F.; Passler, T.; Huber, L.; Walz, P.H.; Thoresen, M.; Raithel, G.; Silvis, S.; Stockler, R.; Woolums, A.R. Local and systemic antibody responses in beef calves vaccinated with a modified-live virus bovine respiratory syncytial virus (BRSV) vaccine at birth following BRSV infection. Vet. Sci. 2022, 10, 20. [Google Scholar] [CrossRef]
- Willis, E.; Pardi, N.; Parkhouse, K.; Mui, B.L.; Tam, Y.K.; Weissman, D.; Hensley, S.E. Nucleoside-modified mRNA vaccination partially overcomes maternal antibody inhibition of de novo immune responses in mice. Sci. Transl. Med. 2020, 12, eaav5701. [Google Scholar] [CrossRef]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef]
- Victor, M.P.; Acharya, D.; Begum, T.; Ghosh, T.C. The optimization of mRNA expression level by its intrinsic properties-insights from codon usage pattern and structural stability of mRNA. Genomics 2019, 111, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Lin, A.; Xu, C.; Li, Z.; Liu, K.; Liu, B.; Ma, X.; Zhao, F.; Jiang, H.; et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 2023, 621, 396–403. [Google Scholar] [CrossRef]
- Fanzo, J. From big to small: The significance of smallholder farms in the global food system. Lancet Planet. Health 2017, 1, e15–e16. [Google Scholar] [CrossRef]
- Erickson, N.E.N.; Berenik, A.; Lardner, H.; Lacoste, S.; Campbell, J.; Gow, S.; Waldner, C.; Ellis, J. Evaluation of bovine respiratory syncytial virus (BRSV) and bovine herpesvirus (BHV) specific antibody responses between heterologous and homologous prime-boost vaccinated western Canadian beef calves. Can. Vet. J. 2021, 62, 37–44. [Google Scholar] [PubMed]
- Liang, R.; van den Hurk, J.V.; Landi, A.; Lawman, Z.; Deregt, D.; Townsend, H.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. DNA prime protein boost strategies protect cattle from bovine viral diarrhea virus type 2 challenge. J. Gen. Virol. 2008, 89, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.M.J.; Molinari, M.P.; Gravisaco, M.J.; Paoletta, M.S.; Montenegro, V.N.; Wilkowsky, S.E. Evaluation of different heterologous prime-boost immunization strategies against Babesia bovis using viral vectored and protein-adjuvant vaccines based on a chimeric multi-antigen. Vaccine 2016, 34, 3913–3919. [Google Scholar] [CrossRef]
- Park, H.J.; Bang, Y.J.; Kwon, S.P.; Kwak, W.; Park, S.I.; Roh, G.; Bae, S.H.; Kim, J.Y.; Kwak, H.W.; Kim, Y.; et al. Analyzing immune responses to varied mRNA and protein vaccine sequences. NPJ Vaccines 2023, 8, 84. [Google Scholar] [CrossRef]
- Durel, L.; Rose, C.; Bainbridge, T.; Roubert, J.; Dressel, K.U.; Bennemann, J.; Ruckner, A.; Vahlenkamp, T.; Maillard, R. Immune response of mature cows subjected to annual booster vaccination against neonatal calf diarrhoea with two different commercial vaccines: A non-inferiority study. Livest. Sci. 2017, 204, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Miyazaki, T.; Muto, H.; Kubara, K.; Mukai, Y.; Watari, R.; Sato, S.; Kondo, K.; Tsukumo, S.I.; Yasutomo, K.; et al. Design and lyophilization of lipid nanoparticles for mRNA vaccine and its robust immune response in mice and nonhuman primates. Mol. Ther. Nucleic Acids 2022, 30, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, Y.; Zhou, M.; Xu, S.; Varley, A.J.; Golubovic, A.; Lu, R.X.Z.; Wang, K.C.; Yeganeh, M.; Vosoughi, D.; et al. Combinatorial design of ionizable lipid nanoparticles for muscle-selective mRNA delivery with minimized off-target effects. Proc. Natl. Acad. Sci. USA 2023, 120, e2309472120. [Google Scholar] [CrossRef]
- Boley, P.A.; Lee, C.M.; Schrock, J.; Yadav, K.K.; Patil, V.; Suresh, R.; Lu, S.; Feng, M.M.; Hanson, J.; Channappanavar, R.; et al. Enhanced mucosal immune responses and reduced viral load in the respiratory tract of ferrets to intranasal lipid nanoparticle-based SARS-COV-2 proteins and mRNA vaccines. J. Nanobiotechnol. 2023, 21, 60. [Google Scholar] [CrossRef]
