

Immune Checkpoint Inhibitors in HBV-Caused Hepatocellular Carcinoma Therapy

Abstract
1. Introduction
2. Molecular Characterization and Typing of HBV-HCC
2.1. Molecular Characterization of HBV-HCC
2.2. Typing of HBV-HCC
3. Occurrence and Development of HBV-HCC
3.1. Direct Oncogenic Mechanism of HBV
3.1.1. Insertion Mutations
3.1.2. HBV Causes Genomic Instability
3.1.3. Prolonged Expression of Viral Proteins Affects Cell Functioning
3.2. Immune-Mediated Indirect Oncogenic Mechanisms of HBV
4. Current Status of Treatment of HBV-HCC
4.1. Antiviral Therapy
4.2. Surgical Excision and Radiofrequency Therapy
4.3. Liver Transplantation
4.4. Radiotherapy and Chemotherapy
4.5. Tyrosine Kinase Inhibitor Therapy
4.6. Immunotherapy
5. Immune Checkpoint Inhibitors for the Treatment of HBV-HCC
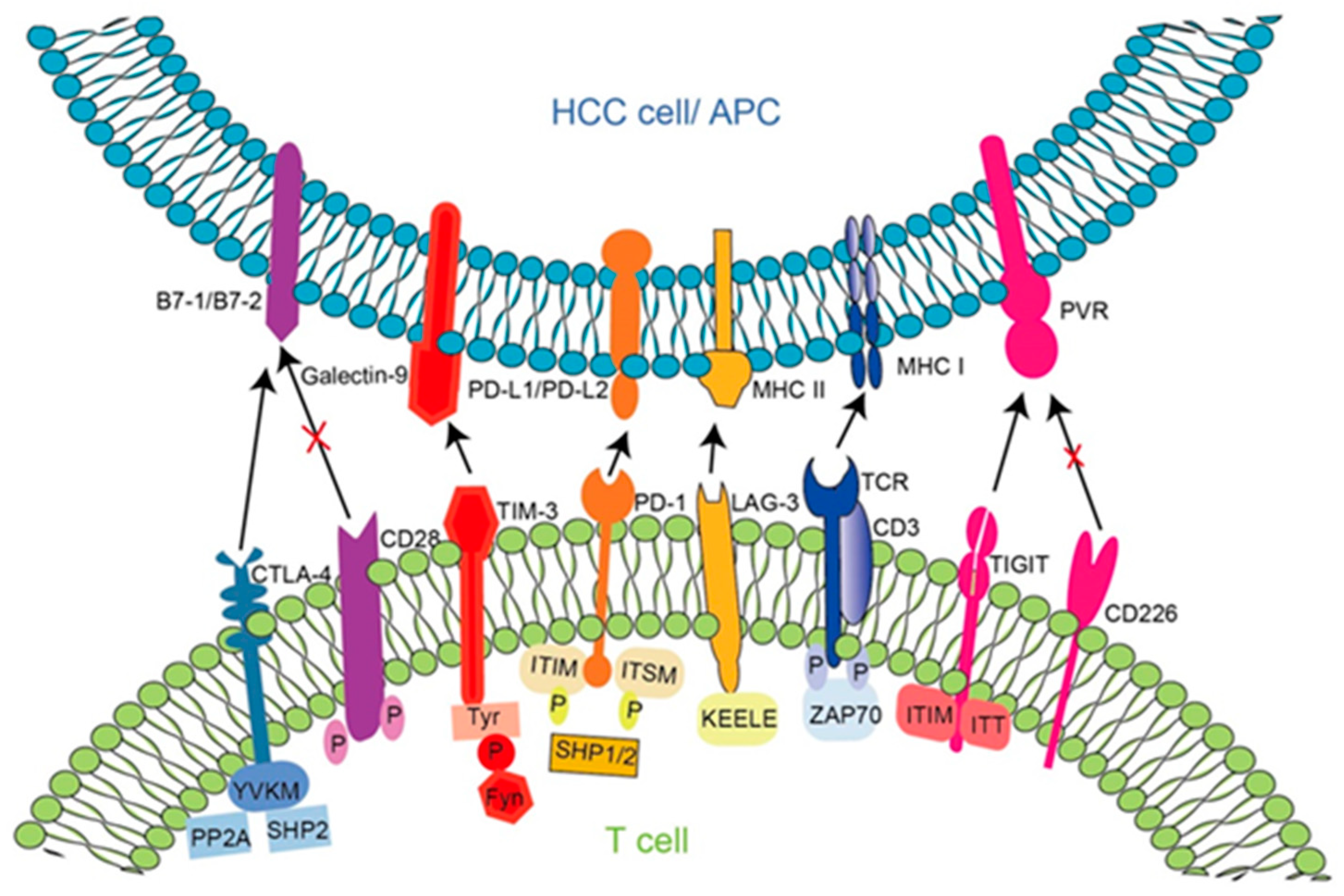
5.1. Principle of Action of Immune Checkpoint Molecules in HBV-HCC and Targeted Inhibitors
5.1.1. Programmed Cell Death Protein 1 (PD-1)
5.1.2. Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA-4)
5.1.3. Lymphocyte Activation Gene 3 (LAG3)
5.1.4. T Cell Immunoglobulins and ITIM Domains (TIGIT)
5.1.5. T Cell Immunoglobulin Mucin 3 (TIM-3)
5.2. Advantages of Immune Checkpoint Inhibitors in the Treatment of HBV-HCC
5.3. Efficacy of Immune Checkpoint Inhibitors in the Treatment of HCC with Different Etiologies
6. Potential Immune Checkpoint Therapeutic Targets
6.1. HHLA2
6.2. CCR4
6.3. DDRs
6.4. IL-27/IL-27R
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [PubMed]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Thomas, J.A.; Kendall, B.J.; Dalais, C.; Macdonald, G.A.; Thrift, A.P. Hepatocellular and extrahepatic cancers in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Eur. J. Cancer 2022, 173, 250–262. [Google Scholar] [CrossRef]
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Roudot-Thoraval, F. Epidemiology of hepatitis C virus infection. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101596. [Google Scholar] [CrossRef]
- Seto, W.K.; Lo, Y.R.; Pawlotsky, J.M.; Yuen, M.F. Chronic hepatitis B virus infection. Lancet 2018, 392, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Hepatitis B Collaborators. Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 796–829. [Google Scholar] [CrossRef]
- Shunsuke, U.; Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Sunakozaka, H.; Sakai, Y.; Horimoto, K.; Kaneko, S. Differential Microrna Expression between Hepatitis B and Hepatitis C Leading Disease Progression to Hepatocellular Carcinoma. Hepatology 2009, 49, 1098–1112. [Google Scholar]
- De Battista, D.; Zamboni, F.; Gerstein, H.; Sato, S.; Markowitz, T.E.; Lack, J.; Engle, R.E.; Farci, P. Molecular Signature and Immune Landscape of HCV-Associated Hepatocellular Carcinoma (HCC): Differences and Similarities with HBV-HCC. J. Hepatocell. Carcinoma 2021, 8, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Amaddeo, G.; Cao, Q.; Ladeiro, Y.; Imbeaud, S.; Nault, J.C.; Jaoui, D.; Gaston Mathe, Y.; Laurent, C.; Laurent, A.; Bioulac-Sage, P.; et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut 2015, 64, 820–829. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Miyanishi, K.; Osuga, T.; Tanaka, S.; Ito, R.; Sakamoto, H.; Kubo, T.; Ohnuma, H.; Murase, K.; Takada, K.; et al. Association between Hepatic Oxidative Stress Related Factors and Activation of Wnt/β-Catenin Signaling in NAFLD-Induced Hepatocellular Carcinoma. Cancers 2022, 14, 2066. [Google Scholar]
