
A Monovalent Mt10-CVB3 Vaccine Prevents CVB4-Accelerated Type 1 Diabetes in NOD Mice

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Virus Propagation and Titration
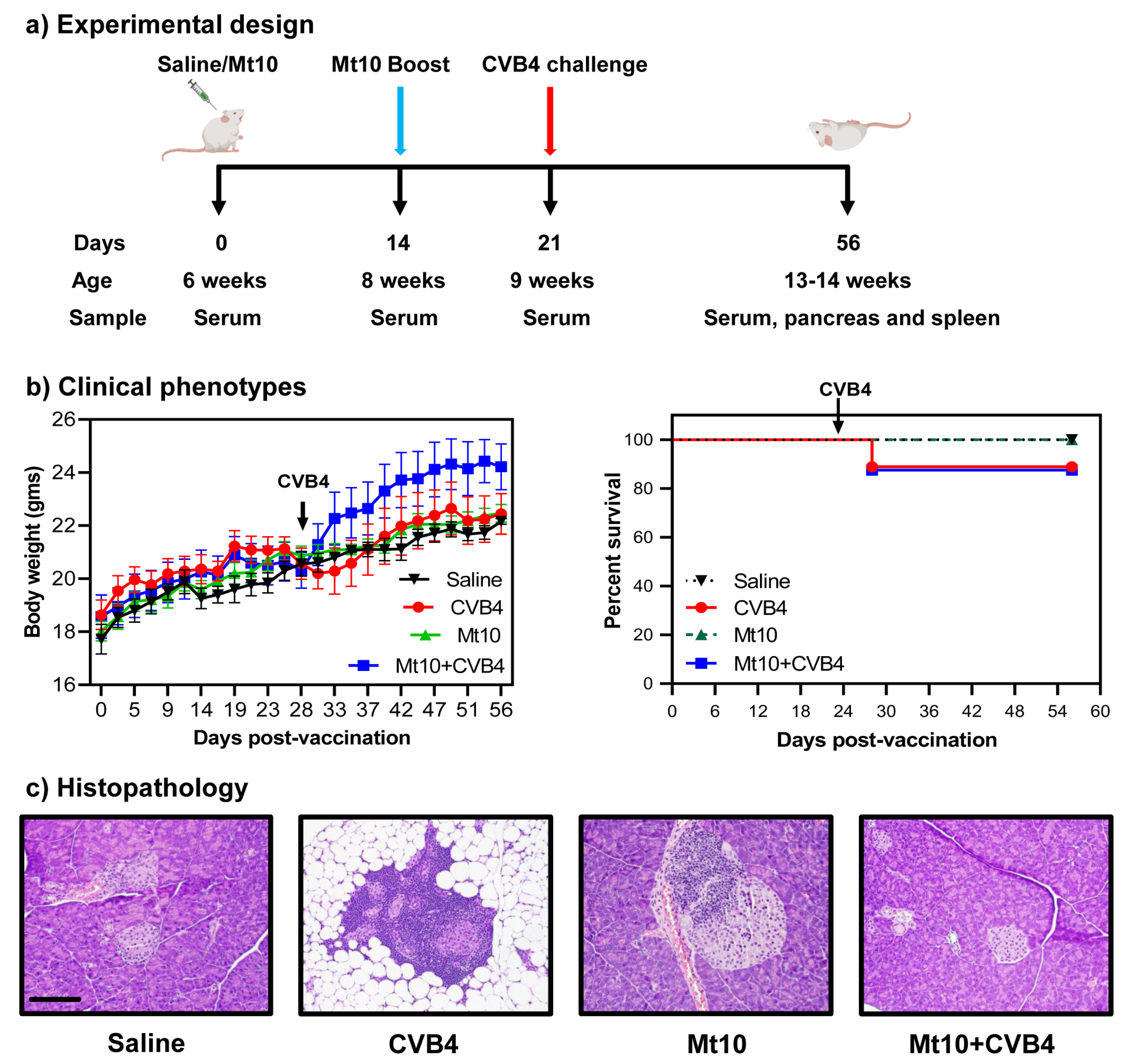
2.3. Vaccination and Challenge Studies
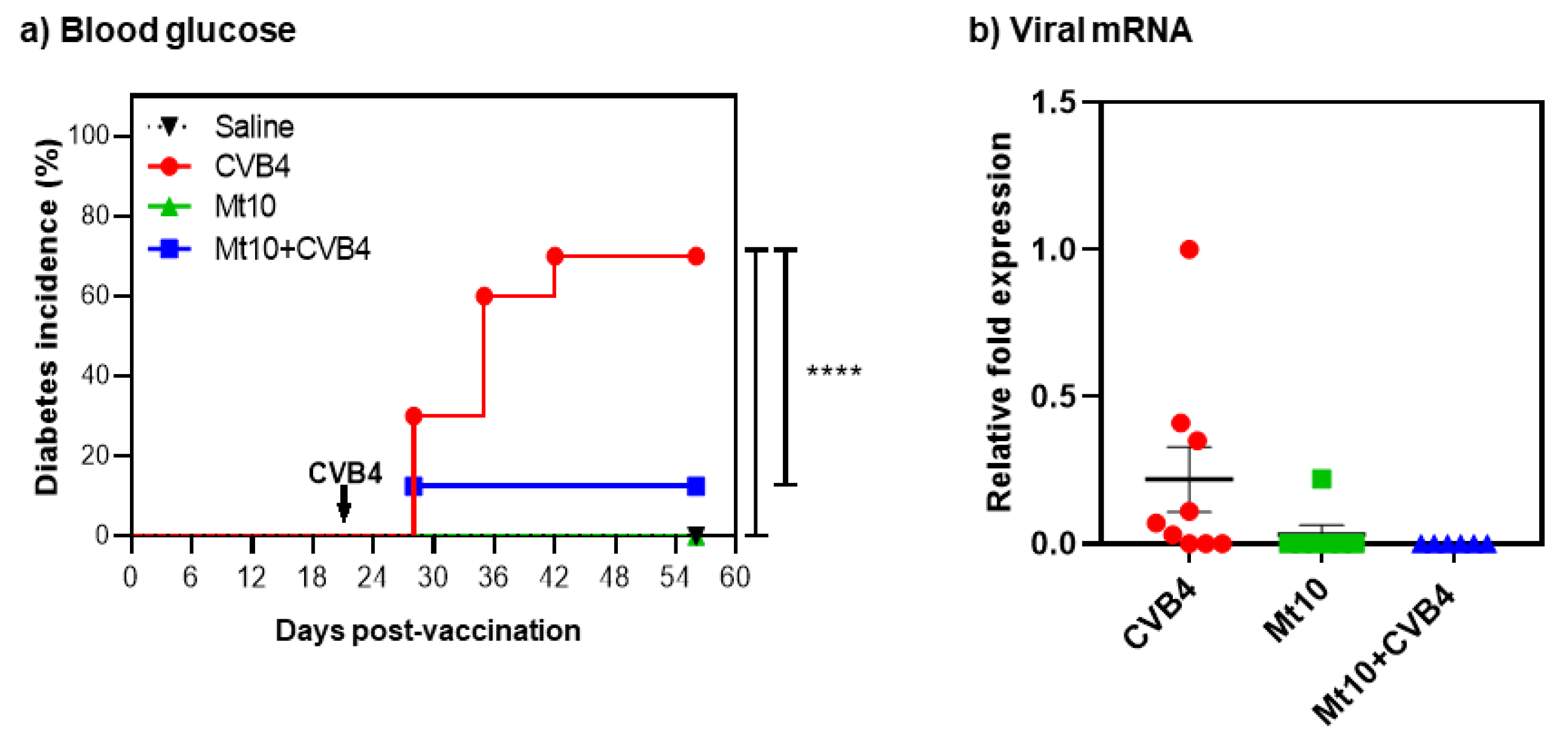
2.4. Blood Glucose and Diabetes Monitoring
2.5. Histopathology
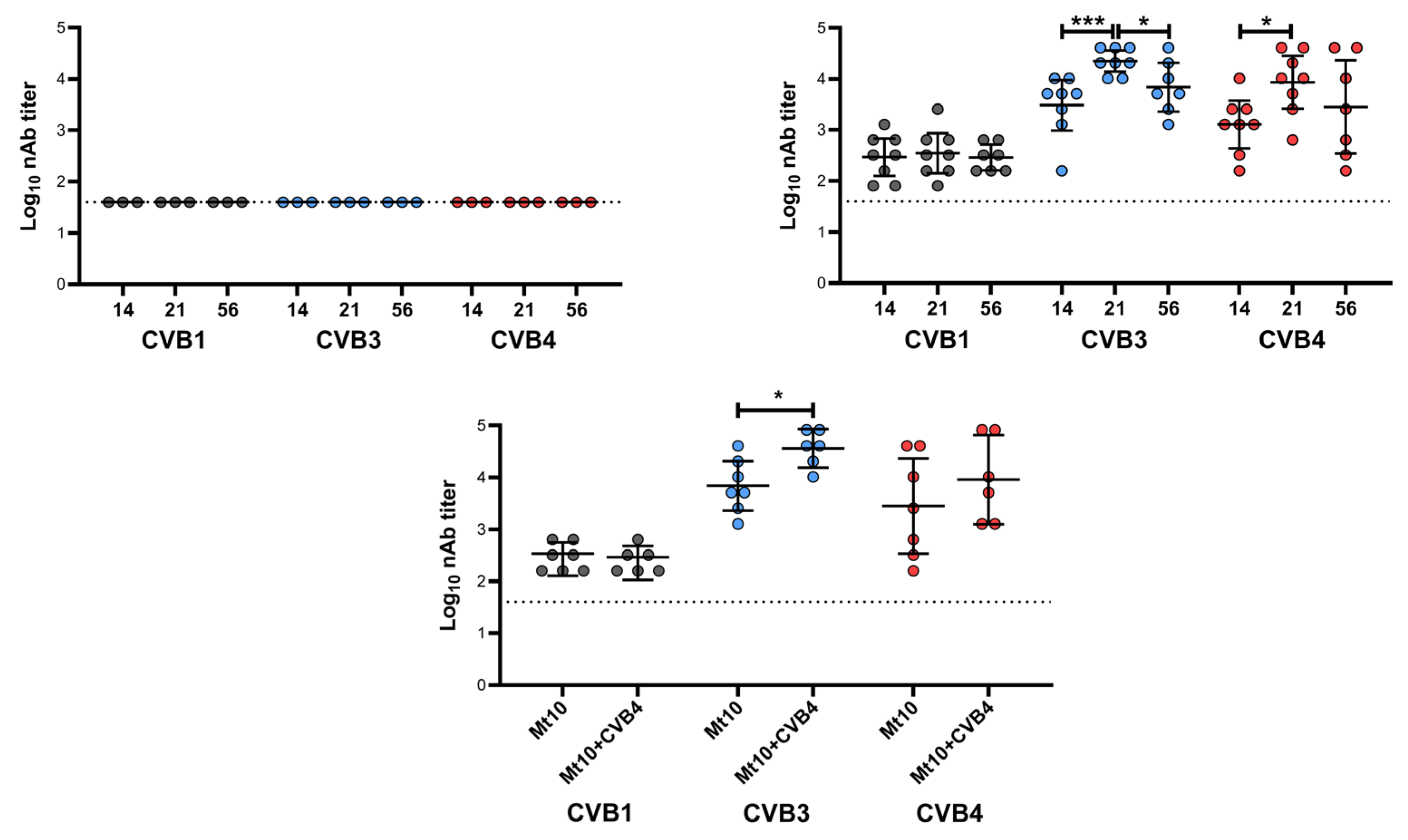
2.6. Virus Neutralization Assay
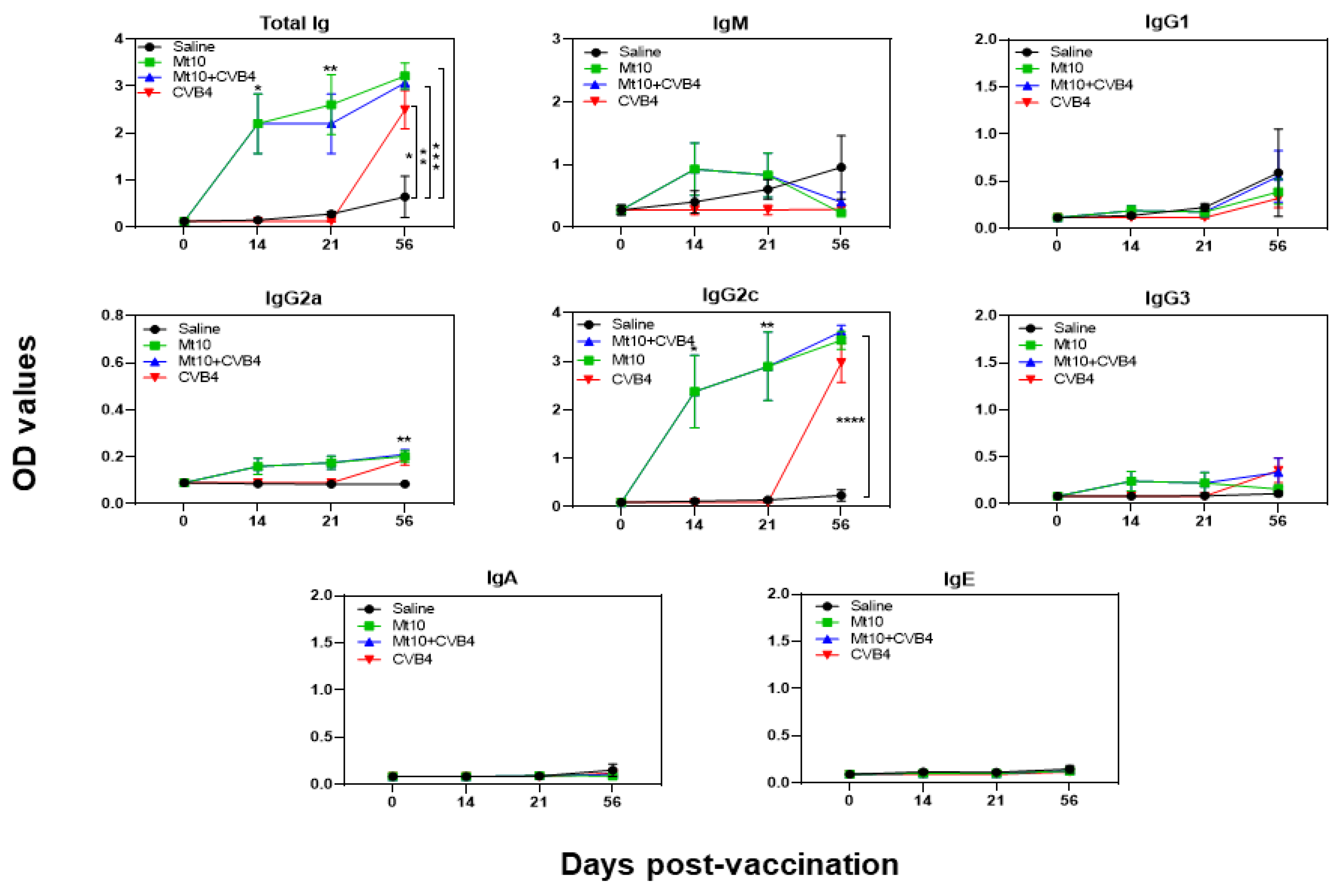
2.7. Determination of CVB-Reactive Antibodies
2.8. Determination of Anti-Insulin Antibodies
2.9. Flow Cytometry
2.10. Cytokine Analysis
2.11. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.12. Statistical Analysis
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2019. Diabetes Care 2019, 42 (Suppl. S1), S13–S28. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Pathak, V.; Pathak, N.M.; O’Neill, C.L.; Guduric-Fuchs, J.; Medina, R.J. Therapies for Type 1 Diabetes: Current Scenario and Future Perspectives. Clin. Med. Insights Endocrinol. Diabetes 2019, 12, 1179551419844521. [Google Scholar] [CrossRef]
- Von Scholten, B.J.; Kreiner, F.F.; Gough, S.C.L.; von Herrath, M. Current and future therapies for type 1 diabetes. Diabetologia 2021, 64, 1037–1048. [Google Scholar] [CrossRef]
- Ikegami, H.; Noso, S.; Babaya, N.; Kawabata, Y. Genetics and pathogenesis of type 1 diabetes: Prospects for prevention and intervention. J. Diabetes Investig. 2011, 2, 415–420. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Alidjinou, E.K.; Hober, D. Persistent coxsackievirus B infection and pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 503–516. [Google Scholar] [CrossRef]
- Lernmark, A.; Larsson, H.E. Immune therapy in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 92–103. [Google Scholar] [CrossRef]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef]
- TEDDY Study Group. The Environmental Determinants of Diabetes in the Young (TEDDY) Study. Ann. N. Y. Acad. Sci. 2008, 1150, 1–13. [Google Scholar] [CrossRef]
- Quinn, L.M.; Wong, F.S.; Narendran, P. Environmental Determinants of Type 1 Diabetes: From Association to Proving Causality. Front. Immunol. 2021, 12, 737964. [Google Scholar] [CrossRef]
- Fathallah, N.; Slim, R.; Larif, S.; Hmouda, H.; Ben Salem, C. Drug-Induced Hyperglycaemia and Diabetes. Drug Saf. 2015, 38, 1153–1168. [Google Scholar] [CrossRef]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef]
- Taplin, C.E.; Barker, J.M. Autoantibodies in type 1 diabetes. Autoimmunity 2008, 41, 11–18. [Google Scholar] [CrossRef]
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef]
- Richardson, S.J.; Morgan, N.G. Enteroviral infections in the pathogenesis of type 1 diabetes: New insights for therapeutic intervention. Curr. Opin. Pharmacol. 2018, 43, 11–19. [Google Scholar] [CrossRef]
