Abstract
Adaptive immune response modulation has taken a central position in cancer therapy in recent decades. Treatment with immune checkpoint inhibitors (ICIs) is now indicated in many cancer types with exceptional results. The two major inhibitory pathways involved are cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and programmed cell death protein 1 (PD-1). Unfortunately, immune activation is not tumor-specific, and as a result, most patients will experience some form of adverse reaction. Most immune-related adverse events (IRAEs) involve the skin and gastrointestinal (GI) tract; however, any organ can be involved. Cardiotoxicity ranges from arrhythmias to life-threatening myocarditis with very high mortality rates. To date, most treatments of ICI cardiotoxicity include immune suppression, which is also not cardiac-specific and may result in hampering of tumor clearance. Understanding the mechanisms behind immune activation in the heart is crucial for the development of specific treatments. Histological data and other models have shown mainly CD4 and CD8 infiltration during ICI-induced cardiotoxicity. Inhibition of CTLA4 seems to result in the proliferation of more diverse T0cell populations, some of which with autoantigen recognition. Inhibition of PD-1 interaction with PD ligand 1/2 (PD-L1/PD-L2) results in release from inhibition of exhausted self-recognizing T cells. However, CTLA4, PD-1, and their ligands are expressed on a wide range of cells, indicating a much more intricate mechanism. This is further complicated by the identification of multiple co-stimulatory and co-inhibitory signals, as well as the association of myocarditis with antibody-driven myasthenia gravis and myositis IRAEs. In this review, we focus on the recent advances in unraveling the complexity of the mechanisms driving ICI cardiotoxicity and discuss novel therapeutic strategies for directly targeting specific underlying mechanisms to reduce IRAEs and improve outcomes.
1. Introduction
Immune evasion is a fundamental process crucial for tumor survival and growth. Tumor cells have been shown to manipulate the tumor microenvironment (TME) and to alter the expression of immune inhibitory proteins to achieve immune evasion [1]. In the past few decades, advances in our understanding of the adaptive immune system activation processes have allowed the development of a new class of therapies. Immune checkpoint inhibitors (ICIs) target inhibitory signals, allowing immune cell activation and clearance of tumor cells. These therapies have proven remarkably effective in a growing number of cancer types (melanoma, non-small cell lung cancer (NSCLC), colorectal cancer, renal cell carcinoma (RCC)) and are indicated for as many as 43% of cancer patients in the United States (US) in 2018 [2]. The hallmark of these therapies is the blockade of the cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and programmed cell death protein 1 (PD-1) inhibitory pathways in CD4/CD8 lymphocytes (Table 1).

Table 1.
List of available immune checkpoint inhibitors licensed by the FDA.
Unfortunately, ICI immune cell activation is not specific or selective to the TME. Systemic immune activation results in a very high incidence of immune-related adverse events (IRAE). Indeed, IRAEs are expected in up to 60% of treated patients [3], most commonly affecting the skin, gastrointestinal (GI) tract, thyroid, and lungs; however, any organ can be affected. Cardiovascular adverse events (CVAEs) from ICIs can manifest in multiple ways, ranging from conduction abnormalities to severe myocarditis [4,5], which is fatal in ~50% of cases [6]. The severity of these reactions, together with substantially increased use of ICIs, make understanding the processes governing IRAEs of great importance. Indeed, protocols have been devised for active surveillance of ICI-treated patients for the early detection of CVAEs to allow early treatment and reduce mortality. These include routine electrocardiogram (ECG) and biomarkers (mainly troponin and N-terminal (NT)-pro-B-type natriuretic peptide (NT-proBNP)), screening during the first weeks of treatment, followed by echocardiography and cardiac magnetic resonance imaging (CMR) in suspected cases [7]. Endomyocardial biopsy, though considered the gold standard for definitive diagnosis of myocarditis, is far from being ideal both in terms of its sensitivity and safety. Sensitivity may be as low as 60% [8] due to the patchy nature of myocarditis, a phenomenon also seen with ICI therapy [9]. The relatively low sensitivity and risks involved combined with the increasing diagnostic performance of other modalities, such as CMR, may make endomyocardial biopsy less necessary in many cases. Although troponin T/I and NT-proBNP were shown to be sensitive [10,11], they are far from being specific. This led to a search of new markers of ICI-related myocarditis, among which are increased neutrophile-to-lymphocyte ratio and decreased absolute lymphocyte count [12]. Additionally, the search for risk factors has shown associations of other IRAEs, such as myositis (odds ratio (OR) 25.5, 95% confidence interval (CI): 18.7–34.9; p < 0.001) and hepatitis (OR 2.9, 95% CI: 1.9–4.5; p < 0.001), pre-existing heart conditions (OR 1.26, 95% CI: 1.2–1.5; p < 0.001), and increased age with the development of cardiotoxicity [13,14]. In this review, we describe the emerging range of ICI-induced cardiotoxicities and address the recent advances in understanding the fundamental processes driving these events.
2. Adaptive Immune Response Modulation
The hallmark of T-cell activation is the specific interaction between T-cell antigen receptor (TCR) and major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). However, this interaction is insufficient to elicit T-cell activation and co-stimulatory interactions are also needed. To our knowledge, the main co-stimulatory interaction is between CD28 on T cells and CD80/CD86 (B7-1, B7-1) on APCs. The binding of both TCR and CD28 creates the immune synapse necessary for activating exquisitely timed signaling cascades necessary for achieving specificity and coordinated T-cell activation (Figure 1) [15]. In addition to effector T cells, regulatory T cells have been shown to have major roles in tumor immune evasion [16,17]. Of these, the populations of CD4+, CD25+, Forkhead box protein P3+ (Foxp3) regulatory cells (Treg cells) have major roles. Foxp3 expression allows these cells to exert their inhibitory effects by multiple mechanisms—namely, sequestering free IL2 in the TME by expressing the high-affinity IL2-receptor, high-level expression of multiple immune checkpoint inhibitor (CTLA-4; PD-L1/2; TIGIT; GITR, OX-40; TIM-3) [18], and TGF-β and IL-10 production [19]. Indeed, Treg numbers and Treg/Teff ratios have been implicated in the poor prognosis of several cancer types [16].
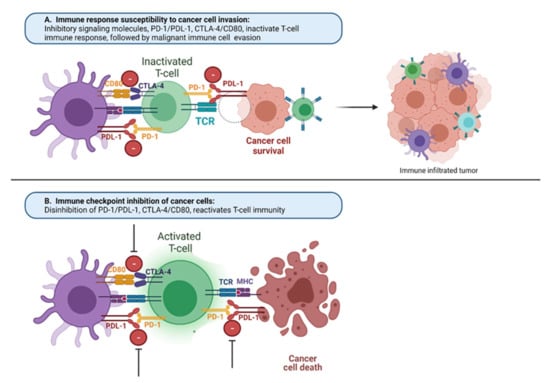
Figure 1.
Mechanisms of the immune response under malignancy and with immune checkpoint inhibitors: (A) immune response susceptibility to cancer cell invasion. Common ligands, MHC and PDL-1, found on immune macrophages (purple) and cancer cells (brown) compete with binding to receptors on T cells (green) TCR and PD-1, which normally regulate the immune response. When immunologic homeostasis is compromised between positive (TCR/MHC) and negative (CTLA-4/CD80, PD-1/PDL-1) signaling of T-cell activation, cancer cells can avoid the immune response of the host and escape from immune cell attack, providing cancer cell growth and proliferation uncontrollably; (B) in presence of immune checkpoint inhibitors toward CTLA-4/CD80, PD-1/PDL-1 signaling, there is disinhibition of these pathways and reactivation of T cells toward cancer cell death. MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TCR, T-cell receptor.
3. Additional Stimulatory Molecules
Recent advances have identified multiple other co-stimulatory interactions [20], including Glucocorticoid-induced tumor necrosis factor (TNF) receptor family-related protein (GITR), expressed on intra-tumoral Treg cells, as well as on natural killer (NK) cells and tumor-infiltrating lymphocytes (TILs) [21,22,23]. Evidence from agonist antibody studies has shown that GITR activation results in increased numbers of effector T cells and reduced Treg cells [24]. TNF receptor superfamily member 4 (TNFRSF4, OX40) is expressed on Treg and transiently on CD4 and CD8 cells [25]. Its activation results in Treg gain of pro-inflammatory traits, secreting interferon-gamma (INFγ), TNFα, and granzyme B [26]. TNF receptor superfamily member 9 (TNFRSF9, 4-1BB) can be induced on CD4 and CD8, NK, as well as other APCs [20,27]. It is used in CAR constructs and was shown to promote T-cell proliferation [28]. Inducible T-cell co-stimulatory (ICOS) is constitutively expressed on Treg cells and is induced in CD4 and CD8 cells following TCR/CD28 activation [29]. ICOS activation has been shown to stimulate effector T cells (Figure 2) [20,30].
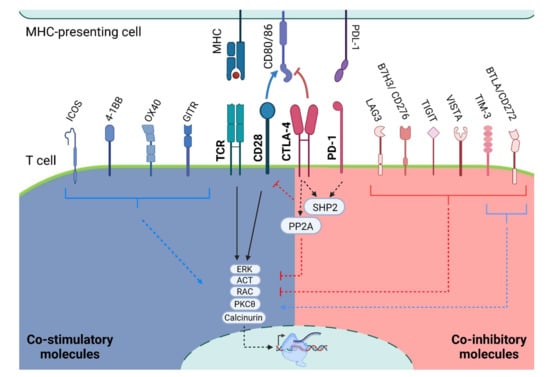
Figure 2.
Co-stimulatory and Co-inhibitory signaling. Schematic representation of TCR activation with the co-stimulatory and co-inhibitory molecules. TIM-3 and BTLA have shown both inhibitory and stimulatory activity. It is worth noting that the T-cell signaling depicts a general interaction of T cells with any MHC-presenting cells and is not specific to one cell type: MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TCR, T-cell receptor; GITR, glucocorticoid-induced tumor necrosis factor (TNF) receptor family-related protein; OX40 is also known as TNF receptor superfamily member 4 (TNFRSF4); 4-1BB is also known as TNF receptor superfamily member 9 (TNFRSF9); ICOS, inducible T cell co-stimulatory; LAG3, lymphocyte activation gene 3; TIM-3, T-cell immunoglobulin, and mucin-domain containing-3; TIGIT, T-Cell immunoglobulin and ITIM domain; VISTA, V-domain Ig suppressor of T-cell activation; B7H3 or CD276, B7 homolog 3; BTLA or CD272, B- and T-lymphocyte attenuator.