- Mulroney, T.E.; Poyry, T.; Yam-Puc, J.C.; Rust, M.; Harvey, R.F.; Kalmar, L.; Horner, E.; Booth, L.; Ferreira, A.P.; Stoneley, M.; et al. N(1)-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting. Nature 2023, 625, 189–194. [Google Scholar] [CrossRef]
- Mikl, M.; Pilpel, Y.; Segal, E. High-throughput interrogation of programmed ribosomal frameshifting in human cells. Nat. Commun. 2020, 11, 3061. [Google Scholar] [CrossRef]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sur, J.H.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. The genomes of sheeppox and goatpox viruses. J. Virol. 2002, 76, 6054–6061. [Google Scholar] [CrossRef]
- Hillen, H.S.; Bartuli, J.; Grimm, C.; Dienemann, C.; Bedenk, K.; Szalay, A.A.; Fischer, U.; Cramer, P. Structural basis of poxvirus transcription: Transcribing and capping vaccinia complexes. Cell 2019, 179, 1525–1536.e1512. [Google Scholar] [CrossRef]
- Grimm, C.; Bartuli, J.; Fischer, U. Cytoplasmic gene expression: Lessons from poxviruses. Trends Biochem. Sci. 2022, 47, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Carn, V.M.; Kitching, R.P. An investigation of possible routes of transmission of lumpy skin disease virus (neethling). Epidemiol. Infect. 1995, 114, 219–226. [Google Scholar] [CrossRef]
- Aleksandr, K.; Olga, B.; David, W.B.; Pavel, P.; Yana, P.; Svetlana, K.; Alexander, N.; Vladimir, R.; Dmitriy, L.; Alexander, S. Non-vector-borne transmission of lumpy skin disease virus. Sci. Rep. 2020, 10, 7436. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.G. Lumpy skin disease, an African capripox virus disease of cattle. Br. Vet. J. 1991, 147, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, M.; Klement, E. The effect of vaccination with live attenuated neethling lumpy skin disease vaccine on milk production and mortality-an analysis of 77 dairy farms in Israel. Vaccines 2020, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Broglia, A.; Antoniou, S.E.; Tsiamadis, V.; Plevraki, E.; Petrovic, T.; Polacek, V.; Debeljak, Z.; Miteva, A.; Alexandrov, T.; et al. Neethling vaccine proved highly effective in controlling lumpy skin disease epidemics in the Balkans. Prev. Vet. Med. 2020, 181, 104595. [Google Scholar] [CrossRef]
- Kumar, N.; Barua, S.; Kumar, R.; Khandelwal, N.; Kumar, A.; Verma, A.; Singh, L.; Godara, B.; Chander, Y.; Kumar, G.; et al. Evaluation of the safety, immunogenicity and efficacy of a new live-attenuated lumpy skin disease vaccine in India. Virulence 2023, 14, 2190647. [Google Scholar] [CrossRef]
- Matsiela, M.S.; Naicker, L.; Dibakwane, V.S.; Ntombela, N.; Khoza, T.; Mokoena, N. Improved safety profile of inactivated neethling strain of the lumpy skin disease vaccine. Vaccine X 2022, 12, 100209. [Google Scholar] [CrossRef]
- Babiuk, S. Immunity. In Lumpy Skin Disease; Tuppurainen, E.S.M., Babiuk, S., Klement, E., Eds.; Springer: Cham, Switzerland, 2018; pp. 47–51. [Google Scholar]
- Kar, P.P.; Araveti, P.B.; Kuriakose, A.; Srivastava, A. Design of a multi-epitope protein as a subunit vaccine against lumpy skin disease using an immunoinformatics approach. Sci. Rep. 2022, 12, 19411. [Google Scholar] [CrossRef]
- Chervyakova, O.; Issabek, A.; Sultankulova, K.; Bopi, A.; Kozhabergenov, N.; Omarova, Z.; Tulendibayev, A.; Aubakir, N.; Orynbayev, M. Lumpy skin disease virus with four knocked out genes was attenuated in vivo and protects cattle from infection. Vaccines 2022, 10, 1705. [Google Scholar] [CrossRef]
- Muylkens, B.; Meurens, F.; Schynts, F.; Farnir, F.; Pourchet, A.; Bardiau, M.; Gogev, S.; Thiry, J.; Cuisenaire, A.; Vanderplasschen, A.; et al. Intraspecific bovine herpesvirus 1 recombinants carrying glycoprotein E deletion as a vaccine marker are virulent in cattle. J. Gen. Virol. 2006, 87, 2149–2154. [Google Scholar] [CrossRef]