- Beyerlein, A.; Wehweck, F.; Ziegler, A.G.; Pflueger, M. Respiratory infections in early life and the development of islet autoimmunity in children at increased type 1 diabetes risk: Evidence from the BABYDIET study. JAMA Pediatr. 2013, 167, 800–807. [Google Scholar] [CrossRef]
- Frisk, G.; Nilsson, E.; Tuvemo, T.; Friman, G.; Diderholm, H. The possible role of Coxsackie A and echo viruses in the pathogenesis of type I diabetes mellitus studied by IgM analysis. J. Infect 1992, 24, 13–22. [Google Scholar] [CrossRef]
- Hyoty, H. Viruses in type 1 diabetes. Pediatr. Diabetes 2016, 17 (Suppl. S22), 56–64. [Google Scholar] [CrossRef]
- Romero, J.R. Pediatric group B coxsackievirus infections. Curr. Top. Microbiol. Immunol. 2008, 323, 223–239. [Google Scholar]
- Spickard, A.; Evans, H.; Knight, V.; Johnson, K. Acute respiratory disease in normal volunteers associated with Coxsackie A-21 viral infection. III. Response to nasopharyngeal and enteric inoculation. J. Clin. Investig. 1963, 42, 840–852. [Google Scholar] [CrossRef]
- Tariq, N.; Kyriakopoulos, C. Group B Coxsackie Virus; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Fairweather, D.; Stafford, K.A.; Sung, Y.K. Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 2012, 24, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Lasrado, N.; Yalaka, B.; Reddy, J. Triggers of Inflammatory Heart Disease. Front. Cell Dev. Biol. 2020, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Tschope, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Alhazmi, A.; Nekoua, M.P.; Michaux, H.; Sane, F.; Halouani, A.; Engelmann, I.; Alidjinou, E.K.; Martens, H.; Jaidane, H.; Geenen, V.; et al. Effect of Coxsackievirus B4 Infection on the Thymus: Elucidating Its Role in the Pathogenesis of Type 1 Diabetes. Microorganisms 2021, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; Andre, P.; Harju, R.; et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef]
- Serreze, D.V.; Ottendorfer, E.W.; Ellis, T.M.; Gauntt, C.J.; Atkinson, M.A. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 2000, 49, 708–711. [Google Scholar] [CrossRef]
- Martino, T.A.; Petric, M.; Weingartl, H.; Bergelson, J.M.; Opavsky, M.A.; Richardson, C.D.; Modlin, J.F.; Finberg, R.W.; Kain, K.C.; Willis, N.; et al. The coxsackie-adenovirus receptor (CAR) is used by reference strains and clinical isolates representing all six serotypes of coxsackievirus group B and by swine vesicular disease virus. Virology 2000, 271, 99–108. [Google Scholar] [CrossRef]
- Jenson, A.B.; Rosenberg, H.S.; Notkins, A.L. Pancreatic islet-cell damage in children with fatal viral infections. Lancet 1980, 2, 354–358. [Google Scholar]