To date, several attempts to harness these stimulatory effectors have shown limited effects in clinical settings [25,31,32,33,34]. One reason is the use of antibodies as agonists, resulting in antibody-dependent cellular cytotoxicity (ADCC) of target cells. The use of low-affinity antibodies or the development of small molecule agonists may prove more beneficial in the future [35]. However, current high-affinity antibodies may still be of use by driving ADCC removal of inhibitory Treg cells.
Opposing the stimulatory interaction is a set of inhibitory molecules that are believed to be important for cessation of the immune response and avoidance of self-recognition. Many inhibitory receptors were found to be upregulated subsequent to T-cell activation. This allows fast activation, followed by a shift toward a more balanced response and finally rapid cessation of the immune response after antigen removal. This is further emphasized by the exquisite timing and intensity-sensitive intracellular signaling orchestrating T-cell activation [15]. Indeed, chronic T-cell activation can result in reduced activation over time, i.e., “exhaustion” [36,37,38,39], making the inhibitory signals important for self-avoidance as well as proper timely response.
CTLA-4 and PD-1, discovered by the Nobel Prize laureates James P. Allison and Tasuku Honjo, are two important inhibitory pathways. However, several other interactions have been revealed. These molecules are expressed on different cell types, allowing multiple mechanisms for immune modulation.
4. CTLA4 Pathway
CTLA4 is present in intracellular vesicles in naïve T cells and is translocated to the cell membrane upon activation [15]. CTLA4 also competes with CD28 for B7-1/2 binding albeit with greater affinity [40]. In CD8 cells, CTLA4 binding results in the recruitment and activation of an SH2 domain-containing tyrosine phosphatase SYP and PP2A phosphatase, which remove TCR phosphorylation and deactivate PIK3K signaling needed for CD28 function [41,42]. This results in reduced production of IL-2 and INFγ [43,44,45] and increases the exhausted CD8 population [40]. In CD4 cells, CTLA4 activation drives differentiation into inhibitory Treg (FOXP3) rather than excitatory Th1 cells expressing inducible costimulatory molecule (ICOS) [46,47,48]. Indeed, CTLA4 is constitutively expressed on Treg cells participating in their ability to suppress immune activation and tolerance [49,50]. In B cells, CTLA4-induced immune suppression occurs predominantly via intrinsic STAT3 activation, and CTLA4 is critical for B-cell lymphoma proliferation and survival. Consequently, CTLA4 can exert its suppressive effect by utilizing multiple steps in the immune cascade, resulting in a shift toward inhibitory Treg cells rather than activating Th1 cells and reduced numbers/clones of effector CD8, NK, and B cells.
5. PD-1 Pathway
PD-1 receptor is an inhibitory molecule found to be upregulated following T-cell activation [51]. In turn, PD-1 binds PD ligand 1 (PD-L1) or PD-L2, which are expressed on multiple cell types, both immune and non-immune (including tumor cells) [52,53,54]. PD-1 exerts its inhibitory effect by activating two particular SH2-containing protein tyrosine phosphatases: SHP-1 and SHP-2 [55]. Similar to CTLA4 signaling, SHP-2 can dephosphorylate and thus block CD28 signaling [56] as well as the ERK and AKT pathways [57,58]. PD-1 activation also results in reduced IL-2, INFγ secretion [59], as well as inhibiting cell cycle mediators [60,61,62].
In B cells, PD-1 activation results in inhibition of B-cell antigen receptor (BCR) signaling by recruiting SHP-2 to its phosphotyrosine and dephosphorylating key signal transducers of BCR signaling [55,63], including reduced IL-6 and antibody secretion [64]. In Treg cells, PD1-PD-L1 interaction results in increased inhibitory effects on CD8 cells. Currently, the expression and the exact role of PD-1 signaling in NK-cell activation are still under investigation [51,65,66]. Taken together, these data provide strong evidence that PD-1 has a major role predominantly in T-cell inhibition via blocking the CD28 and TCR signaling pathways, resulting in anergy or exhaustion of T cells (Figure 2).
6. Additional Inhibitory Checkpoint Molecules
Following the discovery of CTLA4 and PD-1, additional inhibitory signals were identified: Lymphocyte activation gene 3 (LAG3) was found on activated T cells, B cells, and NK cells [51] and competes with CD4 for MHC-II binding with greater affinity [65,67]. In CD4 cells, LAG3 activation reduces IL-2, IL-7, IL-12, and INFγ secretion [67,68], and evidence shows its possible inhibitory effect on CD8 cells [69]. LAG3 can be cleaved by metalloproteases, resulting in an inactive soluble form (sLAG-3), which may be part of the mechanism driven by PD-1 blockade [70]. T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) was shown to reduce CD8 response [71,72,73] and may be part of the exhausted state of T cells [74]. Additionally, it may also play a suppressive role in Th1 and Th17 cell ability to mount a response [75,76]. In contrast to these inhibitory effects, TIM-3 may also have activation effects [77,78], but its exact stimulatory role needs further investigation. T Cell immunoglobulin and ITIM domain (TIGIT) exerts its effect mainly on Treg, memory T cells, and NK cells [79]. TIGIT blocks the positive stimulator DNAM-1 signaling by having a higher affinity to CD155 ligand; however, it also harbors its own negative signaling, resulting in reduced IL-12 and increased IL-10 secretion [80,81,82,83]. V-domain Ig suppressor of T-cell activation (VISTA) is a PD-1 homolog able to exert both inhibitory and excitatory effects [84,85], mainly on T cells, macrophages, and neutrophils [86]. Initial evidence suggests that VISTA blockade results in increased T-cell activation [87]. B7 Homolog 3 (B7H3 or CD276) expression correlates with inhibition of T-cell function and proliferation [88,89,90]. It is expressed on T cells, NK cells, and other immune cells. Although its binding partner and mode of action have not yet been identified, initial data suggest the involvement of CD8 and NK cells [91]. B- and T-lymphocyte attenuator (BTLA or CD272) binding to herpes virus entry mediator (HVEM) results in SHP-2 phosphatase recruitment, similar to PD-1 signaling [92,93,94]. There is growing evidence suggesting its important role in suppressing autoimmunity [95], thus making it a promising therapeutic candidate [70,96] (Figure 2).
The discovery of multiple co-stimulatory and co-inhibitory molecules emphasizes how tightly controlled the immune response needs to be in order to achieve maximal antigen clearance with minimal collateral damage. Of the various co-inhibitory signals, CTLA4 and PD-1 are best understood with current therapies focusing on their blockage. Multiple clinical trials utilizing other co-inhibitory molecules in combination with PD-1 or CTLA4 inhibitors are underway showing promising preliminary results [25,31,32,33,34,81,97,98,99].
7. Innate and Antibody-Directed Tumor-Immune Modulation
As delineated above, some of the immune-modulatory molecules are expressed on a variety of immune cells, including B, NK, dendritic and myeloid cells. Apart from T-cell expression, the upregulation of co-inhibitory molecules, such as PD-1/CTLA4 and others, on APCs plays a crucial role in tumor immune avoidance. Additionally, CD73 expression on myeloid cells results in T-cell inhibition and seems independent of PD-1 blockade [100]. Furthermore, M2 macrophages may also contribute to tumor immune escape [92]. Further investigation of the effects of modulating these signals is necessary to develop additional immune-modulating therapies [101,102,103].
8. Mechanisms Driving IRAEs
The understanding that tumor cells utilize co-inhibitory signals to evade immune clearance drove the effort to create anti-PD-1 and Anti-CTLA4 therapies. Data show that CTLA4 blockade results in both increased CD4 cell number in the TME and a shift towards Th1 active cell type rather than the suppressive Treg cell type [46,47,48,104]. Additionally, an increased CD8 clone diversity has been observed [1,105,106,107,108,109]. On the other hand, PD-1 blockade seems to increase CD4/CD8 activity in the TME [110], probably by inhibiting entry into the exhaustion of CD8 cells or reactivating already exhausted cells [111]. Treg cells play crucial roles in TME immune suppression and show high expression levels of CTLA-4/PD1 and other co-modulatory molecules. Evidence suggests that anti-CTLA4 immune activation is dependent on ADCC removal of Treg cells [47,112,113,114]. PD1 signaling was found to promote Treg inhibitory function [115], and that loss of PD1 signaling results in Treg cells shifting to a pro-inflammatory state [116,117]. As loss of Foxp3 Treg cells results in autoimmune phenotypes in animal models, and in humans [118,119], CTLA4/PD1 inhibition may drive IRAEs by systemic inhibition of Treg cells.
Collectively, CTLA4 and PD-1 blockade seems to exert major effects primarily on T-cell immunity. Indeed, studies have demonstrated that the amount of T lymphocytes in the TME is correlated with ICI treatment [120]. However, the wide range of cells expressing co-inhibitory molecules indicates that there are multiple mechanisms involved. Evidence suggests antibody diversity of both antitumor and autoantibodies are increased [121] following ICI blockade and that the antibody levels are correlated with the IRAE severity [122,123]. Furthermore, recent findings are supportive of specific antibodies implicated in ICI hypophysitis, pneumonitis, diabetes mellitus, and hypothyroidism [124,125,126,127]. The association between ICI myocarditis and myositis and myasthenia gravis [5,6] further supports the role of antibodies in IRAE, including in the heart. Additional cell types, such as neutrophils, have been shown to be activated during CTAL4 blockade [128,129]. Following ICI treatment monocytes were found to be recruited to the pancreas and liver by increased INFγ, resulting in islet cell damage [130,131], and M2 macrophages were found in autopsies of IRAE patients [132]. Elevated cytokine production identified in concordance with an activated immune response may drive further damage, similar to that seen in autoinflammatory diseases. Indeed, IL-1 levels were found to be elevated in patients suffering from IRAEs [133,134]. Additionally, TNFα, IL-17, INFγ, and IL-6 are all involved in autoimmune reactions and were shown to be in correlation with IRAEs or overexpressed following PD-1 inhibition [105,135]. Finally, the expression of inhibitory or excitatory molecules on non-immune cell types may drive cell-type-specific IRAEs, as seen with CTLA4 expression on hypothalamic and pituitary cells [136].