- Zhang, Y.; Jiang, H.-H.; Wang, Z.-Y.; Zhai, B.; Lin, M.-B. Alcohol dehydrogenase 4 is a TP53-associated gene signature for the prediction of prognosis in hepatocellular carcinoma. Oncol. Lett. 2023, 25, 3. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and Genetic Variability of Hepatitis B Virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- McMahon, B.J.; Nolen, L.D.; Snowball, M.; Homan, C.; Negus, S.; Roik, E.; Spradling, P.R.; Bruden, D. HBV Genotype: A Significant Risk Factor in Determining Which Patients with Chronic HBV Infection Should Undergo Surveillance for HCC: The Hepatitis B Alaska Study. Hepatology 2021, 74, 2965–2973. [Google Scholar] [CrossRef]
- Wen, J.; Song, C.; Jiang, D.; Jin, T.; Dai, J.; Zhu, L.; An, J.; Liu, Y.; Ma, S.; Qin, N.; et al. Hepatitis B virus genotype, mutations, human leukocyte antigen polymorphisms and their interactions in hepatocellular carcinoma: A multi-centre case-control study. Sci. Rep. 2015, 5, 16489. [Google Scholar] [CrossRef]
- Araujo, N.M.; Teles, S.A.; Spitz, N. Comprehensive Analysis of Clinically Significant Hepatitis B Virus Mutations in Relation to Genotype, Subgenotype and Geographic Region. Front. Microbiol. 2020, 11, 616023. [Google Scholar] [CrossRef]
- Kew, M.C. Hepatocellular carcinoma in African Blacks: Recent progress in etiology and pathogenesis. World J. Hepatol. 2010, 2, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Lin, S.Y.; Zhang, A.; Lian, J.; Wang, J.; Chang, T.T.; Lin, Y.J.; Song, W.; Su, Y.H. Recurrent HBV Integration Targets as Potential Drivers in Hepatocellular Carcinoma. Cells 2021, 10, 1294. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Bachtiar, M.; Choo, C.C.S.; Lee, C.G. Comprehensive review of Hepatitis B Virus-associated hepatocellular carcinoma research through text mining and big data analytics. Biol. Rev. 2019, 94, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Bousali, M.; Karamitros, T. Hepatitis B Virus Integration into Transcriptionally Active Loci and HBV-Associated Hepatocellular Carcinoma. Microorganisms 2022, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Bouchard, M.J. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M. Epigenetic modulation in chronic hepatitis B virus infection. Semin. Immunopathol. 2020, 42, 173–185. [Google Scholar] [CrossRef]
- Sartorius, K.; An, P.; Winkler, C.; Chuturgoon, A.; Li, X.; Makarova, J.; Kramvis, A. The Epigenetic Modulation of Cancer and Immune Pathways in Hepatitis B Virus-Associated Hepatocellular Carcinoma: The Influence of HBx and miRNA Dysregulation. Front. Immunol. 2021, 12, 661204. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liang, X.; Liu, Y.; Qu, Z.; Gao, L.; Han, L.; Liu, S.; Cui, M.; Shi, Y.; Zhang, Z.; et al. Hepatitis B virus core protein inhibits TRAIL-induced apoptosis of hepatocytes by blocking DR5 expression. Cell Death Differ. 2009, 16, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Quiroga, J.A.; Carreño, V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J. Gen. Virol. 2003, 84, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cai, S.; Pu, R.; Li, Z.; Liu, D.; Zhou, X.; Yin, J.; Chen, X.; Chen, L.; Wu, J.; et al. HBV preS Mutations Promote Hepatocarcinogenesis by Inducing Endoplasmic Reticulum Stress and Upregulating Inflammatory Signaling. Cancers 2022, 14, 3274. [Google Scholar] [CrossRef]
- Liang, Y.J.; Teng, W.; Chen, C.L.; Sun, C.P.; Teng, R.D.; Huang, Y.H.; Liang, K.H.; Chen, Y.W.; Lin, C.C.; Su, C.W.; et al. Clinical Implications of HBV PreS/S Mutations and the Effects of PreS2 Deletion on Mitochondria, Liver Fibrosis, and Cancer Development. Hepatology 2021, 74, 641–655. [Google Scholar] [CrossRef]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune Pathogenesis of Hepatocellular Carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef]