- Ylipaasto, P.; Klingel, K.; Lindberg, A.M.; Otonkoski, T.; Kandolf, R.; Hovi, T.; Roivainen, M. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 2004, 47, 225–239. [Google Scholar] [CrossRef]
- Ifie, E.; Russell, M.A.; Dhayal, S.; Leete, P.; Sebastiani, G.; Nigi, L.; Dotta, F.; Marjomaki, V.; Eizirik, D.L.; Morgan, N.G.; et al. Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic beta cells. Diabetologia 2018, 61, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Lasrado, N.; Arumugam, R.; Rasquinha, M.T.; Sur, M.; Steffen, D.; Reddy, J. Mt10-CVB3 Vaccine Virus Protects against CVB4 Infection by Inducing Cross-Reactive, Antigen-Specific Immune Responses. Microorganisms 2021, 9, 2323. [Google Scholar] [CrossRef]
- Lasrado, N.; Gangaplara, A.; Massilamany, C.; Arumugam, R.; Shelbourn, A.; Rasquinha, M.T.; Basavalingappa, R.H.; Delhon, G.; Xiang, S.-H.; Pattnaik, A.K.; et al. Attenuated strain of CVB3 with a mutation in the CAR-interacting region protects against both myocarditis and pancreatitis. Sci. Rep. 2021, 11, 12432. [Google Scholar] [CrossRef]
- Dougherty, R.; Harris, R. Techniques in Experimental Virology; Academic Press: Cambridge, MA, USA, 1964; Volume 169, pp. 183–186. [Google Scholar]
- Korstanje, R.; Ryan, J.L.; Savage, H.S.; Lyons, B.L.; Kane, K.G.; Sukoff Rizzo, S.J. Continuous Glucose Monitoring in Female NOD Mice Reveals Daily Rhythms and a Negative Correlation With Body Temperature. Endocrinology 2017, 158, 2707–2712. [Google Scholar] [CrossRef]
- Zhang, C.; Todorov, I.; Lin, C.L.; Atkinson, M.; Kandeel, F.; Forman, S.; Zeng, D. Elimination of insulitis and augmentation of islet beta cell regeneration via induction of chimerism in overtly diabetic NOD mice. Proc. Natl. Acad. Sci. USA 2007, 104, 2337–2342. [Google Scholar] [CrossRef]
- Babaya, N.; Liu, E.; Miao, D.; Li, M.; Yu, L.; Eisenbarth, G.S. Murine high specificity/sensitivity competitive europium insulin autoantibody assay. Diabetes Technol. Ther. 2009, 11, 227–233. [Google Scholar] [CrossRef]
- Lehmann, J.S.; Rughwani, P.; Kolenovic, M.; Ji, S.; Sun, B. LEGENDplex: Bead-assisted multiplex cytokine profiling by flow cytometry. Methods Enzymol. 2019, 629, 151–176. [Google Scholar]
- Lehmann, J.S.; Zhao, A.; Sun, B.; Jiang, W.; Ji, S. Multiplex Cytokine Profiling of Stimulated Mouse Splenocytes Using a Cytometric Bead-based Immunoassay Platform. J. Vis. Exp. 2017, 129, e56440. [Google Scholar] [CrossRef]
- Basavalingappa, R.H.; Arumugam, R.; Lasrado, N.; Yalaka, B.; Massilamany, C.; Gangaplara, A.; Riethoven, J.-J.; Xiang, S.-H.; Steffen, D.; Reddy, J. Viral myocarditis involves the generation of autoreactive T cells with multiple antigen specificities that localize in lymphoid and non-lymphoid organs in the mouse model of CVB3 infection. Mol. Immunol. 2020, 124, 218–228. [Google Scholar] [CrossRef]