Based on this understanding, immune checkpoint inhibition damage in non-tumor sites can result from several mechanisms (Figure 3):
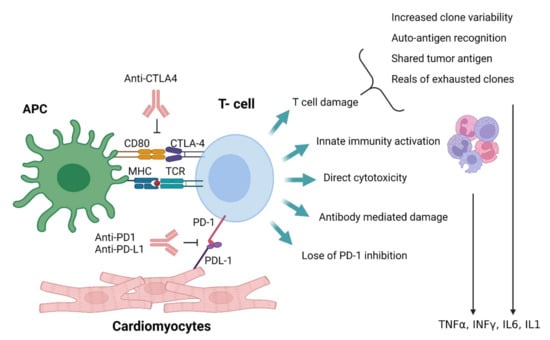
Figure 3.
Depiction of possible IRAE mechanisms: APC, antigen-presenting cell; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TCR, T-cell receptor; TNFα, tumor necrosis factor-alpha; INFγ, interferon-gamma; IL1, interleukin 1; IL6, interleukin 6.
- Increased T-cell clone expansion results in the appearance of new self-recognizing effector cells [4,137,138,139];
- The release from inhibition of preexisting self-recognizing effector T cells [140];
- Tumor damage exposure of autoantigens, resulting in expansion of auto-recognizing clones [4,141,142,143,144];
- Antibody-mediated autoimmunity [124,125];
- A shift toward a more active and less selective innate immune response;
- Bystander damage from increased levels of cytokines produced by the active antitumor immune response [133,134];
- Direct damage resulting from specific molecule expression on target cells.
Ironically, the appearance of IRAEs seems to corollate with better antitumor response [145,146,147,148,149,150,151,152], making ICIs a double-edged sword with a need for balancing the antitumor effect against the severity of IRAEs.
9. Cardiotoxicity of ICIs
The increased use of ICI therapy has revealed the extent and importance of ICI-induced cardiotoxicity (Figure 4). The severity of cardiac IRAEs is classified according to the American Society of Clinical Oncology [153] and ranges from benign ECG changes to life-threatening myocarditis. Recently, analysis of the Pharmacovigilance database and a meta-analysis of published papers and randomized controlled trials (RCTs) has identified cardiac events in 1.3–5.8% of patients receiving ICIs (single and dual therapy) [154]. Another large meta-analysis of RCTs showed increased odds ratios for developing myocarditis and pericarditis (4.4 and 2.2, respectively) in ICI-treated patients, with increased associated risks of heart failure and myocardial infarction (MI) [155]. Surprisingly, a meta-analysis of RCTs from phase II and III trials did not find any difference in CVAE between ICI-treated and control groups [156]. This may be explained by including highly selected patients in these trials (selection bias), reporting bias, and the type of analysis [155].
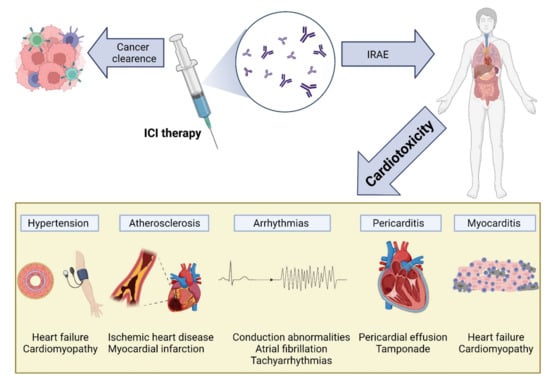
Figure 4.
Cardiovascular toxicity induced by immune checkpoint inhibitor (ICI) therapy. The reactivation of T cells in response to ICIs is mediated by inflammation and fibrosis, leading to various cardiac manifestations, including myocarditis, pericarditis and pericardial effusion, arrhythmias, atherosclerosis, and hypertension: IRAEs, immune-related adverse events; ICI, immune checkpoint inhibitor.
The immune landscape of the resting heart is mostly dominated by macrophages [157,158], with low numbers of monocytes, dendritic cells, and mast cells present [159,160]. In addition, low numbers of T and B cells can be found [161,162]. Myocarditis, inflammation of the heart muscle, can be triggered by multiple etiologies and is characterized by lymphocyte-predominant infiltration (such as in viral myocarditis), eosinophilic infiltration (such as in hypereosinophilic syndrome), or the presence of giant cells (giant cell myocarditis). Like other IRAEs, ICI cardiotoxicity is also driven by lymphocyte, predominant immunity with CTLA4 and PD-L1 expression in the myocardium playing an important role in T-cell activity regulation [163,164,165,166].
10. Myocarditis
The most serious cardiac manifestation of ICI cardiotoxicity is the development of myocarditis. Although it may be silent with merely cardiac biomarker elevation, it may present as a fulminant disease, leading to cardiogenic shock and death [11]. A meta-analysis has reported that myocarditis may represent up to 50% of cardiac IRAEs (34 of 61 cardiac IRAEs among 4751 patients) [154], with mortality rates ranging from 20% to 50% [5,6,10,11,14]. Cardiac damage seems to start early after ICI treatment, with myocarditis reported to occur approximately 17 days (range: 13–64 days) following treatment initiation [4], but delayed appearance as far as 32 weeks after treatment initiation was reported [167]. PD-1 pathway inhibition seems to elicit more cardiotoxicity than CTLA4, and combinational therapy harbors the highest risk of myocarditis [4,10,168]. Interestingly, female patients are more likely to develop IRAEs and myocarditis [169,170]. This may be explained, at least partially, by PD-1 and PD-1L upregulation by estrogen [171,172], and is supported by an observation from CTLA4(+/−)/PD-1(−/−) deficient mice showing that female mice died at significantly higher rates than their male, age-matched counterparts [173].
Blockade of PD-1 and CTLA4 was shown to result in increased lymphocyte presence in the heart and other tissues [174]. This is supported by histological data from ICI-induced myocarditis patients showing increased CD4 and CD8 lymphocytes and, to a lesser degree, macrophages [4,175,176]. Interestingly, these T cells seem to have lower stimulatory thresholds for self-antigens [139]. Mouse models have shown that PD-1L is upregulated in endothelial and myocardial cells in response to inflammation or ischemia, presumably reducing the inflammatory response and tissue damage [4,177,178,179]. Several approaches have been perused to elucidate the mechanisms driving myocardial damage during ICI treatment. Initial mouse models showed varied effects, depending on the genetic background. PD-1 knockout in C57BL/6 mice displayed a normal phenotype [180], whereas deleting PD-1 in BALB/c background resulted in increased anti-troponin-I antibodies and dilated cardiomyopathy rather than myocarditis [143,163,180,181]. However, the transfer of CD8+ PD-1-deficient cell population to mice already experiencing myocarditis resulted in enhanced inflammation [182]. In MRL-lpr−/− mice (lacking FAS and predisposed to the development of systemic lupus erythematosus (SLE)-like phenotype), deletion of PD-1/PD-1L or the administration of neutralizing antibodies resulted in autoimmune myocarditis and CD4/8 infiltration; however, unlike in human ICI myocarditis, substantial levels of autoantibody formation against myosin were detected [173,183]. Interestingly, blocking PD-1 or CTLA4 in MRL mice results in increased lymphocyte infiltration without the development of overt myocarditis [173]. These differences between early mouse models and human ICI myocarditis may be explained by the fact that in mice most T cells are naïve, as opposed to most human T cells [173]. This notion is supported by studies examining myocarditis developing in melanoma mouse models treated with PD-1 blockade [166]. In contrast to solitary PD-1 deficiency, mouse models of CTLA-4 deficiency led to systemic inflammation involving T lymphocytes and resulted in early multiorgan failure and death [164,165]. This included myocarditis with CD8 cells infiltration [164,184]; however, the global activation of T lymphocytes also differs from ICI myocarditis. In light of these shortcomings of early mouse models, a primate model was developed showing CD4/CD8 T cell and low numbers of macrophage infiltration into cardiac tissues, with increased troponin-I and NT-proBNP levels [175]. Another approach to recapitulate ICI myocarditis in mice employed removing multiple participating genes. Deletion of both PD-1 and LAG-3 in BALB/c mice resulted in myocarditis with CD8 and CD4 cell infiltration, in addition to increased TNFα secretion [185,186]. More recently, a mouse model harboring a compound loss of CTLA4 and PD-1 was created. Ctla4+/− Pdcd1−/− mice showed increased troponin levels with cardiac infiltration of CD3, CD8, and CD4 cells, and reduced numbers of Treg cells, similar to that seen in human ICI myocarditis [173]. A more “physiologic” approach was employed by Lars et al., who injected immune-competent mice with melanoma cells, followed by anti-PD-1 antibodies treatment, resulting in functionally noticeable myocarditis with increased CD4 and CD8 cell infiltration [166]. Both Ctla4+/− Pdcd1−/− and melanoma models were used to assess treatment options (CTLA4-IgG1 and anti-TNFα, respectively) paving the way for a more comprehensive search for novel and more specific therapeutic options.
One reason for T-cell infiltration within the myocardium was elucidated by the recognition of similar clonal populations of T cells recognizing antigens expressed in both the TME and cardiac tissues [4]. Shared antigens between myeloma cells and cardiomyocytes have been later discovered [144]. This mechanism is further supported by the finding of similar TME and skin T cells in NSCLC presenting with skin IRAEs [142], suggesting a common underlying IRAE mechanism. In addition to antigen recognition, ICI treatment results in upregulation of CXCR3–CXCL9/CXCL10 and CCR5/CCL5 chemokines necessary for T-cell activation [175,187]. Following activation, T cells overexpress TNFα, INFγ, and granzyme B, thus promoting local cell damage and death [188,189,190]. This is also supported by in vitro co-culture experiments of cardiomyocytes exposed to lymphocytes and anti-PD-1/CTLA4 antibodies, demonstrating increased levels of leukotriene B4 [188]. The surprisingly high incidence of myasthenia gravis with ICI myocarditis suggests that, in addition to T cells, antibody-mediated damage might also contribute to ICI autoimmunity [191,192,193,194,195]. Antibodies against AChR and MuSK are commonly present (~66% (30 of 45 patients) and 5% (1 of 19 patients), respectively) in ICI-induced myasthenia gravis cases [196]. However, as mentioned, increased anti-troponin-I and anti-myosin antibodies seen in PD-1 mouse models result in dilated cardiomyopathy or myocarditis that differs from ICI myocarditis. Further investigation is needed to elucidate this aspect.