- d’Offay, J.M.; Fulton, R.W.; Fishbein, M.; Eberle, R.; Dubovi, E.J. Isolation of a naturally occurring vaccine/wild-type recombinant bovine herpesvirus type 1 (BoHV-1) from an aborted bovine fetus. Vaccine 2019, 37, 4518–4524. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Vaseghi, H.R.; Qian, L.; Liu, J. Systematic comparison of 2a peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Beer, M.; Hoffmann, B. Cross-protection of an inactivated and a live-attenuated lumpy skin disease virus vaccine against sheeppox virus infections in sheep. Vaccines 2023, 11, 763. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunova, G.A.; Shchelkunov, S.N. Smallpox, monkeypox and other human orthopoxvirus infections. Viruses 2022, 15, 103. [Google Scholar] [CrossRef]
- Shchelkunov, S.N.; Shchelkunova, G.A. Genes that control vaccinia virus immunogenicity. Acta Nat. 2020, 12, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Domi, A.; Moss, B. Engineering of a vaccinia virus bacterial artificial chromosome in Escherichia coli by bacteriophage lambda-based recombination. Nat. Methods 2005, 2, 95–97. [Google Scholar] [CrossRef]
- Roth, S.J.; Hoper, D.; Beer, M.; Feineis, S.; Tischer, B.K.; Osterrieder, N. Recovery of infectious virus from full-length cowpox virus (CPXV) DNA cloned as a bacterial artificial chromosome (BAC). Vet. Res. 2011, 42, 3. [Google Scholar] [CrossRef]
- Noyce, R.S.; Lederman, S.; Evans, D.H. Construction of an infectious horsepox virus vaccine from chemically synthesized DNA fragments. PLoS ONE 2018, 13, e0188453. [Google Scholar] [CrossRef]
- Yuan, M.; Zhang, W.; Wang, J.; Al Yaghchi, C.; Ahmed, J.; Chard, L.; Lemoine, N.R.; Wang, Y. Efficiently editing the vaccinia virus genome by using the CRISPR-Cas9 system. J. Virol. 2015, 89, 5176–5179. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.E.; Meers, J.; Gravel, J.L.; McCarthy, F.M.; Mahony, T.J. The essential and non-essential genes of bovine herpesvirus 1. J. Gen. Virol. 2008, 89, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Zhang, Z.; Liu, F.; Lu, H.; Yu, C.; Sun, H.; Long, J.; Cao, Y.; Mai, J.; Miao, Y.; et al. Monkeypox virus quadrivalent mRNA vaccine induces immune response and protects against vaccinia virus. Signal Transduct. Target. Ther. 2023, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Li, Y.; Jiang, L.; Luo, L.; Wang, Y.; Wang, H.; Han, X.; Zhao, J.; Gu, G.; Fang, M.; et al. Mpox multi-antigen mRNA vaccine candidates by a simplified manufacturing strategy afford efficient protection against lethal orthopoxvirus challenge. Emerg. Microbes Infect. 2023, 12, 2204151. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Khristova, M.L.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Complete genome analysis of 33 ecologically and biologically diverse rift valley fever virus strains reveals widespread virus movement and low genetic diversity due to recent common ancestry. J. Virol. 2007, 81, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Kwasnik, M.; Rozek, W.; Rola, J. Rift valley fever—A growing threat to humans and animals. J. Vet. Res. 2021, 65, 7–14. [Google Scholar] [CrossRef]
- Muller, R.; Poch, O.; Delarue, M.; Bishop, D.H.; Bouloy, M. Rift valley fever virus l segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994, 75 Pt 6, 1345–1352. [Google Scholar] [CrossRef]
- Kitandwe, P.K.; McKay, P.F.; Kaleebu, P.; Shattock, R.J. An overview of rift valley fever vaccine development strategies. Vaccines 2022, 10, 1794. [Google Scholar] [CrossRef]
- Ikegami, T. Molecular biology and genetic diversity of rift valley fever virus. Antivir. Res. 2012, 95, 293–310. [Google Scholar] [CrossRef]
- Caplen, H.; Peters, C.J.; Bishop, D.H. Mutagen-directed attenuation of rift valley fever virus as a method for vaccine development. J. Gen. Virol. 1985, 66 Pt 10, 2271–2277. [Google Scholar] [CrossRef]
- Ikegami, T. Rift valley fever vaccines: An overview of the safety and efficacy of the live-attenuated MP-12 vaccine candidate. Expert Rev. Vaccines 2017, 16, 601–611. [Google Scholar] [CrossRef]
- Muller, R.; Saluzzo, J.F.; Lopez, N.; Dreier, T.; Turell, M.; Smith, J.; Bouloy, M. Characterization of clone 13, a naturally attenuated avirulent isolate of rift valley fever virus, which is altered in the small segment. Am. J. Trop. Med. Hyg. 1995, 53, 405–411. [Google Scholar] [CrossRef]