- Drescher, K.M.; Kono, K.; Bopegamage, S.; Carson, S.D.; Tracy, S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: Insulitis determines susceptibility of pancreatic islets to virus infection. Virology 2004, 329, 381–394. [Google Scholar] [CrossRef]
- Gangaplara, A.; Massilamany, C.; Brown, D.M.; Delhon, G.; Pattnaik, A.K.; Chapman, N.; Rose, N.; Steffen, D.; Reddy, J. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-alpha-reactive CD4 T cells in A/J mice. Clin. Immunol. 2012, 144, 237–249. [Google Scholar] [CrossRef]
- Leipner, C.; Grun, K.; Schneider, I.; Gluck, B.; Sigusch, H.H.; Stelzner, A. Coxsackievirus B3-induced myocarditis: Differences in the immune response of C57BL/6 and Balb/c mice. Med. Microbiol. Immunol. 2004, 193, 141–147. [Google Scholar] [CrossRef]
- Neu, N.; Rose, N.R.; Beisel, K.W.; Herskowitz, A.; Gurri-Glass, G.; Craig, S.W. Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 1987, 139, 3630–3636. [Google Scholar]
- Wolfgram, L.J.; Beisel, K.W.; Herskowitz, A.; Rose, N.R. Variations in the susceptibility to Coxsackievirus B3-induced myocarditis among different strains of mice. J. Immunol. 1986, 136, 1846–1852. [Google Scholar]
- Honkimaa, A.; Kimura, B.; Sioofy-Khojine, A.B.; Lin, J.; Laiho, J.; Oikarinen, S.; Hyoty, H. Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells. Microorganisms 2020, 8, 1790. [Google Scholar]
- Huber, S.; Ramsingh, A.I. Coxsackievirus-induced pancreatitis. Viral. Immunol. 2004, 17, 358–369. [Google Scholar] [CrossRef]
- Lasrado, N.; Gangaplara, A.; Arumugam, R.; Massilamany, C.; Pokal, S.; Zhou, Y.; Xiang, S.H.; Steffen, D.; Reddy, J. Identification of Immunogenic Epitopes That Permit the Detection of Antigen-Specific T Cell Responses in Multiple Serotypes of Group B Coxsackievirus Infections. Viruses 2020, 12, 347. [Google Scholar] [CrossRef]
- Filippi, C.; von Herrath, M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol. 2005, 233, 125–132. [Google Scholar] [CrossRef]
- Mathews, C.E.; Xue, S.; Posgai, A.; Lightfoot, Y.L.; Li, X.; Lin, A.; Wasserfall, C.; Haller, M.J.; Schatz, D.; Atkinson, M.A. Acute Versus Progressive Onset of Diabetes in NOD Mice: Potential Implications for Therapeutic Interventions in Type 1 Diabetes. Diabetes 2015, 64, 3885–3890. [Google Scholar] [CrossRef]
- Chen, D.; Thayer, T.C.; Wen, L.; Wong, F.S. Mouse Models of Autoimmune Diabetes: The Nonobese Diabetic (NOD) Mouse. Methods Mol. Biol. 2020, 2128, 87–92. [Google Scholar]
- Chen, Y.G.; Mathews, C.E.; Driver, J.P. The Role of NOD Mice in Type 1 Diabetes Research: Lessons from the Past and Recommendations for the Future. Front. Endocrinol. 2018, 9, 51. [Google Scholar] [CrossRef]