Taken together, myocarditis seems to develop by multiple steps including enhanced recruitment of T lymphocytes, reduced PD-1/PD-1L protection, reduced Treg activity, and T-cell recognition of shared epitopes. The development of novel mouse models will potentially allow better elucidation of other mechanisms driving ICI myocarditis and drive better-targeted treatment options (Figure 3).
11. Pericarditis
Pericardial effusion was reported following ICI treatment, with some developing overt pericarditis [197,198]. The incidence of pericardial effusion ranges from 0.3% (95/31321 cardiovascular IRAEs) but may be as high as 7% in NSCLC (4/60 patients receiving ICIs), and this has been associated with worse outcomes [6,199]. As pericardial effusion is present in multiple tumor types, it is important to differentiate the etiology driving pericardial effusion accumulation; however, the association of pericardial effusion with higher mortality in ICI-treated patients suggests a possible cardiotoxic effect. Indeed, pericardial samples from ICI-treated patients suffering from pericarditis showed significant lymphocytic infiltrates [200].
12. Arrhythmias
Atrial fibrillation (AF), conduction delays, and ventricular arrhythmias have all been observed following ICI treatment [4,11,139,201], and cardiac arrhythmias may represent a common cardiac IRAE [6,11,202,203] that may occur in up to 10% of patients receiving ICIs, according to one study [202]. This may represent isolated conduction toxicity or be part of a widespread manifestation of myocarditis [11]. Interestingly, CD4 PD-1 expression and PD-1L myeloid dendritic cells were found to be reduced in patients with AF [204]. Additionality, mouse models of myocarditis showed an increased incidence of arrhythmias [173]. The significantly worse outcome of patients presenting with conduction abnormalities (80% vs. 16% mortality) [11] following ICI treatment emphasizes its significance and the importance of ECG surveillance.
13. Atherosclerosis
Atherosclerosis has emerged as a new target of ICI-induced toxicity [205]. Indeed, acute coronary syndrome (ACS) incidence among ICI patients was reported to be around 0.5% (2/402 of ICI-treated patients) in a large metanalysis [206], but other reports of incidences as high as 2.4% (80/3326) to 3.6% (102/2842) of patients receiving ICI have emerged [14,207,208]. Although macrophages play major roles in the development of the atherosclerotic lesion, T lymphocytes were shown to occupy the atherosclerotic plaque [209] ], and their activation and secretion of INFγ lead to macrophage activation [210]. Interestingly, patients with coronary artery disease have reduced expression of PD-1 and PD-L1 on peripheral mononuclear cells [211]. The role of PD-1 in atherosclerosis is further supported by observations of larger atherosclerotic lesions in PD-1 knockout mice [212] with a more prominent T-cell response and larger necrotic cores [213]. As for CTLA-4, studies in mice have shown increased plaque size, following anti-CTLA-4 antibody treatment. Concurrently, treatment with abatacept (a CLTA-4 analog) or overexpression of CTLA-4 reduced plaque size [214,215,216]. The proatherosclerotic effect of PD-1 and CTLA4 inhibition seems to be, at least in part, mediated by the increase in INFγ and TNFα secretion, which are known mediators promoting coronary thrombosis [217]. As new immunomodulatory targets are discovered, it is interesting to see whether it can be possible to activate the immune response without increasing atherosclerotic risk [205].
Events of coronary spasm following ICI administration have been reported [218,219], but the full extent of this phenomenon warrants further investigation.
14. Hypertension
In addition to direct cardiotoxicity, ICI-related hypertension may add to cardiac morbidity and mortality. Hypertension seems to be a rear IRAE and was not reported in phase III trials [220,221,222]. However, more recent data show that hypertension may develop in as high as 18% (13/70) of treated patients and is reported to account for 0.63% of ICI-related IRAEs [6,223].
15. Thromboembolism
Venous thromboembolism (VTE), although indirect, has a profound effect on the cardiovascular system. As cancer and certain chemotherapies are known to induce a pro-thrombotic state, and as ICIs are commonly used as second-line therapies, it is difficult to assess the isolated contribution of ICI treatment to thromboembolism formation. Initial data, including RCT and metanalysis data, showed that approximately 2.7% (449/20273 ICI-treated patients) developed VTE during ICI therapy [224,225,226]. However, this analysis included studies in which VTE was not the primary outcome, nor was standardly reported, resulting in possible underrepresentation. New emerging data show an increased incidence of pulmonary embolism (PE) and deep vein thrombosis (DVT) [227], suggesting the prothrombotic effects of ICIs. Indeed, multiple retrospective studies have shown a much higher VTE incidence of 6–24% in ICI-treated patients, with risk increasing after prolonged treatment [228,229,230,231,232]. Furthermore, these studies have found that the main risk factors for VTE in ICI-treated patients include the presence of metastatic disease and previous VTE and that these risk factors are also associated with a worse prognosis. Interestingly, no correlation was found in these studies between ICI type or cancer type and VTE risk. Despite a potentially increased risk of VTE, it is worth noting that these studies were all retrospective and did not include non-ICI-treated control groups. Furthermore, several studies have shown that VTE risk was similar between patients treated with ICIs and those treated with chemotherapy [225,233].
Mechanistically, ICIs may induce VTE by causing vasculitis and endothelial damage secondary to local and systemic activation of immune cells [228]. A more specific role of PD1/PD-L1 signaling has been suggested in one study showing that T-cell activation in ICI-treated patients induces production of tissue factor in peripheral monocytes highly expressing PD-L1, which can initiate the thrombotic cascade [234]. Taken together, it seems VTE is emerging as an important IRAE. However, a better understanding of the underlying mechanisms, as well as controlled clinical trials, is necessary to explore the extent and risk factors of VTE associated with ICI use.
16. Emerging Treatment of Cardiac IRAEs
To date, the hallmark of IRAE management is based on steroid therapy and escalation to other immunosuppressive regiments in non-responders [7,153]. Immune suppression using the inosine-5′-monophosphate dehydrogenase (IMPDH) inhibitor, mycophenolate mofetil (MMF), results primarily in B- and T-cell inhibition [235,236]. Calcineurin inhibitors, such as tacrolimus, exert immunosuppressive properties through inhibiting T-cell activity [167,237,238,239]. Lymphopenia may be induced by antithymocyte globulin (ATG) [167,237,238], resulting in the cessation of autoimmunity. Other strategies include the use of intravenous immunoglobulin (IVIG) [191,192,193,240,241,242,243], and plasmapheresis [194,195,240,244,245,246] to incapacitate or remove ICIs (which have a long half-life of up to 27 days in some cases [201] can be employed.
Recent advances in the understanding of IRAE mechanisms have allowed the employment of targeted therapies. These include CTLA4-Ig (abatacept) to reimpose inhibition of activated T cells in myocarditis, as shown in mouse models [173] and case reports in humans [173,247]. Anti-CD52 (alemtuzumab), commonly used in CLL and multiple myeloma, has shown T-cell depletion capabilities and has been used in a case report with encouraging results [248]. Anti-TNFα (infliximab) has also shown promising results [153,166,240,249], although its utility in heart failure patients may be limited. It is worth noting that any treatment resulting in global quenching of the immune response also has the potential to allow tumor escape from immunosurveillance and disease progression. A better understanding of both the stimulatory and inhibitory molecules implicated in intracellular signaling pathways may allow better control of adverse events while allowing the tenacious antitumoral activity.
17. Conclusions
Harnessing the immune response to facilitate tumor clearance has opened up a new avenue in cancer treatment. The achievement of successful treatment results has made PD-1/CTLA4 inhibition available to an increasing number of cancer patients. With this increase came the realization of various adverse events accompanying this treatment type. Cardiac toxicity is emerging to be far more common, with grave consequences even in more benign manifestations. Recent advances in understanding the mechanisms behind ICI treatment have allowed new treatment options such as abatacept and anti-TNFα agents that can reduce drug toxicity and allow continuation of treatment to achieve tumor clearance. Furthermore, the discovery of new stimulatory and inhibitory molecules may allow the development of ICIs with safer adverse event profiles.