- Dungu, B.; Louw, I.; Lubisi, A.; Hunter, P.; von Teichman, B.F.; Bouloy, M. Evaluation of the efficacy and safety of the rift valley fever clone 13 vaccine in sheep. Vaccine 2010, 28, 4581–4587. [Google Scholar] [CrossRef]
- von Teichman, B.; Engelbrecht, A.; Zulu, G.; Dungu, B.; Pardini, A.; Bouloy, M. Safety and efficacy of Rift Valley fever Smithburn and Clone 13 vaccines in calves. Vaccine 2011, 29, 5771–5777. [Google Scholar] [CrossRef]
- Wichgers Schreur, P.J.; Oymans, J.; Kant, J.; van de Water, S.; Kollar, A.; Dehon, Y.; Soos, P.; Penzes, Z.; van Keulen, L.; Kortekaas, J. A single vaccination with four-segmented rift valley fever virus prevents vertical transmission of the wild-type virus in pregnant ewes. NPJ Vaccines 2021, 6, 8. [Google Scholar] [CrossRef]
- Ly, H.J.; Lokugamage, N.; Nishiyama, S.; Ikegami, T. Risk analysis of inter-species reassortment through a rift valley fever phlebovirus mp-12 vaccine strain. PLoS ONE 2017, 12, e0185194. [Google Scholar] [CrossRef]
- Ly, H.J.; Nishiyama, S.; Lokugamage, N.; Smith, J.K.; Zhang, L.; Perez, D.; Juelich, T.L.; Freiberg, A.N.; Ikegami, T. Attenuation and protective efficacy of Rift Valley fever phlebovirus rMP12-GM50 strain. Vaccine 2017, 35, 6634–6642. [Google Scholar] [CrossRef]
- Jenkin, D.; Wright, D.; Folegatti, P.M.; Platt, A.; Poulton, I.; Lawrie, A.; Tran, N.; Boyd, A.; Turner, C.; Gitonga, J.N.; et al. Safety and immunogenicity of a chadox1 vaccine against Rift Valley fever in UK adults: An open-label, non-randomised, first-in-human phase 1 clinical trial. Lancet Infect. Dis. 2023, 23, 956–964. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Heise, M.T.; Ross, T.M. Vaccination with DNA plasmids expressing Gn coupled to C3d or alphavirus replicons expressing gn protects mice against Rift Valley fever virus. PLoS Negl. Trop. Dis. 2010, 4, e725. [Google Scholar] [CrossRef]
- Kortekaas, J.; Antonis, A.F.; Kant, J.; Vloet, R.P.; Vogel, A.; Oreshkova, N.; de Boer, S.M.; Bosch, B.J.; Moormann, R.J. Efficacy of three candidate Rift Valley fever vaccines in sheep. Vaccine 2012, 30, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Faburay, B.; Lebedev, M.; McVey, D.S.; Wilson, W.; Morozov, I.; Young, A.; Richt, J.A. A glycoprotein subunit vaccine elicits a strong Rift Valley fever virus neutralizing antibody response in sheep. Vector Borne Zoonotic Dis. 2014, 14, 746–756. [Google Scholar] [CrossRef]
- Faburay, B.; Wilson, W.C.; Gaudreault, N.N.; Davis, A.S.; Shivanna, V.; Bawa, B.; Sunwoo, S.Y.; Ma, W.; Drolet, B.S.; Morozov, I.; et al. A recombinant Rift Valley fever virus glycoprotein subunit vaccine confers full protection against Rift Valley fever challenge in sheep. Sci. Rep. 2016, 6, 27719. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.C.; Faburay, B.; Trujillo, J.D.; Ragan, I.; Sunwoo, S.Y.; Morozov, I.; Shivanna, V.; Balogh, A.; Urbaniak, K.; McVey, D.S.; et al. Preliminary evaluation of a recombinant Rift Valley fever virus glycoprotein subunit vaccine providing full protection against heterologous virulent challenge in cattle. Vaccines 2021, 9, 748. [Google Scholar] [CrossRef]
- Rahman, A.U.; Dhama, K.; Ali, Q.; Hussain, I.; Oneeb, M.; Chaudhary, U.; Wensman, J.J.; Shabbir, M.Z. Peste des petits ruminants in large ruminants, camels and unusual hosts. Vet. Q. 2020, 40, 35–42. [Google Scholar] [CrossRef]
- Lefevre, P.C.; Diallo, A. Peste des petits ruminants. Rev. Sci. Tech. 1990, 9, 935–981. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H.; Kundu, P.; Begum, J.A.; Chowdhury, T.; Rahman, M.; Khatun, A.; Saha, S.S.; Nooruzzaman, M.; Parvin, R.; Islam, M.R. Peste des petits ruminants virus antibodies in domestic large ruminants in Bangladesh. J. Infect. Dev. Ctries. 2022, 16, 369–373. [Google Scholar] [CrossRef]
- OIE; World Organization for Animal Health. Resolutions Adopted by the World Assembly Of delegates of the Oie during Its 81st General Session; OIE: Paris, France, 2013. [Google Scholar]