- Tracy, S.; Drescher, K.M.; Chapman, N.M.; Kim, K.S.; Carson, S.D.; Pirruccello, S.; Lane, P.H.; Romero, J.R.; Leser, J.S. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: Inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J. Virol. 2002, 76, 12097–12111. [Google Scholar] [CrossRef][Green Version]
- Ostrowski, S.E.; Reilly, A.A.; Collins, D.N.; Ramsingh, A.I. Progression or resolution of coxsackievirus B4-induced pancreatitis: A genomic analysis. J. Virol. 2004, 78, 8229–8237. [Google Scholar] [CrossRef]
- Serreze, D.V.; Wasserfall, C.; Ottendorfer, E.W.; Stalvey, M.; Pierce, M.A.; Gauntt, C.; O’Donnell, B.; Flanagan, J.B.; Campbell-Thompson, M.; Ellis, T.M.; et al. Diabetes acceleration or prevention by a coxsackievirus B4 infection: Critical requirements for both interleukin-4 and gamma interferon. J. Virol. 2005, 79, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.P.; Asplin, C.M.; Clemons, P.; Lyen, K.; Tatpati, O.; Raghu, P.K.; Paquette, T.L. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983, 222, 1337–1339. [Google Scholar] [CrossRef] [PubMed]
- Sioofy-Khojine, A.B.; Lehtonen, J.; Nurminen, N.; Laitinen, O.H.; Oikarinen, S.; Huhtala, H.; Pakkanen, O.; Ruokoranta, T.; Hankaniemi, M.M.; Toppari, J.; et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018, 61, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, E.; Atkinson, M.; Eisenbarth, G.; Serreze, D.; Kay, T.W.; Lee-Chan, E.; Singh, B. International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: Identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes 2001, 50, 2451–2458. [Google Scholar] [CrossRef]
- Bonifacio, E.; Atkinson, M.; Eisenbarth, G.; Serreze, D.; Kay, T.W.; Lee-Chan, E.; Singh, B. International Workshop on Lessons from Animal Models for Human Type 1 Diabetes: Analyzing target autoantigens of humoral immunity in nonobese diabetic mice. Ann. N. Y. Acad. Sci. 2002, 958, 1–2. [Google Scholar] [CrossRef]
- Kuglin, B.; Kolb, H.; Greenbaum, C.; Maclaren, N.K.; Lernmark, A.; Palmer, J.P. The Fourth International Workshop on the Standardisation of Insulin Autoantibody Workshop. Diabetologia 1990, 33, 638–639. [Google Scholar]
- Yu, L.; Eisenbarth, G.; Bonifacio, E.; Thomas, J.; Atkinson, M.; Wasserfall, C. The second murine autoantibody workshop: Remarkable interlaboratory concordance for radiobinding assays to identify insulin autoantibodies in nonobese diabetic mice. Ann. N. Y. Acad. Sci. 2003, 1005, 1–12. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef]