Author Contributions
D.R., A.B. and R.A. conceptualized the review; D.R., A.B. and R.A. wrote the first draft; M.L., S.A., M.Y., O.A. and R.A. provided critical editing and review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Salem, J.-E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef] [Green Version]
- Salem, J.-E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.-P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef]
- Hauck, A.J.; Kearney, D.L.; Edwards, W.D. Evaluation of postmortem endomyocardial biopsy specimens from 38 patients with lymphocytic myocarditis: Implications for role of sampling error. Mayo Clin. Proc. 1989, 64, 1235–1245. [Google Scholar] [CrossRef]
- Agrawal, N.; Khunger, A.; Vachhani, P.; Colvin, T.A.; Hattoum, A.; Spangenthal, E.; Curtis, A.B.; Dy, G.K.; Ernstoff, M.S.; Puzanov, I. Cardiac Toxicity Associated with Immune Checkpoint Inhibitors: Case Series and Review of the Literature. Case Rep. Oncol. 2019, 12, 260–276. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated with Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef]
- Escudier, M.; Cautela, J.; Malissen, N.; Ancedy, Y.; Orabona, M.; Pinto, J.; Monestier, S.; Grob, J.-J.; Scemama, U.; Jacquier, A.; et al. Clinical Features, Management, and Outcomes of Immune Checkpoint Inhibitor-Related Cardiotoxicity. Circulation 2017, 136, 2085–2087. [Google Scholar] [CrossRef] [PubMed]
- Drobni, Z.D.; Zafar, A.; Zubiri, L.; Zlotoff, D.A.; Alvi, R.M.; Lee, C.; Hartmann, S.; Gilman, H.K.; Villani, A.; Nohria, A.; et al. Decreased Absolute Lymphocyte Count and Increased Neutrophil/Lymphocyte Ratio with Immune Checkpoint Inhibitor-Associated Myocarditis. J. Am. Heart Assoc. 2020, 9, e018306. [Google Scholar] [CrossRef] [PubMed]
- Guha, A.; Al-Kindi, S.; Jain, P.; Tashtish, N.; ElAmm, C.; Oliveira, G.H. Association between myocarditis and other immune-related adverse events secondary to immune checkpoint inhibitor use. Int. J. Cancer 2020, 147, 1753–1754. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Yang, E.H.; Molina, J.R.; Bailey, K.R.; Blumenthal, R.S.; Kopecky, S.L. Cardiovascular Health and Outcomes in Cancer Patients Receiving Immune Checkpoint Inhibitors. Am. J. Cardiol. 2020, 125, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Kreileder, M.; Barrett, I.; Bendtsen, C.; Brennan, D.; Kolch, W. Signaling Dynamics Regulating Crosstalks between T-Cell Activation and Immune Checkpoints. Trends Cell Biol. 2021, 31, 224–235. [Google Scholar] [CrossRef]
- González-Navajas, J.M.; Fan, D.D.; Yang, S.; Yang, F.M.; Lozano-Ruiz, B.; Shen, L.; Lee, J. The Impact of Tregs on the Anticancer Immunity and the Efficacy of Immune Checkpoint Inhibitor Therapies. Front. Immunol. 2021, 12, 625783. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- de Simone, M.; Arrigoni, A.; Rossetti, G.; Gruarin, P.; Ranzani, V.; Politano, C.; Bonnal, R.J.P.; Provasi, E.; Sarnicola, M.L.; Panzeri, I.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016, 45, 1135–1147. [Google Scholar] [CrossRef] [Green Version]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y.; et al. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef]
- Jeong, S.; Park, S.-H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 2020, 20, e3. [Google Scholar] [CrossRef]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vence, L.; Bucktrout, S.L.; Fernandez Curbelo, I.; Blando, J.; Smith, B.M.; Mahne, A.E.; Lin, J.C.; Park, T.; Pascua, E.; Sai, T.; et al. Characterization and Comparison of GITR Expression in Solid Tumors. Clin. Cancer Res. 2019, 25, 6501–6510. [Google Scholar] [CrossRef] [Green Version]
- Tran, B.; Carvajal, R.D.; Marabelle, A.; Patel, S.P.; LoRusso, P.M.; Rasmussen, E.; Juan, G.; Upreti, V.V.; Beers, C.; Ngarmchamnanrith, G.; et al. Dose escalation results from a first-in-human, phase 1 study of glucocorticoid-induced TNF receptor-related protein agonist AMG 228 in patients with advanced solid tumors. J. Immunother. Cancer 2018, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, D.A.; Budhu, S.; Liu, C.; Bryson, C.; Malandro, N.; Cohen, A.; Zhong, H.; Yang, X.; Houghton, A.N.; Merghoub, T.; et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol. Res. 2013, 1, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Polesso, F.; Sarker, M.; Weinberg, A.D.; Murray, S.E.; Moran, A.E. OX40 Agonist Tumor Immunotherapy Does Not Impact Regulatory T Cell Suppressive Function. J. Immunol. 2019, 203, 2011–2019. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Etxeberría, I.; Otano, I.; Melero, I. Twists and turns to translating 4-1BB cancer immunotherapy. Sci. Transl. Med. 2019, 11, eaax4738. [Google Scholar] [CrossRef]
- He, Y.; Vlaming, M.; van Meerten, T.; Bremer, E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers 2022, 14, 299. [Google Scholar] [CrossRef]
- Amatore, F.; Gorvel, L.; Olive, D. Role of Inducible Co-Stimulator (ICOS) in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 141–150. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Carlino, M.; Joerger, M.; di Nicola, M.; Meniawy, T.; Rottey, S.; Moreno, V.; Gazzah, A.; Delord, J.-P.; Paz-Ares, L.; et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination with Nivolumab for Patients with Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2020, 6, 100–107. [Google Scholar]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.E.W.; Pishvaian, M.J.; Shepard, D.R.; Wang, D.; Weiss, J.; Johnson, M.L.; Chung, C.H.; Chen, Y.; Huang, B.; Davis, C.B.; et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J. Immunother. Cancer 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Gu-Trantien, C.; Willard-Gallo, K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open 2020, 5, e000544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraehenbuehl, L.; Weng, C.-H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef]
- Speiser, D.E.; Utzschneider, D.T.; Oberle, S.G.; Münz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Gallimore, A.; Hengartner, H.; Zinkernagel, R. Hierarchies of antigen-specific cytotoxic T-cell responses. Immunol. Rev. 1998, 164, 29–36. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining “T cell exhaustion”. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Marengère, L.E.; Waterhouse, P.; Duncan, G.S.; Mittrücker, H.W.; Feng, G.S.; Mak, T.W. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar] [CrossRef]
- Chuang, E.; Fisher, T.S.; Morgan, R.W.; Robbins, M.D.; Duerr, J.M.; vander Heiden, M.G.; Gardner, J.P.; Hambor, J.E.; Neveu, M.J.; Thompson, C.B. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 2000, 13, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Lingel, H.; Wissing, J.; Arra, A.; Schanze, D.; Lienenklaus, S.; Klawonn, F.; Pierau, M.; Zenker, M.; Jänsch, L.; Brunner-Weinzierl, M.C. CTLA-4-mediated posttranslational modifications direct cytotoxic T-lymphocyte differentiation. Cell Death Differ. 2017, 24, 1739–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, D.; Betge, S.; Windisch, A.; Pistulli, R.; Rohm, I.; Fritzenwanger, M.; Jung, C.; Schubert, K.; Theis, B.; Petersen, I.; et al. Recruitment of circulating dendritic cell precursors into the infarcted myocardium and pro-inflammatory response in acute myocardial infarction. Clin. Sci. 2012, 123, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Chen, H.; Liakou, C.I.; Kamat, A.; Pettaway, C.; Ward, J.F.; Tang, D.N.; Sun, J.; Jungbluth, A.A.; Troncoso, P.; Logothetis, C.; et al. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [Green Version]
- Carthon, B.C.; Wolchok, J.D.; Yuan, J.; Kamat, A.; Ng Tang, D.S.; Sun, J.; Ku, G.; Troncoso, P.; Logothetis, C.J.; Allison, J.; et al. Preoperative CTLA-4 blockade: Tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin. Cancer Res. 2010, 16, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef]
- Jost, S.; Altfeld, M. Control of human viral infections by natural killer cells. Annu. Rev. Immunol. 2013, 31, 163–194. [Google Scholar] [CrossRef]
- Judge, S.J.; Dunai, C.; Aguilar, E.G.; Vick, S.C.; Sturgill, I.R.; Khuat, L.T.; Stoffel, K.M.; Van Dyke, J.; Longo, D.L.; Darrow, M.A.; et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig. 2020, 130, 3051–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, P.; Guethlein, L.A. Genetics of Natural Killer Cells in Human Health, Disease, and Survival. Annu. Rev. Immunol. 2018, 36, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [Green Version]
- Youngnak, P.; Kozono, Y.; Kozono, H.; Iwai, H.; Otsuki, N.; Jin, H.; Omura, K.; Yagita, H.; Pardoll, D.M.; Chen, L.; et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem. Biophys. Res. Commun. 2003, 307, 672–677. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry R v Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Molawi , K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Xu, W.; Guo, Q.; Jiang, Z.; Wang, P.; Yue, Y.; Xiong, S. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 2009, 105, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Thibult, M.-L.; Mamessier, E.; Gertner-Dardenne, J.; Pastor, S.; Just-Landi, S.; Xerri, L.; Chetaille, B.; Olive, D. PD-1 is a novel regulator of human B-cell activation. Int. Immunol. 2013, 25, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-M.; Li, C.-W.; Lai, Y.-J.; Hung, M.-C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-C.; Chen, W.-D.; Alvarez, J.B.; Jia, K.; Shi, L.; Wang, Q.; Zou, N.; He, K.; Zhu, H. Cancer immune checkpoint blockade therapy and its associated autoimmune cardiotoxicity. Acta Pharmacol. Sin. 2018, 39, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Scala, E.; Carbonari, M.; del Porto, P.; Cibati, M.; Tedesco, T.; Mazzone, A.M.; Paganelli, R.; Fiorilli, M. Lymphocyte activation gene-3 (LAG-3) expression and IFN-gamma production are variably coregulated in different human T lymphocyte subpopulations. J. Immunol. 1998, 161, 489–493. [Google Scholar]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Takamura, S.; Tsuji-Kawahara, S.; Yagita, H.; Akiba, H.; Sakamoto, M.; Chikaishi, T.; Kato, M.; Miyazawa, M. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J. Immunol. 2010, 184, 4696–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. Available online: https://pubmed.ncbi.nlm.nih.gov/25363763/ (accessed on 1 January 2022). [CrossRef] [PubMed] [Green Version]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef]