- Dundon, W.G.; Diallo, A.; Cattoli, G. Peste des petits ruminants in Africa: A review of currently available molecular epidemiological data, 2020. Arch. Virol. 2020, 165, 2147–2163. [Google Scholar] [CrossRef]
- Couacy-Hymann, E.; Berete, K.; Odoom, T.; Zerbo, L.H.; Mathurin, K.Y.; Kouakou, V.K.; Doumbouya, M.I.; Balde, A.; Ababio, P.T.; Ouoba, L.B.; et al. The spread of peste des petits ruminants virus lineage IV in West Africa. Animals 2023, 13, 1268. [Google Scholar] [CrossRef]
- Hodgson, S.; Moffat, K.; Hill, H.; Flannery, J.T.; Graham, S.P.; Baron, M.D.; Darpel, K.E. Comparison of the immunogenicities and cross-lineage efficacies of live attenuated peste des petits ruminants virus vaccines PPRV/Nigeria/75/1 and PPRV/Sungri/96. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Aziz, M.H.; Shabbir, M.Z.; Ali, M.M.; Asif, Z.; Ijaz, M.U. Immunoinformatics approach for epitope mapping of immunogenic regions (N, F and H gene) of small ruminant morbillivirus and its comparative analysis with standard vaccinal strains for effective vaccine development. Vaccines 2022, 10, 2179. [Google Scholar] [CrossRef]
- Selvaraj, M.; Mahapatra, M.; Parida, S. Exchange of C-terminal variable sequences within morbillivirus nucleocapsid protein are tolerated: Development and evaluation of two marker (DIVA) vaccines (Sungri/96 diva, Nigeria/75/1 DIVA) against PPR. Viruses 2021, 13, 2320. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Sevilla, N.; Martin, V. A new look at vaccine strategies against PPRV focused on adenoviral candidates. Front. Vet. Sci. 2021, 8, 729879. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, G.; Shi, L.; Li, W.; Li, C.; Chen, Z.; Jin, H.; Xu, B.; Li, G. Immune responses in mice vaccinated with a suicidal DNA vaccine expressing the hemagglutinin glycoprotein from the peste des petits ruminants virus. J. Virol. Methods 2013, 193, 525–530. [Google Scholar] [CrossRef]
- Wang, Y.; Yue, X.; Jin, H.; Liu, G.; Pan, L.; Wang, G.; Guo, H.; Li, G.; Li, Y. A suicidal DNA vaccine expressing the fusion protein of peste des petits ruminants virus induces both humoral and cell-mediated immune responses in mice. J. Virol. Methods 2015, 225, 35–40. [Google Scholar] [CrossRef]
- Norval, R.A.I.; Perry, B.D.; Young, A. The Epidemiology of Theileriosis in Africa; Academic Press: London, UK, 1992. [Google Scholar]
- Perry, B.D. The control of east coast fever of cattle by live parasite vaccination: A science-to-impact narrative. One Health 2016, 2, 103–114. [Google Scholar] [CrossRef]
- Nene, V.; Morrison, W.I. Approaches to vaccination against Theileria parva and Theileria annulata. Parasite Immunol. 2016, 38, 724–734. [Google Scholar] [CrossRef]
- Musoke, A.; Rowlands, J.; Nene, V.; Nyanjui, J.; Katende, J.; Spooner, P.; Mwaura, S.; Odongo, D.; Nkonge, C.; Mbogo, S.; et al. Subunit vaccine based on the p67 major surface protein of Theileria parva sporozoites reduces severity of infection derived from field tick challenge. Vaccine 2005, 23, 3084–3095. [Google Scholar] [CrossRef]
- Lacasta, A.; Mwalimu, S.; Kibwana, E.; Saya, R.; Awino, E.; Njoroge, T.; Poole, J.; Ndiwa, N.; Pelle, R.; Nene, V.; et al. Immune parameters to p67C antigen adjuvanted with ISA206VG correlate with protection against East Coast fever. Vaccine 2018, 36, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Lacasta, A.; Mody, K.T.; De Goeyse, I.; Yu, C.; Zhang, J.; Nyagwange, J.; Mwalimu, S.; Awino, E.; Saya, R.; Njoroge, T.; et al. Synergistic effect of two nanotechnologies enhances the protective capacity of the Theileria parva sporozoite p67c antigen in cattle. J. Immunol. 2021, 206, 686–699. [Google Scholar] [CrossRef]
- Lacasta, A.; Kim, H.C.; Kepl, E.; Gachogo, R.; Chege, N.; Ojuok, R.; Muriuki, C.; Mwalimu, S.; Touboul, G.; Stiber, A.; et al. Design and immunological evaluation of two-component protein nanoparticle vaccines for East Coast fever. Front. Immunol. 2022, 13, 1015840. [Google Scholar] [CrossRef]
