
A Simple Epidemiologic Model for Predicting Impaired Neutralization of New SARS-CoV-2 Variants




Abstract
1. Introduction
2. Materials and Methods
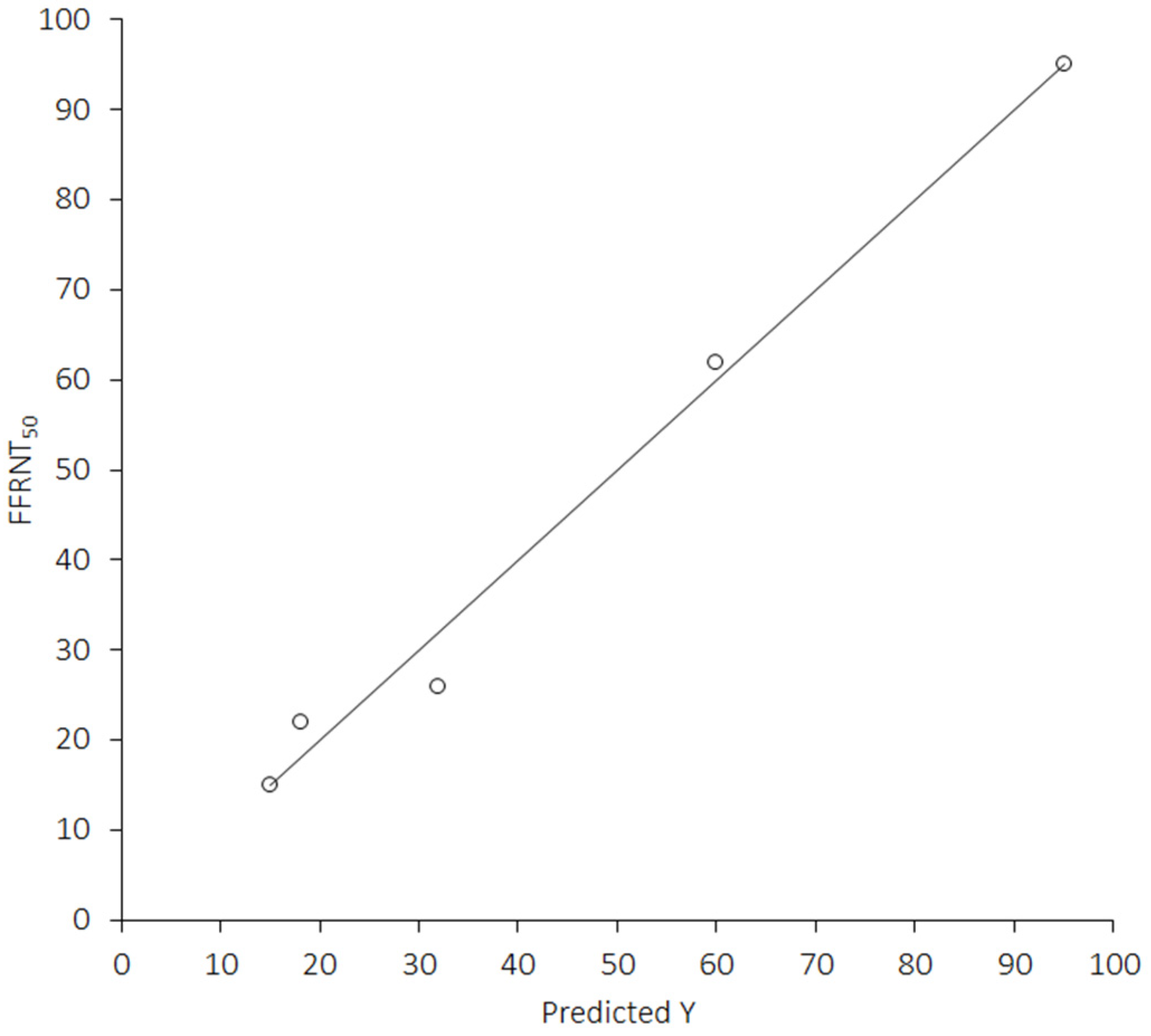
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 23 November 2022).