- Sabatos, C.A.; Chakravarti, S.; Cha, E.; Schubart, A.; Sánchez-Fueyo, A.; Zheng, X.X.; Coyle, A.J.; Strom, T.B.; Freeman, G.J.; Kuchroo, V.K. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003, 4, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-T.; Chen, C.-H.; Ku, K.-L.; Hsiao, M.; Chiang, C.-P.; Hsu, T.-L.; Chen, M.H.; Wong, C.H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Tekle, C.; Nygren, M.K.; Chen, Y.-W.; Dybsjord, I.; Nesland, J.M.; Maelandsmo, G.M.; Fodstad, O. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int. J. Cancer 2012, 130, 2282–2290. [Google Scholar] [CrossRef]
- Chen, C.; Shen, Y.; Qu, Q.-X.; Chen, X.-Q.; Zhang, X.-G.; Huang, J.-A. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp. Cell Res. 2013, 319, 96–102. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Toor, S.M.; Sasidharan Nair, V.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2020, 65, 1–12. [Google Scholar] [CrossRef]
- Gavrieli, M.; Watanabe, N.; Loftin, S.K.; Murphy, T.L.; Murphy, K.M. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem. Biophys. Res. Commun. 2003, 312, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.W.; Gill, D.M.; Pal, S.K.; Agarwal, N. The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy 2017, 9, 681–692. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. Available online: https://pubmed.ncbi.nlm.nih.gov/31873309/ (accessed on 1 January 2022). [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Chin, R.; Chen, C.-H.; Budhani, P.; Ai, M.; Reilley, M.J.; Sebastian, M.M.; Hong, D.S.; et al. Activation of 4-1BB on Liver Myeloid Cells Triggers Hepatitis via an Interleukin-27-Dependent Pathway. Clin. Cancer Res. 2018, 24, 1138–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfahani, K.; Elkrief, A.; Calabrese, C.; Lapointe, R.; Hudson, M.; Routy, B.; Miller, W.H., Jr.; Calabrese, L. Moving towards personalized treatments of immune-related adverse events. Nat. Rev. Clin. Oncol. 2020, 17, 504–515. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 437, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, X.J.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity 2019, 51, 1043–1058.e4. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855.e5. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl. Acad. Sci. USA 2016, 113, 8490–8495. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Granito, A.; Muratori, L.; Lalanne, C.; Quarneti, C.; Ferri, S.; Guidi, M.; Lenzi, M.; Muratori, P. Hepatocellular carcinoma in viral and autoimmune liver diseases: Role of CD4+ CD25+ Foxp3+ regulatory T cells in the immune microenvironment. World J. Gastroenterol. 2021, 27, 2994–3009. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- da Gama Duarte, J.; Parakh, S.; Andrews, M.C.; Woods, K.; Pasam, A.; Tutuka, C.; Ostrouska, S.; Blackburn, J.M.; Behren, A.; Cebon, J. Autoantibodies May Predict Immune-Related Toxicity: Results from a Phase I Study of Intralesional Bacillus Calmette-Guérin followed by Ipilimumab in Patients with Advanced Metastatic Melanoma. Front. Immunol. 2018, 9, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Bar, N.; Ferreira, M.; Newman, A.M.; Zhang, L.; Bailur, J.K.; Bacchiocchi, A.; Kluger, H.; Wei, W.; Halaban, R.; et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J. Clin. Investig. 2018, 128, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Gowen, M.F.; Giles, K.M.; Simpson, D.; Tchack, J.; Zhou, H.; Moran, U.; Dawood, Z.; Pavlick, A.C.; Hu, S.; Wilson, M.A.; et al. Baseline antibody profiles predict toxicity in melanoma patients treated with immune checkpoint inhibitors. J. Transl. Med. 2018, 16, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, S.A.; Gao, J.; Miura, Y.; Blando, J.; Tidwell, R.S.S.; Zhao, H.; Subudhi, S.K.; Tawbi, H.; Keung, E.; Wargo, J.; et al. Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc. Natl. Acad. Sci. USA 2019, 116, 22246–22251. [Google Scholar] [CrossRef]
- Iwama, S.; de Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci. Transl. Med. 2014, 6, 230ra45. [Google Scholar] [CrossRef]
- Esfahani, K.; Meti, N.; Miller, W.H.; Hudson, M. Adverse events associated with immune checkpoint inhibitor treatment for cancer. CMAJ Can. Med. Assoc. J. 2019, 191, E40–E46. [Google Scholar] [CrossRef] [Green Version]
- Kurimoto, C.; Inaba, H.; Ariyasu, H.; Iwakura, H.; Ueda, Y.; Uraki, S.; Takeshima, K.; Furukawa, Y.; Morita, S.; Yamamoto, Y.; et al. Predictive and sensitive biomarkers for thyroid dysfunctions during treatment with immune-checkpoint inhibitors. Cancer Sci. 2020, 111, 1468–1477. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, P.; Wood, K.; Wassmann, K.; Christenfeld, A.M.; Bhardwaj, N.; Oh, W.K. A whole-blood RNA transcript-based gene signature is associated with the development of CTLA-4 blockade-related diarrhea in patients with advanced melanoma treated with the checkpoint inhibitor tremelimumab. J. Immunother. Cancer 2018, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Hu, H.; Zakharov, P.N.; Peterson, O.J.; Unanue, E.R. Cytocidal macrophages in symbiosis with CD4 and CD8 T cells cause acute diabetes following checkpoint blockade of PD-1 in NOD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 31319–31330. [Google Scholar] [CrossRef]
- Gudd, C.L.C.; Au, L.; Triantafyllou, E.; Shum, B.; Liu, T.; Nathwani, R.; Kumar, N.; Mukherjee, S.; Dhar, A.; Woollard, K.J.; et al. Activation and transcriptional profile of monocytes and CD8+ T cells are altered in checkpoint inhibitor-related hepatitis. J. Hepatol. 2021, 75, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Mihic-Probst, D.; Reinehr, M.; Dettwiler, S.; Kolm, I.; Britschgi, C.; Kudura, K.; Maggio, E.M.; Lenggenhager, D.; Rushing, E.J. The role of macrophages type 2 and T-regs in immune checkpoint inhibitor related adverse events. Immunobiology 2020, 225, 152009. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Naidoo, J.; Zhong, Q.; Xiong, Y.; Mammen, J.; de Flores, M.V.; Cappelli, L.; Balaji, A.; Palmer, A.; Forde, P.M.; et al. The alveolar immune cell landscape is dysregulated in checkpoint inhibitor pneumonitis. J. Clin. Investig. 2019, 129, 4305–4315. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; H Yang, J.Y.; Ghazanfar, S.; Kefford, R.F.; et al. Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1-Based Immunotherapy. Clin. Cancer Res. 2019, 25, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Gerber, D.E. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin. Cancer Biol. 2020, 64, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Albarel, F.; Castinetti, F.; Brue, T. Management of endocrine disease: Immune check point inhibitors-induced hypophysitis. Eur. J. Endocrinol. 2019, 181, R107–R118. [Google Scholar] [CrossRef] [Green Version]
- Nishino, M.; Sholl, L.M.; Hodi, F.S.; Hatabu, H.; Ramaiya, N.H. Anti-PD-1-Related Pneumonitis during Cancer Immunotherapy. N. Engl. J. Med. 2015, 373, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Rapisuwon, S.; Izar, B.; Batenchuk, C.; Avila, A.; Mei, S.; Sorger, P.; Parks, J.M.; Cooper, S.J.; Wagner, D.; Zeck, J.C.; et al. Exceptional response and multisystem autoimmune-like toxicities associated with the same T cell clone in a patient with uveal melanoma treated with immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Berner, F.; Bomze, D.; Diem, S.; Ali, O.H.; Fässler, M.; Ring, S.; Niederer, R.; Ackermann, C.J.; Baumgaertner, P.; Pikor, N.; et al. Association of Checkpoint Inhibitor-Induced Toxic Effects With Shared Cancer and Tissue Antigens in Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, A.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Calle, N.; Rodriguez-Otero, P.; Villar, S.; Mejías, L.; Melero, I.; Prosper, F.; Marinello, P.; Paiva, B.; Idoate, M.; San-Miguel, J. Anti-PD1 associated fulminant myocarditis after a single pembrolizumab dose: The role of occult pre-existing autoimmunity. Haematologica 2018, 103, e318–e321. [Google Scholar] [CrossRef]
- Maher, V.E.; Fernandes, L.L.; Weinstock, C.; Tang, S.; Agarwal, S.; Brave, M.; Ning, Y.M.; Singh, H.; Suzman, D.; Xu, J.; et al. Analysis of the Association Between Adverse Events and Outcome in Patients Receiving a Programmed Death Protein 1 or Programmed Death Ligand 1 Antibody. J. Clin. Oncol. 2019, 37, 2730–2737. [Google Scholar] [CrossRef]
- Indini, A.; di Guardo, L.; Cimminiello, C.; Prisciandaro, M.; Randon, G.; de Braud, F.; del Vecchio, M. Immune-related adverse events correlate with improved survival in patients undergoing anti-PD1 immunotherapy for metastatic melanoma. J. Cancer Res. Clin. Oncol. 2019, 145, 511–521. [Google Scholar] [CrossRef]
- Sato, K.; Akamatsu, H.; Murakami, E.; Sasaki, S.; Kanai, K.; Hayata, A.; Tokudome, N.; Akamatsu, K.; Koh, Y.; Ueda, H.; et al. Correlation between immune-related adverse events and efficacy in non-small cell lung cancer treated with nivolumab. Lung Cancer 2018, 115, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Quandt, Z.; Bluestone, J.A. The Balancing Act between Cancer Immunity and Autoimmunity in Response to Immunotherapy. Cancer Immunol. Res. 2018, 6, 1445–1452. [Google Scholar] [CrossRef] [Green Version]
- Xing, P.; Zhang, F.; Wang, G.; Xu, Y.; Li, C.; Wang, S.; Guo, Y.; Cai, S.; Wang, Y.; Li, J. Incidence rates of immune-related adverse events and their correlation with response in advanced solid tumours treated with NIVO or NIVO+IPI: A systematic review and meta-analysis. J. Immunother. Cancer 2019, 7, 341. [Google Scholar] [CrossRef]
- Ricciuti, B.; Genova, C.; de Giglio, A.; Bassanelli, M.; Dal Bello, M.G.; Metro, G.; Brambilla, M.; Baglivo, S.; Grossi, F.; Chiari, R. Impact of immune-related adverse events on survival in patients with advanced non-small cell lung cancer treated with nivolumab: Long-term outcomes from a multi-institutional analysis. J. Cancer Res. Clin. Oncol. 2019, 145, 479–485. [Google Scholar] [CrossRef]