- Kolakowski, J.; Connelley, T.; Lukacik, P.; Pfuhl, M.; Werling, D. East coast fever, a neglected tropical disease with an outdated vaccine approach? Trends Parasitol. 2022, 38, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Yakubu, R.R.; Weiss, L.M.; Silmon de Monerri, N.C. Post-translational modifications as key regulators of apicomplexan biology: Insights from proteome-wide studies. Mol. Microbiol. 2018, 107, 1–23. [Google Scholar] [CrossRef]
- Atchou, K.; Ongus, J.; Machuka, E.; Juma, J.; Tiambo, C.; Djikeng, A.; Silva, J.C.; Pelle, R. Comparative transcriptomics of the bovine apicomplexan parasite Theileria parva developmental stages reveals massive gene expression variation and potential vaccine antigens. Front. Vet. Sci. 2020, 7, 287. [Google Scholar] [CrossRef]
- Johnson, K.K.; Pendell, D.L. Market impacts of reducing the prevalence of bovine respiratory disease in united states beef cattle feedlots. Front. Vet. Sci. 2017, 4, 189. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.E.; Barnes, T.S.; Morton, J.M.; Clements, A.C.; Mahony, T.J. Risk factors for bovine respiratory disease in australian feedlot cattle: Use of a causal diagram-informed approach to estimate effects of animal mixing and movements before feedlot entry. Prev. Vet. Med. 2014, 117, 160–169. [Google Scholar] [CrossRef]
- Hay, K.E.; Ambrose, R.C.; Morton, J.M.; Horwood, P.F.; Gravel, J.L.; Waldron, S.; Commins, M.A.; Fowler, E.V.; Clements, A.C.; Barnes, T.S.; et al. Effects of exposure to bovine viral diarrhoea virus 1 on risk of bovine respiratory disease in australian feedlot cattle. Prev. Vet. Med. 2016, 126, 159–169. [Google Scholar] [CrossRef]
- Hay, K.E.; Barnes, T.S.; Morton, J.M.; Gravel, J.L.; Commins, M.A.; Horwood, P.F.; Ambrose, R.C.; Clements, A.C.; Mahony, T.J. Associations between exposure to viruses and bovine respiratory disease in Australian feedlot cattle. Prev. Vet. Med. 2016, 127, 121–133. [Google Scholar] [CrossRef]
- Bowland, S.L.; Shewen, P.E. Bovine respiratory disease: Commercial vaccines currently available in Canada. Can. Vet. J. 2000, 41, 33–48. [Google Scholar] [PubMed]
- Theurer, M.E.; Larson, R.L.; White, B.J. Systematic review and meta-analysis of the effectiveness of commercially available vaccines against bovine herpesvirus, bovine viral diarrhea virus, bovine respiratory syncytial virus, and parainfluenza type 3 virus for mitigation of bovine respiratory disease complex in cattle. J. Am. Vet. Med. Assoc. 2015, 246, 126–142. [Google Scholar] [PubMed]
- Ambrose, R.K.; Blakebrough-Hall, C.; Gravel, J.L.; Gonzalez, L.A.; Mahony, T.J. Characterisation of the upper respiratory tract virome of feedlot cattle and its association with bovine respiratory disease. Viruses 2023, 15, 455. [Google Scholar] [CrossRef] [PubMed]
- Esnault, G.; Earley, B.; Cormican, P.; Waters, S.M.; Lemon, K.; Cosby, S.L.; Lagan, P.; Barry, T.; Reddington, K.; McCabe, M.S. Assessment of rapid minion nanopore DNA virus meta-genomics using calves experimentally infected with bovine herpes virus-1. Viruses 2022, 14, 1859. [Google Scholar] [CrossRef] [PubMed]
- Brito, B.P.; Frost, M.J.; Anantanawat, K.; Jaya, F.; Batterham, T.; Djordjevic, S.P.; Chang, W.S.; Holmes, E.C.; Darling, A.E.; Kirkland, P.D. Expanding the range of the respiratory infectome in Australian feedlot cattle with and without respiratory disease using metatranscriptomics. Microbiome 2023, 11, 158. [Google Scholar] [CrossRef]
- Lamb, H.J.; Nguyen, L.T.; Briody, T.E.; Ambrose, R.K.; Hayes, B.J.; Mahony, T.J.; Ross, E.M. Skim-nanopore sequencing for routine genomic evaluation and bacterial pathogen detection in cattle. Anim. Prod. Sci. 2023, 63, 1074–1085. [Google Scholar] [CrossRef]
- Baldeon Vaca, G.; Meyer, M.; Cadete, A.; Hsiao, C.J.; Golding, A.; Jeon, A.; Jacquinet, E.; Azcue, E.; Guan, C.M.; Sanchez-Felix, X.; et al. Intranasal mRNA-LNP vaccination protects hamsters from SARS-CoV-2 infection. Sci. Adv. 2023, 9, eadh1655. [Google Scholar] [CrossRef]