- Mattiuzzi, C.; Lippi, G. Nationwide analysis of COVID-19 death rate throughout the pandemic in Italy. J. Lab. Precis. Med. 2023. [Google Scholar] [CrossRef]
- Mallapaty, S. Surprising Omicron origins study comes under scrutiny. Nature 2022, 612, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.M.; Wolf, L.M.; Bello, G.L.; Maccari, J.G.; Nasi, L.A. Molecular evolution of SARS-CoV-2 from December 2019 to August 2022. J. Med. Virol. 2022, 95, 28366. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi, C.; Henry, B.M.; Lippi, G. Regional Association between Mean Air Temperature and Case Numbers of Multiple SARS-CoV-2 Lineages throughout the Pandemic. Viruses 2022, 14, 1913. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Chakravarty, N.; Jeyachandran, A.V.; Jayakarunakaran, A.; Sinha, S.; Mishra, R.; Arumugaswami, V.; Ramaiah, A. In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics. Viruses 2022, 14, 2461. [Google Scholar] [CrossRef]
- Cao, L.; Lou, J.; Chan, S.Y.; Zheng, H.; Liu, C.; Zhao, S.; Li, Q.; Mok, C.K.P.; Chan, R.W.Y.; Chong, M.K.C.; et al. Rapid evaluation of COVID-19 vaccine effectiveness against symptomatic infection with SARS-CoV-2 variants by analysis of genetic distance. Nat. Med. 2022, 28, 1715–1722. [Google Scholar] [CrossRef]
- Arora, P.; Kempf, A.; Nehlmeier, I.; Schulz, S.R.; Jäck, H.M.; Pöhlmann, S.; Hoffmann, M. Omicron sublineage BQ.1.1 resistance to monoclonal antibodies. Lancet Infect Dis. 2022, 23, 22–23, Erratum in: Lancet Infect Dis. 2022. [Google Scholar] [CrossRef]
- Imai, M.; Ito, M.; Kiso, M.; Yamayoshi, S.; Uraki, R.; Fukushi, S.; Watanabe, S.; Suzuki, T.; Maeda, K.; Sakai-Tagawa, Y.; et al. Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N. Engl. J. Med. 2022. [Google Scholar] [CrossRef]
- Hoffmann, M.; Behrens, G.M.N.; Arora, P.; Kempf, A.; Nehlmeier, I.; Cossmann, A.; Manthey, L.; Dopfer-Jablonka, A.; Pöhlmann, S. Effect of hybrid immunity and bivalent booster vaccination on omicron sublineage neutralisation. Lancet Infect Dis. 2022, 23, 25–28. [Google Scholar] [CrossRef]
- Sullivan, D.J.; Franchini, M.; Senefeld, J.W.; Joyner, M.J.; Casadevall, A.; Focosi, D. Plasma after both SARS-CoV-2 boosted vaccination and COVID-19 potently neutralizes BQ.1.1 and XBB.1. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C.; Henry, B.M. Uncontrolled confounding in COVID-19 epidemiology. Diagnosis 2022. [Google Scholar] [CrossRef]
- Banerjee, S. Dynamics of the COVID-19 pandemic: Nonlinear approaches on the modelling, prediction and control. Eur. Phys. J. Spec. Top. 2022, 231, 3275–3280. [Google Scholar] [CrossRef]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1, and XBB.1 by parental mRNA vaccine or a BA.5-bivalent booster. Nat. Med. 2022. [Google Scholar] [CrossRef]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 7 December 2022).
- Bacterial and Viral Bioinformatics Resource Center. SARS-CoV-2 Variants and Lineages of Concern. Available online: https://www.bv-brc.org/view/VariantLineage/#view%5Ftab=overview (accessed on 7 December 2022).