- Dahlgren, M.W.; Plumb, A.W.; Niss, K.; Lahl, K.; Brunak, S.; Johansson-Lindbom, B. Type I Interferons Promote Germinal Centers Through B Cell Intrinsic Signaling and Dendritic Cell Dependent Th1 and Tfh Cell Lineages. Front. Immunol. 2022, 13, 932388. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.J.; Natsume-Kitatani, Y.; Temizoz, B.; Fujita, Y.; Konishi, A.; Matsuda, K.; Igari, Y.; Tsukui, T.; Kobiyama, K.; Kuroda, E.; et al. B cell-intrinsic MyD88 signaling controls IFN-gamma-mediated early IgG2c class switching in mice in response to a particulate adjuvant. Eur. J. Immunol. 2019, 49, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; Estes, E.A.; Oldham, J.E.; von Herrath, M.G. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J. Clin. Investig. 2009, 119, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.J.; Straka, N.; Fang, D.; Shanina, I.; Horwitz, M.S. Regulatory T-cells protect from type 1 diabetes after induction by coxsackievirus infection in the context of transforming growth factor-beta. Diabetes 2008, 57, 1302–1311. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shi, Y.; Fukuoka, M.; Li, G.; Liu, Y.; Chen, M.; Konviser, M.; Chen, X.; Opavsky, M.A.; Liu, P.P. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation 2010, 121, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Boettler, T.; von Herrath, M. Type 1 diabetes vaccine development: Animal models vs. humans. Hum. Vaccin. 2011, 7, 19–26. [Google Scholar] [CrossRef][Green Version]
- Drescher, K.M.; von Herrath, M.; Tracy, S. Enteroviruses, hygiene and type 1 diabetes: Toward a preventive vaccine. Rev. Med. Virol. 2015, 25, 19–32. [Google Scholar] [CrossRef]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jorgensen, K.; Flodstrom-Tullberg, M.; Hyoty, H.; Insel, R.A.; Lernmark, A.; Lloyd, R.E.; et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef]
- Hyoty, H.; Leon, F.; Knip, M. Developing a vaccine for type 1 diabetes by targeting coxsackievirus B. Expert Rev. Vaccines 2018, 17, 1071–1083. [Google Scholar] [CrossRef]
- Kondrashova, A.; Hyoty, H. Role of viruses and other microbes in the pathogenesis of type 1 diabetes. Int. Rev. Immunol. 2014, 33, 284–295. [Google Scholar] [CrossRef]
- Drescher, K.M.; Tracy, S.M. The CVB and etiology of type 1 diabetes. Curr. Top. Microbiol. Immunol. 2008, 323, 259–274. [Google Scholar] [PubMed]
- Eizirik, D.L.; Op de Beeck, A. Coxsackievirus and Type 1 Diabetes Mellitus: The Wolf’s Footprints. Trends Endocrinol. Metab. 2018, 29, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; London, W.T.; Curfman, B.L.; Brown, R.L.; Notkins, A.L. Coxsackie virus B4 produces transient diabetes in nonhuman primates. Diabetes 1986, 35, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.J.; Schatz, D.A. The DIPP project: 20 years of discovery in type 1 diabetes. Pediatr. Diabetes 2016, 17 (Suppl. S22), 5–7. [Google Scholar] [CrossRef]