- Stelma, F.; Willemse, S.B.; Erken, R.; de Niet, A.; Sinnige, M.J.; van Dort, K.; Zaaijer, H.L.; van Leeuwen, E.M.M.; Kootstra, N.A.; Reesink, H.W. Dynamics of the Immune Response in Acute Hepatitis B Infection. Open Forum Infect. Dis. 2017, 4, ofx231. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Z. HBV-Induced Immune Imbalance in the Development of HCC. Front. Immunol. 2019, 10, 2048. [Google Scholar] [CrossRef]
- Baudi, I.; Kawashima, K.; Isogawa, M. HBV-Specific CD8+ T-Cell Tolerance in the Liver. Front. Immunol. 2021, 12, 3039. [Google Scholar] [CrossRef]
- Lim, C.J.; Lee, Y.H.; Pan, L.; Lai, L.; Chua, C.; Wasser, M.; Lim, T.K.H.; Yeong, J.; Toh, H.C.; Lee, S.Y.; et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019, 68, 916–927. [Google Scholar] [CrossRef]
- Xiao, Z.; Yeung, C.L.S.; Yam, J.W.P.; Mao, X. An update on the role of complement in hepatocellular carcinoma. Front. Immunol. 2022, 13, 1007382. [Google Scholar] [CrossRef] [PubMed]
- Jayant, K.; Habib, N.; Huang, K.W.; Warwick, J.; Arasaradnam, R. Recent Advances: The Imbalance of Immune Cells and Cytokines in the Pathogenesis of Hepatocellular Carcinoma. Diagnostics 2020, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, T.; Tang, L.; Li, Y. Cytokines and Chemokines in HBV Infection. Front. Mol. Biosci. 2021, 8, 1188. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guan, J.; Khan, N.U.; Li, G.; Shao, J.; Zhou, Q.; Xu, L.; Huang, C.; Deng, J.; Zhu, H.; et al. Potential capacity of interferon-alpha to eliminate covalently closed circular DNA (cccDNA) in hepatocytes infected with hepatitis B virus. Gut Pathog. 2021, 13, 22. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Singh, L.; Indermun, S.; Govender, M.; Kumar, P.; du Toit, L.C.; Choonara, Y.E.; Pillay, V. Drug Delivery Strategies for Antivirals against Hepatitis B Virus. Viruses 2018, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Sun, X.; Mei, J.; Hu, Z.; Yang, Z.; Hou, J.; Fu, Y.; Wang, X.; Chen, M. Antiviral Treatments Eliminate the Adverse Impacts of High Baseline HBV Loads on the Survival of HBV-Related HCC Patients. J. Hepatocell. Carcinoma 2022, 9, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.Y.; Sun, B.Y.; Wang, Z.T.; Zhou, C.; Yang, Z.F.; Gan, W.; Huang, J.L.; Liu, G.; Zhou, J.; Fan, J.; et al. Antiviral therapy improves postoperative survival of patients with HBV-related hepatocellular carcinoma. Am. J. Surg. 2022, 224, 494–500. [Google Scholar] [CrossRef]
- Vitiello, G.A.; Wang, A.; Lee, R.M.; Russell, M.C.; Yopp, A.; Ryon, E.L.; Goel, N.; Luu, S.; Hsu, C.; Silberfein, E.; et al. Surgical resection of early stage hepatocellular carcinoma improves patient survival at safety net hospitals. J. Surg. Oncol. 2021, 123, 963–969. [Google Scholar] [CrossRef]
- Parikh, N.D.; Cuneo, K.; Mendiratta-Lala, M. Radiation therapies for the treatment of hepatocellular carcinoma. Clin. Liver Dis. 2021, 17, 341. [Google Scholar] [CrossRef]