- Horvat, T.Z.; Adel, N.G.; Dang, T.-O.; Momtaz, P.; Postow, M.A.; Callahan, M.K.; Carvajal, R.D.; Dickson, M.A.; D’Angelo, S.P.; Woo, K.M.; et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J. Clin. Oncol. 2015, 33, 3193–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.H.; Wei, L.; Bertino, E.M.; Edd, T.; Villalona-Calero, M.A.; He, K.; Shields, P.G.; Carbone, D.P.; Otterson, G.A. Incidence, Risk Factors, and Effect on Survival of Immune-related Adverse Events in Patients with Non-Small-cell Lung Cancer. Clin. Lung Cancer 2018, 19, e893–e900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Infante, N.; Ramírez-Flores, Y.A.; Castillo, E.C.; Lozano, O.; García-Rivas, G.; Torre-Amione, G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: A meta-analysis. Eur. J. Heart Fail. 2021, 23, 1739–1747. [Google Scholar] [CrossRef]
- Dolladille, C.; Akroun, J.; Morice, P.-M.; Dompmartin, A.; Ezine, E.; Sassier, M.; Da-Silva, A.; Plane, A.-F.; Legallois, D.; L’Orphelin, J.-M.; et al. Cardiovascular immunotoxicities associated with immune checkpoint inhibitors: A safety meta-analysis. Eur. Heart J. 2021, 42, 4964–4977. [Google Scholar] [CrossRef] [PubMed]
- Agostinetto, E.; Eiger, D.; Lambertini, M.; Ceppi, M.; Bruzzone, M.; Pondé, N.; Plummer, C.; Awada, A.H.; Santoro, A.; Piccart-Gebhart, M.; et al. Cardiotoxicity of immune checkpoint inhibitors: A systematic review and meta-analysis of randomised clinical trials. Eur. J. Cancer 2021, 148, 76–91. [Google Scholar] [CrossRef]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 2012, 7, e36814. [Google Scholar]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.-S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 2009, 206, 497–505. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [PubMed]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1233-42. [Google Scholar] [PubMed]
- Tarrio, M.L.; Grabie, N.; Bu, D.; Sharpe, A.H.; Lichtman, A.H. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J. Immunol. 2012, 188, 4876–4884. [Google Scholar] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar]
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K.; et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Heart J. 2021, 14, 316–329. [Google Scholar]
- Jain, V.; Bahia, J.; Mohebtash, M.; Barac, A. Cardiovascular Complications Associated with Novel Cancer Immunotherapies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 36. [Google Scholar]
- Zimmer, L.; Goldinger, S.M.; Hofmann, L.; Loquai, C.; Ugurel, S.; Thomas, I.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.S.; Göppner, D.; et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur. J. Cancer 2016, 60, 210–225. [Google Scholar]
- Zamami, Y.; Niimura, T.; Okada, N.; Koyama, T.; Fukushima, K.; Izawa-Ishizawa, Y.; Ishizawa, K. Factors Associated With Immune Checkpoint Inhibitor-Related Myocarditis. JAMA Oncol. 2019, 5, 1635–1637. [Google Scholar]
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J. Transl. Med. 2018, 16, 94. [Google Scholar]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378. [Google Scholar] [PubMed]
- Wang, C.; Dehghani, B.; Li, Y.; Kaler, L.J.; Vandenbark, A.A.; Offner, H. Oestrogen modulates experimental autoimmune encephalomyelitis and interleukin-17 production via programmed death 1. Immunology 2009, 126, 329–335. [Google Scholar] [PubMed]
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.-A.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625. [Google Scholar] [PubMed]
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 2019, 58 (Suppl. S7), vii59–vii67. [Google Scholar] [PubMed] [Green Version]
- Ji, C.; Roy, M.D.; Golas, J.; Vitsky, A.; Ram, S.; Kumpf, S.W.; Martin, M.; Barletta, F.; Meier, W.A.; Hooper, A.T.; et al. Myocarditis in Cynomolgus Monkeys Following Treatment with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2019, 25, 4735–4748. [Google Scholar]
- Ganatra, S.; Neilan, T.G. Immune Checkpoint Inhibitor-Associated Myocarditis. Oncologist 2018, 23, 879–886. [Google Scholar]
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Martin, M.; Barletta, F.; Meier, W.A.; Hooper, A.T.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar]
- Keir, M.E.; Freeman, G.J.; Sharpe, A.H. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J. Immunol. 2007, 179, 5064–5070. [Google Scholar]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar]
- Parmacek, M.S.; Solaro, R.J. Biology of the troponin complex in cardiac myocytes. Prog. Cardiovasc. Dis. 2004, 47, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.A.; Menke, J.; Rabacal, W.A.; Schoen, F.J.; Sharpe, A.H.; Kelley, V.R. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J. Immunol. 2008, 181, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okazaki, I.-M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Love, V.A.; Grabie, N.; Duramad, P.; Stavrakis, G.; Sharpe, A.; Lichtman, A. CTLA-4 ablation and interleukin-12 driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ. Res. 2007, 101, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Okazaki, I.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; Baba, M.H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Quagliariello, V.; Passariello, M.; Rea, D.; Barbieri, A.; Iovine, M.; Bonelli, A.; Caronna, A.; Botti, G.; Lorenzo, C.D.; Maurea, N. Evidences of CTLA-4 and PD-1 Blocking Agents-Induced Cardiotoxicity in Cellular and Preclinical Models. J. Pers. Med. 2020, 10, 179. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Galdiero, M.R.; Varricchi, G. Cardiac Toxicity in Patients Treated with Immune Checkpoint Inhibitors: It Is Now Time for Cardio-Immuno-Oncology. J. Am. Coll. Cardiol. 2018, 71, 1765–1767. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, e000247. [Google Scholar] [CrossRef] [Green Version]
- Xing, Q.; Zhang, Z.-W.; Lin, Q.-H.; Shen, L.-H.; Wang, P.-M.; Zhang, S.; Fan, M.; Zhu, B. Myositis-myasthenia gravis overlap syndrome complicated with myasthenia crisis and myocarditis associated with anti-programmed cell death-1 (sintilimab) therapy for lung adenocarcinoma. Ann. Transl. Med. 2020, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Yanase, T.; Moritoki, Y.; Kondo, H.; Ueyama, D.; Akita, H.; Yasui, T. Myocarditis and myasthenia gravis by combined nivolumab and ipilimumab immunotherapy for renal cell carcinoma: A case report of successful management. Urol. Case Rep. 2021, 34, 101508. [Google Scholar] [CrossRef] [PubMed]
- Fazel, M.; Jedlowski, P.M. Severe Myositis, Myocarditis, and Myasthenia Gravis with Elevated Anti-Striated Muscle Antibody following Single Dose of Ipilimumab-Nivolumab Therapy in a Patient with Metastatic Melanoma. Case Rep. Immunol. 2019, 2019, 2539493. [Google Scholar]
- Kimura, T.; Fukushima, S.; Miyashita, A.; Aoi, J.; Jinnin, M.; Kosaka, T.; Ando, Y.; Matsukawa, M.; Inoue, H.; Kiyotani, K.; et al. Myasthenic crisis and polymyositis induced by one dose of nivolumab. Cancer Sci. 2016, 107, 1055–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, E.; Varese, P.; Agosti, S.; Celli, L.; Ghiglione, E.; Pappalardo, I.; Zaccone, G.; Paglia, A.; Morelli, N. Concomitant myasthenia gravis, myositis, myocarditis and polyneuropathy, induced by immune-checkpoint inhibitors: A life-threatening continuum of neuromuscular and cardiac toxicity. eNeurologicalSci 2019, 14, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-T.; Chen, Y.-P.; Lin, W.-C.; Su, W.-C.; Sun, Y.-T. Immune Checkpoint Inhibitor-Induced Myasthenia Gravis. Front. Neurol. 2020, 11, 634. [Google Scholar] [CrossRef] [PubMed]
- Palaskas, N.; Morgan, J.; Daigle, T.; Banchs, J.; Durand, J.-B.; Hong, D.; Naing, A.; Le, H.; Hassan, S.A.; Karimzad, K.; et al. Targeted Cancer Therapies With Pericardial Effusions Requiring Pericardiocentesis Focusing on Immune Checkpoint Inhibitors. Am. J. Cardiol. 2019, 123, 1351–1357. [Google Scholar] [CrossRef]
- Kolla, B.C.; Patel, M.R. Recurrent pleural effusions and cardiac tamponade as possible manifestations of pseudoprogression associated with nivolumab therapy—A report of two cases. J. Immunother. Cancer 2016, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Canale, M.L.; Camerini, A.; Casolo, G.; Lilli, A.; Bisceglia, I.; Parrini, I.; Lestuzzi, C.; Meglio, J.D.; Puccetti, C.; Camerini, L.; et al. Incidence of Pericardial Effusion in Patients with Advanced Non-Small Cell Lung Cancer Receiving Immunotherapy. Adv. Ther. 2020, 37, 3178–3184. [Google Scholar] [CrossRef]
- Altan, M.; Toki, M.I.; Gettinger, S.N.; Carvajal-Hausdorf, D.E.; Zugazagoitia, J.; Sinard, J.H.; Herbst, R.S.; Rimm, D.L. Immune Checkpoint Inhibitor-Associated Pericarditis. J. Thorac. Oncol. 2019, 14, 1102–1108. [Google Scholar] [CrossRef]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef] [PubMed]
- Mir, H.; Alhussein, M.; Alrashidi, S.; Alzayer, H.; Alshatti, A.; Valettas, N.; Mukherjee, S.D.; Nair, V.; Leong, D.P. Cardiac Complications Associated With Checkpoint Inhibition: A Systematic Review of the Literature in an Important Emerging Area. Can. J. Cardiol. 2018, 34, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, M.; Nielsen, D.; Svane, I.M.; Iversen, K.; Rasmussen, P.V.; Madelaire, C.; Fosbøl, E.; Køber, L.; Gustafsson, F.; Andersson, C.; et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: A nationwide Danish study. Eur. Heart J. 2021, 42, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zheng, Q.; Lee, J.; Ma, Z.; Zhu, Q.; Wang, Z. PD-1/PD-L1 expression on CD(4+) T cells and myeloid DCs correlates with the immune pathogenesis of atrial fibrillation. J. Cell. Mol. Med. 2015, 19, 1223–1233. [Google Scholar] [CrossRef]