- Hyoty, H.; Hiltunen, M.; Knip, M.; Laakkonen, M.; Vahasalo, P.; Karjalainen, J.; Koskela, P.; Roivainen, M.; Leinikki, P.; Hovi, T.; et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 1995, 44, 652–657. [Google Scholar] [CrossRef]
- Krogvold, L.; Leete, P.; Mynarek, I.M.; Russell, M.A.; Gerling, I.C.; Lenchik, N.I.; Mathews, C.; Richardson, S.J.; Morgan, N.G.; Dahl-Jorgensen, K. Detection of Antiviral Tissue Responses and Increased Cell Stress in the Pancreatic Islets of Newly Diagnosed Type 1 Diabetes Patients: Results From the DiViD Study. Front. Endocrinol. 2022, 13, 881997. [Google Scholar] [CrossRef]
- Esposito, S.; Mariotti Zani, E.; Torelli, L.; Scavone, S.; Petraroli, M.; Patianna, V.; Predieri, B.; Iughetti, L.; Principi, N. Childhood Vaccinations and Type 1 Diabetes. Front. Immunol. 2021, 12, 667889. [Google Scholar] [CrossRef]
- Wasfy, J.H. Childhood vaccination and type 1 diabetes. N. Engl. J. Med. 2004, 351, 298. [Google Scholar] [CrossRef]
- Haller, M.J.; Atkinson, M.A.; Schatz, D. Type 1 diabetes mellitus: Etiology, presentation, and management. Pediatr. Clin. N. Am. 2005, 52, 1553–1578. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Nepom, G.T. Prediction and pathogenesis in type 1 diabetes. Immunity 2010, 32, 468–478. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, H.; Shao, J.; Jia, Y.; Bao, Q.; Zhu, J.; Zhang, L.; Shen, Y. Different Hepatitis C Virus Infection Statuses Show a Significant Risk of Developing Type 2 Diabetes Mellitus: A Network Meta-Analysis. Dig. Dis. Sci. 2020, 65, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Klitz, W.; Niklasson, B. Extending the Enterovirus Lead: Could a Related Picornavirus be Responsible for Diabetes in Humans? Microorganisms 2020, 8, 1382. [Google Scholar] [CrossRef] [PubMed]
- Baicus, A. History of polio vaccination. World J. Virol. 2012, 1, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. EV71 vaccines: A milestone in the history of global vaccine development. Emerg. Microbes Infect. 2014, 3, e27. [Google Scholar] [CrossRef]
- Stone, V.M.; Hankaniemi, M.M.; Svedin, E.; Sioofy-Khojine, A.; Oikarinen, S.; Hyoty, H.; Laitinen, O.H.; Hytonen, V.P.; Flodstrom-Tullberg, M. A Coxsackievirus B vaccine protects against virus-induced diabetes in an experimental mouse model of type 1 diabetes. Diabetologia 2018, 61, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Davydova, B.; Harkonen, T.; Kaialainen, S.; Hovi, T.; Vaarala, O.; Roivainen, M. Coxsackievirus immunization delays onset of diabetes in non-obese diabetic mice. J. Med. Virol. 2003, 69, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Hankaniemi, M.M.; Laitinen, O.H.; Sioofy-Khojine, A.B.; Lin, A.; Diaz Lozano, I.M.; Mazur, M.A.; Marjomaki, V.; Lore, K.; Hyoty, H.; et al. A hexavalent Coxsackievirus B vaccine is highly immunogenic and has a strong protective capacity in mice and nonhuman primates. Sci. Adv. 2020, 6, eaaz2433. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef]