- Meirelles Junior, R.F.; Salvalaggio, P.; Rezende, M.B.; Evangelista, A.S.; Guardia, B.D.; Matielo, C.E.; Neves, D.B.; Pandullo, F.L.; Felga, G.E.; Alves, J.A.; et al. Liver transplantation: History, outcomes and perspectives. Einstein 2015, 13, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kahn, J.; Terrault, N.A. Liver transplantation as therapy for hepatocellular carcinoma. Liver Int. 2020, 40 (Suppl. S1), 116–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ju, C. Orchestrating liver repair: A newly discovered function of hepatic iNKT cells. Hepatology 2018, 68, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; Regalia, E.; Doci, R.; Andreola, S.; Pulvirenti, A.; Bozzetti, F.; Montalto, F.; Ammatuna, M.; Morabito, A.; Gennari, L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med. 1996, 334, 693–699. [Google Scholar] [CrossRef]
- Hong, S.K.; Lee, K.W.; Yoon, K.C.; Kim, H.S.; Ahn, S.W.; Kim, H.; Lee, J.M.; Cho, J.H.; Yi, N.J.; Suh, K.S. Different prognostic factors and strategies for early and late recurrence after adult living donor liver transplantation for hepatocellular carcinoma. Clin. Transpl. 2019, 33, e13703. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; He, M.; Fu, C.; Feng, K.; Ma, K.; Zhang, L. Radiofrequency ablation in the treatment of hepatocellular carcinoma. Int. J. Hyperth. 2022, 39, 1052–1063. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, M.; Zhang, S. Comparison of radiofrequency ablation and surgical resection for hepatocellular carcinoma conforming to the Milan criteria: A meta-analysis. ANZ J. Surg. 2021, 91, E432–E438. [Google Scholar] [CrossRef]
- Hou, Z.; Liu, J.; Jin, Z.; Qiu, G.; Xie, Q.; Mi, S.; Huang, J. Use of chemotherapy to treat hepatocellular carcinoma. Biosci. Trends 2022, 16, 31–45. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol. Cancer 2018, 17, 43. [Google Scholar] [CrossRef]
- Zhao, L.; Chang, N.; Shi, L.; Li, F.; Meng, F.; Xie, X.; Xu, Z.; Wang, F. Lenvatinib plus sintilimab versus lenvatinib monotherapy as first-line treatment for advanced HBV-related hepatocellular carcinoma: A retrospective, real-world study. Heliyon 2022, 8, e09538. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Sangro, B.; Sarobe, P.; Hervas-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Wang, W.; Zhang, J.; Till, B.G.; Zhao, L.; Huang, H.; Yang, Y.; Li, T.; Han, L.; Zhang, X.; et al. Association of hepatitis B virus DNA levels with overall survival for advanced hepatitis B virus-related hepatocellular carcinoma under immune checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2023, 72, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.N.; Yang, T.C.; Kao, J.T.; Cheng, K.S.; Lee, Y.J.; Wang, Y.M.; Hsieh, C.T.; Lin, C.W.; Wu, Y.Y. Increased PD-1 and decreased CD28 expression in chronic hepatitis B patients with advanced hepatocellular carcinoma. Liver Int. 2010, 30, 1379–1386. [Google Scholar] [CrossRef]
- Xu, C.; Ng, D.T.W. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Sangro, B.; He, A.R.; Yau, T.; Hsu, C.; Kang, Y.-K.; Kim, T.-Y.; Santoro, A.; Melero, I.; Kudo, M.; Ho, M.-M.; et al. Nivolumab + ipilimumab combination therapy in patients with advanced hepatocellular carcinoma: Subgroup analyses from CheckMate 040. J. Hepatol. 2020, 73, S121–S122. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Sangro, B.; Numata, K.; Huang, Y.; Gomez-Martin, C.; Hiraoka, A.; Moriguchi, M.; Shen, Y.; Horvath, A.; Feely, W.; Young, T.; et al. P-61 Relatlimab + nivolumab in patients with advanced hepatocellular carcinoma who are naive to immuno-oncology therapy but progressed on tyrosine kinase inhibitors, a phase 2, randomized, open-label study: RELATIVITY-073. Ann. Oncol. 2021, 32, S117. [Google Scholar] [CrossRef]