- Vuong, J.T.; Stein-Merlob, A.F.; Nayeri, A.; Sallam, T.; Neilan, T.G.; Yang, E.H. Immune Checkpoint Therapies and Atherosclerosis: Mechanisms and Clinical Implications: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 577–593. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Zhang, Q.; Li, H.-J.; Michot, J.M.; Liu, H.-B.; Zhan, P.; Lv, T.-F.; Song, Y. Evaluation of rare but severe immune related adverse effects in PD-1 and PD-L1 inhibitors in non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2017, 6 (Suppl. S1), S8–S20. [Google Scholar] [CrossRef] [Green Version]
- Cautela, J.; Rouby, F.; Salem, J.-E.; Alexandre, J.; Scemama, U.; Dolladille, C.; Cohen, A.; Paganelli, F.; Ederhy, S.; Thuny, F. Acute Coronary Syndrome With Immune Checkpoint Inhibitors: A Proof-of-Concept Case and Pharmacovigilance Analysis of a Life-Threatening Adverse Event. Can. J. Cardiol. 2020, 36, 476–481. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef]
- Stemme, S.; Faber, B.; Holm, J.; Wiklund, O.; Witztum, J.L.; Hansson, G.K. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1995, 92, 3893–3897. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L.; Zheng, Q. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 2009, 46, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poels, K.; van Leent, M.M.T.; Boutros, C.; Tissot, H.; Roy, S.; Meerwaldt, A.E.; Toner, Y.C.A.; Reiche, M.E.; Kusters, P.J.H.; Malinova, T.; et al. Immune Checkpoint Inhibitor Therapy Aggravates T Cell-Driven Plaque Inflammation in Atherosclerosis. JACC CardioOncol. 2020, 2, 599–610. [Google Scholar] [CrossRef]
- Ewing, M.M.; Karper, J.C.; Abdul, S.; de Jong, R.C.M.; Peters, H.A.B.; de Vries, M.R.; Redeker, A.; Kuiper, J.; Toes, R.E.M.; Arens, R.; et al. T-cell co-stimulation by CD28-CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int. J. Cardiol. 2013, 168, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Poels, K.; van Leent, M.M.T.; Reiche, M.E.; Kusters, P.J.H.; Huveneers, S.; de Winther, M.P.J.; Mulder, W.J.M.; Lutgens, E.; Seijkens, T.T.P. Antibody-Mediated Inhibition of CTLA4 Aggravates Atherosclerotic Plaque Inflammation and Progression in Hyperlipidemic Mice. Cells 2020, 9, 1987. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of Cytotoxic T-Lymphocyte-Associated Antigen-4 Prevents Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1141–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foks, A.C.; Kuiper, J. Immune checkpoint proteins: Exploring their therapeutic potential to regulate atherosclerosis. Br. J. Pharmacol. 2017, 174, 3940–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykl, R.; Fischer, O.; Vykoupil, K.; Taborsky, M. A unique reason for coronary spasm causing temporary ST elevation myocardial infarction (inferior STEMI)—systemic inflammatory response syndrome after use of pembrolizumab. Arch. Med. Sci. Atheroscler. Dis. 2017, 2, e100–e102. [Google Scholar] [CrossRef]
- Otsu, K.; Tajiri, K.; Sakai, S.; Ieda, M. Vasospastic angina following immune checkpoint blockade. Eur. Heart J. 2020, 41, 1702. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hu, X.; Li, Q.; Dai, W.; Cheng, X.; Huang, W.; Yu, W.; Chen, M.; Guo, Y.; Yuan, G. Effectiveness and safety of toripalimab, camrelizumab, and sintilimab in a real-world cohort of hepatitis B virus associated hepatocellular carcinoma patients. Ann. Transl. Med. 2020, 8, 1187. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Aguilera, J.V.; Chintakuntlawar, A.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Icht, O.; Darzi, N.; Shimony, S.; Jacobi, O.; Reinhorn, D.; Landman, Y.; Mutai, R.; Averbuch, I.; Shochat, T.; Spectre, G.; et al. Venous thromboembolism incidence and risk assessment in lung cancer patients treated with immune checkpoint inhibitors. J. Thromb. Haemost. JTH 2021, 19, 1250–1258. [Google Scholar] [CrossRef]
- Solinas, C.; Saba, L.; Sganzerla, P.; Petrelli, F. Venous and arterial thromboembolic events with immune checkpoint inhibitors: A systematic review. Thromb. Res. 2020, 196, 444–453. [Google Scholar] [CrossRef]
- Goel, A.; Khorana, A.; Kartika, T.; Gowda, S.; Tao, D.L.; Thawani, R.; Shatzel, J.J. Assessing the risk of thromboembolism in cancer patients receiving immunotherapy. Eur. J. Haematol. 2021, 108, 271–277. [Google Scholar] [CrossRef]
- Roopkumar, J.; Swaidani, S.; Kim, A.S.; Thapa, B.; Gervaso, L.; Hobbs, B.P.; Wei, W.; Alban, T.J.; Funchain, P.; Kundu, S.; et al. Increased Incidence of Venous Thromboembolism with Cancer Immunotherapy. Med 2021, 2, 423–434.e3. [Google Scholar] [CrossRef]
- Moik, F.; Chan, W.-S.E.; Wiedemann, S.; Hoeller, C.; Tuchmann, F.; Aretin, M.-B.; Fuereder, T.; Zöchbauer-Müller, S.; Preusser, M.; Pabinger, I.; et al. Incidence, risk factors, and outcomes of venous and arterial thromboembolism in immune checkpoint inhibitor therapy. Blood 2021, 137, 1669–1678. [Google Scholar] [CrossRef]
- Guven, D.C.; Aksun, M.S.; Sahin, T.K.; Aktepe, O.H.; Yildirim, H.C.; Taban, H.; Ceylan, F.; Kertmen, N.; Arik, Z.; Dizdar, O.; et al. Poorer baseline performance status is associated with increased thromboembolism risk in metastatic cancer patients treated with immunotherapy. Support. Care Cancer 2021, 29, 5417–5423. [Google Scholar] [CrossRef]
- Gutierrez-Sainz, L.; Martinez-Marin, V.; Viñal, D.; Martinez-Perez, D.; Pedregosa, J.; Garcia-Cuesta, J.A.; Villamayor, J.; Zamora, P.; Pinto, A.; Redondo, A.; et al. Incidence of venous thromboembolic events in cancer patients receiving immunotherapy: A single-institution experience. Clin. Transl. Oncol. 2021, 23, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, X.; Sun, D.; Zhao, J.; Xu, Z.; Zhao, P.; Ma, Z.; Zhang, Y. Thromboembolic events associated with immune checkpoint inhibitors: A real-world study of data from the food and drug administration adverse event reporting system (FAERS) database. Int. Immunopharmacol. 2021, 98, 107818. [Google Scholar] [CrossRef]
- Deschênes-Simard, X.; Richard, C.; Galland, L.; Blais, F.; Desilets, A.; Malo, J.; Cvetkovic, L.; Belkaid, W.; Elkrief, A.; Gagné, A.; et al. Venous thrombotic events in patients treated with immune checkpoint inhibitors for non-small cell lung cancer: A retrospective multicentric cohort study. Thromb. Res. 2021, 205, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Imamura, K.; Sakata, S.; Ikeda, T.; Horio, Y.; Iyama, S.; Akaike, K.; Hamada, S.; Jodai, T.; Nakashima, K.; et al. Disorder of Coagulation-Fibrinolysis System: An Emerging Toxicity of Anti-PD-1/PD-L1 Monoclonal Antibodies. J. Clin. Med. 2019, 8, 762. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.; Moudgil, R.; Lopez-Mattei, J.C.; Karimzad, K.; Mouhayar, E.N.; Somaiah, N.; Conley, A.P.; Patel, S.; Giza, D.E.; Iliescu, C. Progressive and Reversible Conduction Disease With Checkpoint Inhibitors. Can. J. Cardiol. 2017, 33, 1335.e13–1335.e15. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-R.; Florido, R.; Lipson, E.J.; Naidoo, J.; Ardehali, R.; Tocchetti, C.G.; Lyon, A.R.; Padera, R.F.; Johnson, D.B.; Moslehi, J. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc. Res. 2019, 115, 854–868. [Google Scholar] [CrossRef] [Green Version]
- Tay, R.Y.; Blackley, E.; McLean, C.; Moore, M.; Bergin, P.; Gill, S.; Haydon, A. Successful use of equine anti-thymocyte globulin (ATGAM) for fulminant myocarditis secondary to nivolumab therapy. Br. J. Cancer 2017, 117, 921–924. [Google Scholar] [CrossRef]
- Jain, V.; Mohebtash, M.; Rodrigo, M.E.; Ruiz, G.; Atkins, M.B.; Barac, A. Autoimmune Myocarditis Caused by Immune Checkpoint Inhibitors Treated with Antithymocyte Globulin. J. Immunother. 2018, 41, 332–335. [Google Scholar] [CrossRef]
- Wang, D.Y.; Okoye, G.D.; Neilan, T.G.; Johnson, D.B.; Moslehi, J.J. Cardiovascular Toxicities Associated with Cancer Immunotherapies. Curr. Cardiol. Rep. 2017, 19, 21. [Google Scholar] [CrossRef]
- Frigeri, M.; Meyer, P.; Banfi, C.; Giraud, R.; Hachulla, A.-L.; Spoerl, D.; Friedlaender, A.; Pugliesi-Rinaldi, A.; Dietrich, P.-Y. Immune Checkpoint Inhibitor-Associated Myocarditis: A New Challenge for Cardiologists. Can. J. Cardiol. 2018, 34, 92.e1–92.e3. [Google Scholar] [CrossRef] [Green Version]
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648, 2648a–2648d. [Google Scholar] [CrossRef] [PubMed]
- Balanescu, D.V.; Donisan, T.; Palaskas, N.; Lopez-Mattei, J.; Kim, P.Y.; Buja, L.M.; McNamara, D.M.; Kobashigawa, J.A.; Durand, J.-B.; Iliescu, C.A. Immunomodulatory treatment of immune checkpoint inhibitor-induced myocarditis: Pathway toward precision-based therapy. Cardiovasc. Pathol. 2020, 47, 107211. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Morimoto, R.; Okumura, T.; Yamashita, Y.; Haga, T.; Kuwayama, T.; Yokoi, T.; Hiraiwa, H.; Kondo, T.; Sugiura, Y.; et al. Late-Onset Fulminant Myocarditis With Immune Checkpoint Inhibitor Nivolumab. Can. J. Cardiol. 2018, 34, e1–e812. [Google Scholar] [CrossRef] [PubMed]
- Compton, F.; He, L.; Sarode, R.; Wodajo, A.; Usmani, A.; Burner, J.; Berlacher, M.; Simone, N.D. Immune checkpoint inhibitor toxicity: A new indication for therapeutic plasma exchange? J. Clin. Apher. 2021, 36, 645–648. [Google Scholar] [CrossRef]
- Schiopu, S.R.I.; Käsmann, L.; Schönermarck, U.; Fischereder, M.; Grabmaier, U.; Manapov, F.; Rauch, J.; Orban, M. Pembrolizumab-induced myocarditis in a patient with malignant mesothelioma: Plasma exchange as a successful emerging therapy-case report. Transl. Lung Cancer Res. 2021, 10, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Yogasundaram, H.; Alhumaid, W.; Chen, J.W.; Church, M.; Alhulaimi, N.; Kimber, S.; Paterson, D.I.; Senaratne, J.M. Plasma Exchange for Immune Checkpoint Inhibitor-Induced Myocarditis. CJC Open 2021, 3, 379–382. [Google Scholar] [CrossRef]
- Salem, J.-E.; Allenbach, Y.; Vozy, A.; Brechot, N.; Johnson, D.B.; Moslehi, J.J.; Kerneis, M. Abatacept for Severe Immune Checkpoint Inhibitor-Associated Myocarditis. N. Engl. J. Med. 2019, 380, 2377–2379. [Google Scholar] [CrossRef]
- Esfahani, K.; Buhlaiga, N.; Thébault, P.; Lapointe, R.; Johnson, N.A.; Miller, W.H. Alemtuzumab for Immune-Related Myocarditis Due to PD-1 Therapy. N. Engl. J. Med. 2019, 380, 2375–2376. [Google Scholar] [CrossRef]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, E447–E458. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).