- Lewis, T.G.; Al Mannai, W.I. Editorial: Modeling Epidemics—Why Are Models Wrong? Front. Public Health 2021, 9, 754746. [Google Scholar] [CrossRef]
- Hoskins, S.; Beale, S.; Nguyen, V.; Boukari, Y.; Yavlinsky, A.; Kovar, J.; Byrne, T.; Fragaszy, E.; Fong, W.L.E.; Geismar, C.; et al. Relative contribution of essential and non-essential activities to SARS-CoV-2 transmission following the lifting of public health restrictions in England and Wales. Epidemiol. Infect. 2022, 1–27. [Google Scholar] [CrossRef]
- Marks, P.W.; Gruppuso, P.A.; Adashi, E.Y. Urgent Need for Next-Generation COVID-19 Vaccines. JAMA 2022. [Google Scholar] [CrossRef]
- Meyer, J.F.C.A.; Lima, M. Relevant mathematical modelling efforts for understanding COVID-19 dynamics: An educational challenge. ZDM 2022, 1–14. [Google Scholar] [CrossRef]
- Snedden, C.E.; Makanani, S.K.; Schwartz, S.T.; Gamble, A.; Blakey, R.V.; Borremans, B.; Helman, S.K.; Espericueta, L.; Valencia, A.; Endo, A.; et al. SARS-CoV-2: Cross-scale Insights from Ecology and Evolution. Trends Microbiol. 2021, 29, 593–605. [Google Scholar] [CrossRef]
- Evolution goes viral. Nat. Ecol. Evol. 2021, 5, 143. [CrossRef]
- Bull, J.J.; Badgett, M.R.; Wichman, H.A.; Huelsenbeck, J.P.; Hillis, D.M.; Gulati, A.; Ho, C.; Molineux, I.J. Exceptional convergent evolution in a virus. Genetics 1997, 147, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Zahradník, J.; Nunvar, J.; Schreiber, G. Perspectives: SARS-CoV-2 Spike Convergent Evolution as a Guide to Explore Adaptive Advantage. Front. Cell Infect. Microbiol. 2022, 12, 748948. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, V.; Patrick, C.; Lucas, A.; Mallela, K.M.G. Convergent Evolution of Multiple Mutations Improves the Viral Fitness of SARS-CoV-2 Variants by Balancing Positive and Negative Selection. Biochemistry 2022, 61, 963–980. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: What new variants are emerging and how are they being investigated? BMJ 2021, 372, n158. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based Covid-19 therapeutics and vaccines. Rev. Med. Virol. 2021, 31, e2231. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. Prediction of Recurrent Mutations in SARS-CoV-2 Using Artificial Neural Networks. Int. J. Mol. Sci. 2022, 23, 14683. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| BA.4/5 | BA.4.6 | BA.2.75.2 | BQ.1.1 | XBB.1 | |
|---|---|---|---|---|---|
| Emergence (date) | 22 January 2002 | 22 April 2002 | 22 June 2002 | 22 July 2002 | 22 August 2002 |
| FFRNT50 | 95 | 62 | 26 | 22 | 15 |
| S protein mutations (n) | 33 | 36 | 38 | 35 | 41 |
| COVID-19 cases (million) | 306.957 | 490.213 | 534.040 | 554.251 | 582.439 |
| Vaccine doses (billion) | 9.20 | 11.31 | 11.85 | 12.10 | 12.35 |
| Univariate | Multivariate | |
|---|---|---|
| Emergence (date) | −0.99 (−1.00 to −0.83); p = 0.001 | 0.003 |
| S protein mutations (n) | −0.76 (−0.98 to 0.38); p = 0.139 | - |
| COVID-19 cases (million) | −0.96 (−1.00 to −0.50); p = 0.001 | 0.004 |
| Vaccine doses (billion) | −0.96 (−1.00 to −0.52); p = 0.001 | 0.004 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippi, G.; Henry, B.M.; Plebani, M. A Simple Epidemiologic Model for Predicting Impaired Neutralization of New SARS-CoV-2 Variants. Vaccines 2023, 11, 128. https://doi.org/10.3390/vaccines11010128
Lippi G, Henry BM, Plebani M. A Simple Epidemiologic Model for Predicting Impaired Neutralization of New SARS-CoV-2 Variants. Vaccines. 2023; 11(1):128. https://doi.org/10.3390/vaccines11010128
Chicago/Turabian StyleLippi, Giuseppe, Brandon M. Henry, and Mario Plebani. 2023. "A Simple Epidemiologic Model for Predicting Impaired Neutralization of New SARS-CoV-2 Variants" Vaccines 11, no. 1: 128. https://doi.org/10.3390/vaccines11010128
APA StyleLippi, G., Henry, B. M., & Plebani, M. (2023). A Simple Epidemiologic Model for Predicting Impaired Neutralization of New SARS-CoV-2 Variants. Vaccines, 11(1), 128. https://doi.org/10.3390/vaccines11010128