- Philips, E.A.; Garcia-Espana, A.; Tocheva, A.S.; Ahearn, I.M.; Adam, K.R.; Pan, R.; Mor, A.; Kong, X.P. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J. Biol. Chem. 2020, 295, 4372–4380. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, H.H.; Fletcher, C.D.M.; Hornick, J.L.; Morgan, E.A.; Freeman, G.J.; Hodi, F.S.; Pinkus, G.S.; Rodig, S.J. Expression of Programmed Cell Death 1 Ligands (PD-L1 and PD-L2) in Histiocytic and Dendritic Cell Disorders. Am. J. Surg. Pathol. 2016, 40, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Riley, J.L.; Frauwirth, K.A.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Parry, R.V. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Blood 2004, 104, 2657. [Google Scholar] [CrossRef]
- Eggert, T.; Greten, T.F. Tumor regulation of the tissue environment in the liver. Pharmacol. Ther. 2017, 173, 47–57. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.-Y.; Qiu, S.-J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.-Z.; Shi, Y.-H.; Xiao, Y.-S.; et al. Overexpression of PD-L1 Significantly Associates with Tumor Aggressiveness and Postoperative Recurrence in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef]
- Fessas, P.; Kaseb, A.; Wang, Y.; Saeed, A.; Szafron, D.; Jun, T.; Dharmapuri, S.; Rafeh Naqash, A.; Muzaffar, M.; Navaid, M.; et al. Post-registration experience of nivolumab in advanced hepatocellular carcinoma: An international study. J. ImmunoTherapy Cancer 2020, 8, e001033. [Google Scholar] [CrossRef] [PubMed]
- Marron, T.U.; Fiel, M.I.; Hamon, P.; Fiaschi, N.; Kim, E.; Ward, S.C.; Zhao, Z.; Kim, J.; Kennedy, P.; Gunasekaran, G.; et al. Neoadjuvant cemiplimab for resectable hepatocellular carcinoma: A single-arm, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2022, 7, 219–229. [Google Scholar] [CrossRef]
- Gaikwad, S.; Agrawal, M.Y.; Kaushik, I.; Ramachandran, S.; Srivastava, S.K. Immune checkpoint proteins: Signaling mechanisms and molecular interactions in cancer immunotherapy. Semin. Cancer Biol. 2022, 86, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, T.; Miyatake, S.; Ohno, H.; Nakaseko, C.; Isono, K.; Bonifacino, J.S.; Saito, T. Tyrosine Phosphorylation Controls Internalization of CTLA-4 by Regulating Its Interaction with Clathrin-Associated Adaptor Complex AP-2. Immunity 1997, 6, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2014, 59, 567–579. [Google Scholar] [CrossRef] [PubMed]
- He, A.R.; Yau, T.; Hsu, C.; Kang, Y.-K.; Kim, T.-Y.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Subgroup analyses from CheckMate 040. J. Clin. Oncol. 2020, 38, 512. [Google Scholar] [CrossRef]
- Akce, M.; El-Rayes, B.F.; Bekaii-Saab, T.S. Frontline therapy for advanced hepatocellular carcinoma: An update. Ther. Adv. Gastroenterol. 2022, 15, 17562848221086126. [Google Scholar] [CrossRef]
- Wang, M.; Du, Q.; Jin, J.; Wei, Y.; Lu, Y.; Li, Q. LAG3 and its emerging role in cancer immunotherapy. Clin. Transl. Med. 2021, 11, e365. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, X.; Zhan, L.; Wang, L.; Wang, X.; Jiang, M. LAG3 and PD1 Regulate CD8+ T Cell in Diffuse Large B-cell Lymphoma Patients. Comput. Math. Methods Med. 2021, 2021, 4468140. [Google Scholar] [CrossRef]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.; Mitrea, D.M.; Chou, P.-C.; Temirov, J.; Vignali, K.M.; Liu, X.; Zhang, H.; Kriwacki, R.; Bruchez, M.P.; Watkins, S.C.; et al. LAG3 associates with TCR–CD3 complexes and suppresses signaling by driving co-receptor–Lck dissociation. Nat. Immunol. 2022, 23, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2021, 11, 3444. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A.A. Negative Regulation of T Cell Homeostasis by Lymphocyte Activation Gene-3 (CD223)1. J. Immunol. 