- Yan, J.; Nielsen, T.B.; Lu, P.; Talyansky, Y.; Slarve, M.; Reza, H.; Novakovic, B.; Netea, M.G.; Keller, A.E.; Warren, T.; et al. A protein-free vaccine stimulates innate immunity and protects against nosocomial pathogens. Sci. Transl. Med. 2023, 15, eadf9556. [Google Scholar] [CrossRef]
- Alexander, A.L.; Doyle, E.; Ingham, A.B.; Colditz, I.; McRae, G.; Alkemade, S.; Cervantes, M.P.; Hine, B.C. The innate immune stimulant Amplimune® is safe to administer to young feedlot cattle. Aust. Vet. J. 2022, 100, 261–270. [Google Scholar] [CrossRef]
- Romanowski, R.; Culbert, R.; Alkemade, S.; Medellin-Peña, M.J.; Bugarski, D.; Milovanovic, A.; Nesic, S.; Masic, A. Mycobacterium cell wall fraction immunostimulant (AMPLIMUNE™) efficacy in the reduction of the severity of ETEC induced diarrhea in neonatal calves. Acta Vet. 2017, 67, 222–237. [Google Scholar] [CrossRef]
- Charlier, J.; van der Voort, M.; Kenyon, F.; Skuce, P.; Vercruysse, J. Chasing helminths and their economic impact on farmed ruminants. Trends Parasitol. 2014, 30, 361–367. [Google Scholar] [CrossRef]
- Adduci, I.; Sajovitz, F.; Hinney, B.; Lichtmannsperger, K.; Joachim, A.; Wittek, T.; Yan, S. Haemonchosis in sheep and goats, control strategies and development of vaccines against Haemonchus contortus. Animals 2022, 12, 2339. [Google Scholar] [CrossRef] [PubMed]
- Lyndal-Murphy, M.; Ehrlich, W.K.; Mayer, D.G. Anthelmintic resistance in ovine gastrointestinal nematodes in inland southern Queensland. Aust. Vet. J. 2014, 92, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Knox, D.P.; Redmond, D.L.; Newlands, G.F.; Skuce, P.J.; Pettit, D.; Smith, W.D. The nature and prospects for gut membrane proteins as vaccine candidates for Haemonchus contortus and other ruminant trichostrongyloids. Int. J. Parasitol. 2003, 33, 1129–1137. [Google Scholar] [CrossRef]
- Kemp, D.H.; Pearson, R.D.; Gough, J.M.; Willadsen, P. Vaccination against Boophilus microplus: Localization of antigens on tick gut cells and their interaction with the host immune system. Exp. Appl. Acarol. 1989, 7, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.N.; Moore, T.; Sriskantha, A.; Spring, K.; Tellam, R.; Willadsen, P.; Cobon, G.S. Cloning and expression of a protective antigen from the cattle tick Boophilus microplus. Proc. Natl. Acad. Sci. USA 1989, 86, 9657–9661. [Google Scholar] [CrossRef]
- Willadsen, P.; Riding, G.A.; McKenna, R.V.; Kemp, D.H.; Tellam, R.L.; Nielsen, J.N.; Lahnstein, J.; Cobon, G.S.; Gough, J.M. Immunologic control of a parasitic arthropod. Identification of a protective antigen from Boophilus microplus. J. Immunol. 1989, 143, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.; Antonopoulos, A.; Haslam, S.M.; Dicker, A.J.; McNeilly, T.N.; Johnston, S.L.; Dell, A.; Knox, D.P.; Britton, C. Novel expression of Haemonchus contortus vaccine candidate aminopeptidase h11 using the free-living nematode Caenorhabditis elegans. Vet. Res. 2013, 44, 111. [Google Scholar] [CrossRef]
- Haslam, S.M.; Coles, G.C.; Munn, E.A.; Smith, T.S.; Smith, H.F.; Morris, H.R.; Dell, A. Haemonchus contortus glycoproteins contain N-linked oligosaccharides with novel highly fucosylated core structures. J. Biol. Chem. 1996, 271, 30561–30570. [Google Scholar] [CrossRef]
- Wang, C.; Liu, L.; Wang, T.; Liu, X.; Peng, W.; Srivastav, R.K.; Zhu, X.Q.; Gupta, N.; Gasser, R.B.; Hu, M. H11-induced immunoprotection is predominantly linked to N-glycan moieties during Haemonchus contortus infection. Front. Immunol. 2022, 13, 1034820. [Google Scholar] [CrossRef]
- Smith, W.D.; Smith, S.K.; Pettit, D. Evaluation of immunization with gut membrane glycoproteins of ostertagia ostertagi against homologous challenge in calves and against Haemonchus contortus in sheep. Parasite Immunol. 2000, 22, 239–247. [Google Scholar] [CrossRef]
- Anonymous. Barbervax—Vaccination Schedle. Available online: https://barbervax.com/how-to-use/ (accessed on 3 August 2023).