- Liu, H.; Luo, H. Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges. Viruses 2021, 13, 1082. [Google Scholar] [CrossRef]
- Koho, T.; Koivunen, M.R.L.; Oikarinen, S.; Kummola, L.; Makinen, S.; Mahonen, A.J.; Sioofy-Khojine, A.; Marjomaki, V.; Kazmertsuk, A.; Junttila, I.; et al. Coxsackievirus B3 VLPs purified by ion exchange chromatography elicit strong immune responses in mice. Antivir. Res. 2014, 104, 93–101. [Google Scholar] [CrossRef]
- Kavanagh, K.; Flynn, D.M.; Nelson, C.; Zhang, L.; Wagner, J.D. Characterization and validation of a streptozotocin-induced diabetes model in the vervet monkey. J. Pharmacol. Toxicol. Methods 2011, 63, 296–303. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rood, P.P.; Bottino, R.; Balamurugan, A.N.; Smetanka, C.; Ezzelarab, M.; Busch, J.; Hara, H.; Trucco, M.; Cooper, D.K. Induction of diabetes in cynomolgus monkeys with high-dose streptozotocin: Adverse effects and early responses. Pancreas 2006, 33, 287–292. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Saline | CVB4 | Mt10 | Mt10 + CVB4 |
|---|---|---|---|---|
| Insulitis | 2/5 (40.0) | 8/9 (88.9) | 4/7 (57.1) | 1/8 (12.5) * |
| Peri-insulitis | 3/5 (60.0) | 7/9 (77.8) | 6/7 (85.7) | 7/8 (87.5) |
| Peri-ductule inflammation | 5/5 (100.0) | 7/9 (77.8) | 4/7 (57.1) | 2/8 (25.0) |
| Destruction/Fatty replacement | 0 (0.0) * | 8/9 (88.9) | 2/7 (28.5) | 1/8 (12.5) * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasquinha, M.T.; Lasrado, N.; Sur, M.; Mone, K.; Qiu, H.; Riethoven, J.-J.; Sobel, R.A.; Reddy, J. A Monovalent Mt10-CVB3 Vaccine Prevents CVB4-Accelerated Type 1 Diabetes in NOD Mice. Vaccines 2023, 11, 76. https://doi.org/10.3390/vaccines11010076
Rasquinha MT, Lasrado N, Sur M, Mone K, Qiu H, Riethoven J-J, Sobel RA, Reddy J. A Monovalent Mt10-CVB3 Vaccine Prevents CVB4-Accelerated Type 1 Diabetes in NOD Mice. Vaccines. 2023; 11(1):76. https://doi.org/10.3390/vaccines11010076
Chicago/Turabian StyleRasquinha, Mahima T., Ninaad Lasrado, Meghna Sur, Kiruthiga Mone, Haowen Qiu, Jean-Jack Riethoven, Raymond A. Sobel, and Jay Reddy. 2023. "A Monovalent Mt10-CVB3 Vaccine Prevents CVB4-Accelerated Type 1 Diabetes in NOD Mice" Vaccines 11, no. 1: 76. https://doi.org/10.3390/vaccines11010076
APA StyleRasquinha, M. T., Lasrado, N., Sur, M., Mone, K., Qiu, H., Riethoven, J.-J., Sobel, R. A., & Reddy, J. (2023). A Monovalent Mt10-CVB3 Vaccine Prevents CVB4-Accelerated Type 1 Diabetes in NOD Mice. Vaccines, 11(1), 76. https://doi.org/10.3390/vaccines11010076