2005, 174, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A.A. Cutting Edge: Molecular Analysis of the Negative Regulatory Function of Lymphocyte Activation Gene-31. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-m.; Shimizu, K.; Maeda, T.K.; Ikubo, J.; Yoshikawa, H.; Maenaka, K.; Ishimaru, N.; Kosako, H.; et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 2022, 55, 912–924.e918. [Google Scholar] [CrossRef]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Chiang, E.Y.; Mellman, I. TIGIT-CD226-PVR axis: Advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004711. [Google Scholar] [CrossRef]
- Inoue, T.; Adachi, K.; Kawana, K.; Taguchi, A.; Nagamatsu, T.; Fujimoto, A.; Tomio, K.; Yamashita, A.; Eguchi, S.; Nishida, H.; et al. Cancer-associated fibroblast suppresses killing activity of natural killer cells through downregulation of poliovirus receptor (PVR/CD155), a ligand of activating NK receptor. Int. J. Oncol. 2016, 49, 1297–1304. [Google Scholar] [CrossRef]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, J.; Cui, J.; Ma, B.; Zhou, Q.; Yang, X.; Lu, Z.; Du, Y.; Su, C. Expression of TIGIT/CD155 and correlations with clinical pathological features in human hepatocellular carcinoma. Mol. Med. Rep. 2019, 20, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y.; Xun, X.; Wang, S.; Xiang, X.; Hu, S.; Cheng, Q.; Guo, J.; Li, Z.; Zhu, J. TIGIT Can Exert Immunosuppressive Effects on CD8+ T Cells by the CD155/TIGIT Signaling Pathway for Hepatocellular Carcinoma In Vitro. J. Immunother. 2020, 43, 236–243. [Google Scholar] [CrossRef]
- Zong, L.; Peng, H.; Sun, C.; Li, F.; Zheng, M.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nat. Commun. 2019, 10, 221. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan Tze, G.; Sefik, E.; Yajnik, V.; et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Li, F.; Li, N.; Sang, J.; Fan, X.; Deng, H.; Zhang, X.; Han, Q.; Lv, Y.; Liu, Z. Highly elevated soluble Tim-3 levels correlate with increased hepatocellular carcinoma risk and poor survival of hepatocellular carcinoma patients in chronic hepatitis B virus infection. Cancer Manag. Res. 2018, 10, 941–951. [Google Scholar] [CrossRef]
- Li, Z.; Li, N.; Li, F.; Zhou, Z.; Sang, J.; Chen, Y.; Han, Q.; Lv, Y.; Liu, Z. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine 2016, 95, e5749. [Google Scholar] [CrossRef]
- Yang, Y.; Wen, F.; Li, J.; Zhang, P.; Yan, W.; Hao, P.; Xia, F.; Bi, F.; Li, Q. A high baseline HBV load and antiviral therapy affect the survival of patients with advanced HBV-related HCC treated with sorafenib. Liver Int. 2015, 35, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Cheng, A.-L.; Meinhardt, G.; Nakajima, K.; De Sanctis, Y.; Llovet, J. Prognostic factors and predictors of sorafenib benefit in patients with hepatocellular carcinoma: Analysis of two phase III studies. J. Hepatol. 2017, 67, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Z.; Ren, Z.; Tang, F.; Li, Y. Obstacles and Coping Strategies of CAR-T Cell Immunotherapy in Solid Tumors. Front. Immunol. 2021, 12, 687822. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The Challenges and Solutions. Front. Oncol. 2021, 11, 794183. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I Trial of a Glypican-3–Derived Peptide Vaccine for Advanced Hepatocellular Carcinoma: Immunologic Evidence and Potential for Improving Overall Survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef]