- Sicard, T.; Kassardjian, A.; Julien, J.P. B cell targeting by molecular adjuvants for enhanced immunogenicity. Expert Rev. Vaccines 2020, 19, 1023–1039. [Google Scholar] [CrossRef]
- Gouglas, D.; Thanh Le, T.; Henderson, K.; Kaloudis, A.; Danielsen, T.; Hammersland, N.C.; Robinson, J.M.; Heaton, P.M.; Rottingen, J.A. Estimating the cost of vaccine development against epidemic infectious diseases: A cost minimisation study. Lancet Glob. Health 2018, 6, e1386–e1396. [Google Scholar] [CrossRef]
- Plotkin, S.; Robinson, J.M.; Cunningham, G.; Iqbal, R.; Larsen, S. The complexity and cost of vaccine manufacturing—An overview. Vaccine 2017, 35, 4064–4071. [Google Scholar] [CrossRef]
- Pronker, E.S.; Weenen, T.C.; Commandeur, H.; Claassen, E.H.; Osterhaus, A.D. Risk in vaccine research and development quantified. PLoS ONE 2013, 8, e57755. [Google Scholar] [CrossRef] [PubMed]
- Lalani, H.S.; Nagar, S.; Sarpatwari, A.; Barenie, R.E.; Avorn, J.; Rome, B.N.; Kesselheim, A.S. Us public investment in development of mrna COVID-19 vaccines: Retrospective cohort study. BMJ 2023, 380, e073747. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Roni, M.A. Challenges of storage and stability of mRNA-based COVID-19 vaccines. Vaccines 2021, 9, 1033. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Animal Health and Sustainability: A Global Data Analysis; Health for Animals—Global Animal Health Association: Brussels, Belgium, 2023. [Google Scholar]

{kind=link}
| Dose Characteristics | |||||
|---|---|---|---|---|---|
| Vaccine Type 1 | Quantity | Scale 2 | Cost | Comment | Reference |
| mRNA | 100µg | N.A. 3 | USD 2.85 | Modern 4 | [28] |
| mRNA | 30 µg | N.A. 3 | USD 1.18 | Pfizer 4 | [28] |
| mRNA | 100 µg | 345 M | USD 2.02 | 30 L | [26] |
| mRNA | 30 µg | 1.15 B | USD 0.61 | 30 L | [26] |
| mRNA | 12 µg | 2.876 B | USD 0.20 | 30 L | [26] |
| saRNA | 1 µg | 8.554 B | USD 0.02 | 30 L | [26] |
| saRNA | 0.1 µg | 12.22 B | USD 0.0043 | 30 L | [26] |
| attBac | N.S. | 20 M | USD 2.62 5 | One vaccine | [27] |
| MLV | N.S. | 20 M | USD 2.36 5 | One vaccine | [27] |
| Rec. Subunit | N.S. | 20 M | USD 2.68 5 | One vaccine | [27] |
| attBac | N.S. | 100 M | USD 2.12 5 | Five vaccines 6 | [27] |
| MLV | N.S. | 100 M | USD 1.95 5 | Five vaccines 6 | [27] |
| Rec. Subunit | N.S. | 100 M | USD 1.84 5 | Five vaccines 6 | [27] |
| AdV vect. | N.S. | 400 M | USD 0.15 | Batch culture | [29] |
| AdV vect. | N.S. | 400 M | USD 0.23 | Perfusion culture | [29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahony, T.J.; Briody, T.E.; Ommeh, S.C. Can the Revolution in mRNA-Based Vaccine Technologies Solve the Intractable Health Issues of Current Ruminant Production Systems? Vaccines 2024, 12, 152. https://doi.org/10.3390/vaccines12020152
Mahony TJ, Briody TE, Ommeh SC. Can the Revolution in mRNA-Based Vaccine Technologies Solve the Intractable Health Issues of Current Ruminant Production Systems? Vaccines. 2024; 12(2):152. https://doi.org/10.3390/vaccines12020152
Chicago/Turabian StyleMahony, Timothy J., Tatiana E. Briody, and Sheila C. Ommeh. 2024. "Can the Revolution in mRNA-Based Vaccine Technologies Solve the Intractable Health Issues of Current Ruminant Production Systems?" Vaccines 12, no. 2: 152. https://doi.org/10.3390/vaccines12020152
APA StyleMahony, T. J., Briody, T. E., & Ommeh, S. C. (2024). Can the Revolution in mRNA-Based Vaccine Technologies Solve the Intractable Health Issues of Current Ruminant Production Systems? Vaccines, 12(2), 152. https://doi.org/10.3390/vaccines12020152