- Ding, Z.; Dong, Z.; Chen, Z.; Hong, J.; Yan, L.; Li, H.; Yao, S.; Yan, Y.; Yang, Y.; Yang, C.; et al. Viral Status and Efficacy of Immunotherapy in Hepatocellular Carcinoma: A Systematic Review with Meta-Analysis. Front. Immunol. 2021, 12, 733530. [Google Scholar] [CrossRef]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Murai, H.; Kodama, T.; Maesaka, K.; Tange, S.; Motooka, D.; Suzuki, Y.; Shigematsu, Y.; Inamura, K.; Mise, Y.; Saiura, A.; et al. Multiomics identifies the link between intratumor steatosis and the exhausted tumor immune microenvironment in hepatocellular carcinoma. Hepatology 2023, 77, 77–91. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Janakiram, M.; Chinai, J.M.; Fineberg, S.; Fiser, A.; Montagna, C.; Medavarapu, R.; Castano, E.; Jeon, H.; Ohaegbulam, K.C.; Zhao, R.; et al. Expression, Clinical Significance, and Receptor Identification of the Newest B7 Family Member HHLA2 Protein. Clin. Cancer Res. 2015, 21, 2359–2366. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, M.; Shah, U.A.; Liu, W.; Zhao, A.; Schoenberg, M.P.; Zang, X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol. Rev. 2017, 276, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Rieder, S.A.; Wang, J.; White, N.; Qadri, A.; Menard, C.; Stephens, G.; Karnell, J.L.; Rudd, C.E.; Kolbeck, R. B7-H7 (HHLA2) inhibits T-cell activation and proliferation in the presence of TCR and CD28 signaling. Cell. Mol. Immunol. 2021, 18, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, C.; Tang, X.; Zhang, T.; Liu, Y.; Yu, H.; Li, Y.; Wang, R. HHLA2 Activates the JAK/STAT Signaling Pathway by Binding to TMIGD2 in Hepatocellular Carcinoma Cells. Inflammation 2022, 45, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.H.; Irvine, A.F.; Jones, R.L.; Samson, A. Immunotherapies for hepatocellular carcinoma. Cancer Med. 2022, 11, 571–591. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Gao, Y.; You, M.; Fu, J.; Tian, M.; Zhong, X.; Du, C.; Hong, Z.; Zhu, Z.; Liu, J.; Markowitz, G.J.; et al. Intratumoral stem-like CCR4+ regulatory T cells orchestrate the immunosuppressive microenvironment in HCC associated with hepatitis B. J. Hepatol. 2022, 76, 148–159. [Google Scholar] [CrossRef]
- Yoshie, O. CCR4 as a Therapeutic Target for Cancer Immunotherapy. Cancers 2021, 13, 5542. [Google Scholar] [CrossRef]
- Leitinger, B. Chapter Two—Discoidin Domain Receptor Functions in Physiological and Pathological Conditions. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 310, pp. 39–87. [Google Scholar]
- Sun, X.; Wu, B.; Chiang, H.-C.; Deng, H.; Zhang, X.; Xiong, W.; Liu, J.; Rozeboom, A.M.; Harris, B.T.; Blommaert, E.; et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature 2021, 599, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Klotzsch, E. Barring the gates to the battleground: DDR1 promotes immune exclusion in solid tumors. Signal Transduct. Target. Ther. 2022, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.M.; Lee, F.Y.F.; Jones, R.T.; Kimball, A.K.; Saravia, E.; Graziano, R.F.; Coleman, B.; Menard, K.; Yan, J.; Michaud, E.; et al. Targeting DDR2 enhances tumor response to anti–PD-1 immunotherapy. Sci. Adv. 2019, 5, eaav2437. [Google Scholar] [CrossRef] [PubMed]
- Bosmann, M.; Ward, P.A. Modulation of inflammation by interleukin-27. J. Leukoc. Biol. 2013, 94, 1159–1165. [Google Scholar] [CrossRef]
- Frangieh, M.; McHenry, A.; Phillips, R.; Ye, C.; Bernier, A.; Laffel, L.; Elyaman, W.; Bradshaw, E.M. IL-27: An endogenous constitutive repressor of human monocytes. Clin. Immunol. 2020, 217, 108498. [Google Scholar] [CrossRef]
- Ficht, X.; Iannacone, M. Immune surveillance of the liver by T cells. Sci. Immunol. 2020, 5, eaba2351. [Google Scholar] [CrossRef] [PubMed]
- Aghayev, T.; Mazitova, A.M.; Fang, J.R.; Peshkova, I.O.; Rausch, M.; Hung, M.; White, K.F.; Masia, R.; Titerina, E.K.; Fatkhullina, A.R.; et al. IL27 Signaling Serves as an Immunologic Checkpoint for Innate Cytotoxic Cells to Promote Hepatocellular Carcinoma. Cancer Discov. 2022, 12, 1960–1983. [Google Scholar] [CrossRef]
- Becquart, O.; Lacotte, J.; Malissart, P.; Nadal, J.; Lesage, C.; Guillot, B.; Du Thanh, A. Myasthenia Gravis Induced by Immune Checkpoint Inhibitors. J. Immunother. 2019, 42, 309–312. [Google Scholar] [CrossRef]
- Astaras, C.; de Micheli, R.; Moura, B.; Hundsberger, T.; Hottinger, A.F. Neurological Adverse Events Associated with Immune Checkpoint Inhibitors: Diagnosis and Management. Curr. Neurol. Neurosci. Rep. 2018, 18, 3. [Google Scholar] [CrossRef]
- Altomonte, J. Liver cancer: Sensitizing hepatocellular carcinoma to oncolytic virus therapy. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 8–10. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| HBV Genotype | A | B | C | D | E | F | G | H | I | J (Putative) |
|---|---|---|---|---|---|---|---|---|---|---|
| Occurrence and development of HBV-HCC | Direct oncogenic mechanism of HBV-HCC | Insertion mutations | ||||||||
| HBV causes genomic instability | ||||||||||
| Prolonged expression of viral proteins affects cell function | ||||||||||
| Indirect oncogenic mechanisms of HBV-HCC | Immune-mediated indirect oncogenic mechanisms of HBV | |||||||||
| Immune Checkpoint | Current FDA-Approved Inhibitors | No. of Ongoing Clinical Trials Related to HCC * | Hyperlinks and References |
|---|---|---|---|
| PD-1 | Nibolumab (FDA-approved for the treatment of HCC following sorafenib) | 88 | ClinicalTrials.gov [68,69] |
| Pembrolizumab (FDA-approved for the treatment of HCC following sorafenib) | 76 | ClinicalTrials.gov [70] | |
| Cemiplimab (FDA has not approved it for HCC treatment) | 4 | ClinicalTrials.gov | |
| Dostarlimab (FDA-approved for treatment of unresectable liver cancer) | 1 | ClinicalTrials.gov | |
| CTLA4 | Tremelimumab (FDA-approved for treatment of unresectable liver cancer) | 20 | ClinicalTrials.gov [71,72] |
| Ipilimumab (FDA-approved for the treatment of hepatocellular carcinoma following sorafenib) | 22 | ClinicalTrials.gov [69] | |
| LAG-3 | Relatlimab (FDA has not approved it for HCC treatment) | 4 | ClinicalTrials.gov [73] |
| TIGIT | Tiragolumab has been granted breakthrough drug status by the FDA, and Vibostolimab has initiated clinical trials related to HCC | 2 | ClinicalTrials.gov1 ClinicalTrials.gov2 |
| TIM-3 | Cobolimab is undergoing a series of clinical trials | 1 | ClinicalTrials.gov |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Hu, C.; Xie, X.; Qi, L.; Li, C.; Li, S. Immune Checkpoint Inhibitors in HBV-Caused Hepatocellular Carcinoma Therapy. Vaccines 2023, 11, 614. https://doi.org/10.3390/vaccines11030614
Zhang J, Hu C, Xie X, Qi L, Li C, Li S. Immune Checkpoint Inhibitors in HBV-Caused Hepatocellular Carcinoma Therapy. Vaccines. 2023; 11(3):614. https://doi.org/10.3390/vaccines11030614
Chicago/Turabian StyleZhang, Jin, Changwei Hu, Xiaoxiao Xie, Linzhi Qi, Chuanzhou Li, and Shangze Li. 2023. "Immune Checkpoint Inhibitors in HBV-Caused Hepatocellular Carcinoma Therapy" Vaccines 11, no. 3: 614. https://doi.org/10.3390/vaccines11030614
APA StyleZhang, J., Hu, C., Xie, X., Qi, L., Li, C., & Li, S. (2023). Immune Checkpoint Inhibitors in HBV-Caused Hepatocellular Carcinoma Therapy. Vaccines, 11(3), 614. https://doi.org/10.3390/vaccines11030614