
Vaccinomics-Aided Development of a Next-Generation Chimeric Vaccine against an Emerging Threat: Mycoplasma genitalium

 , , and
, , and 
Abstract
:1. Introduction
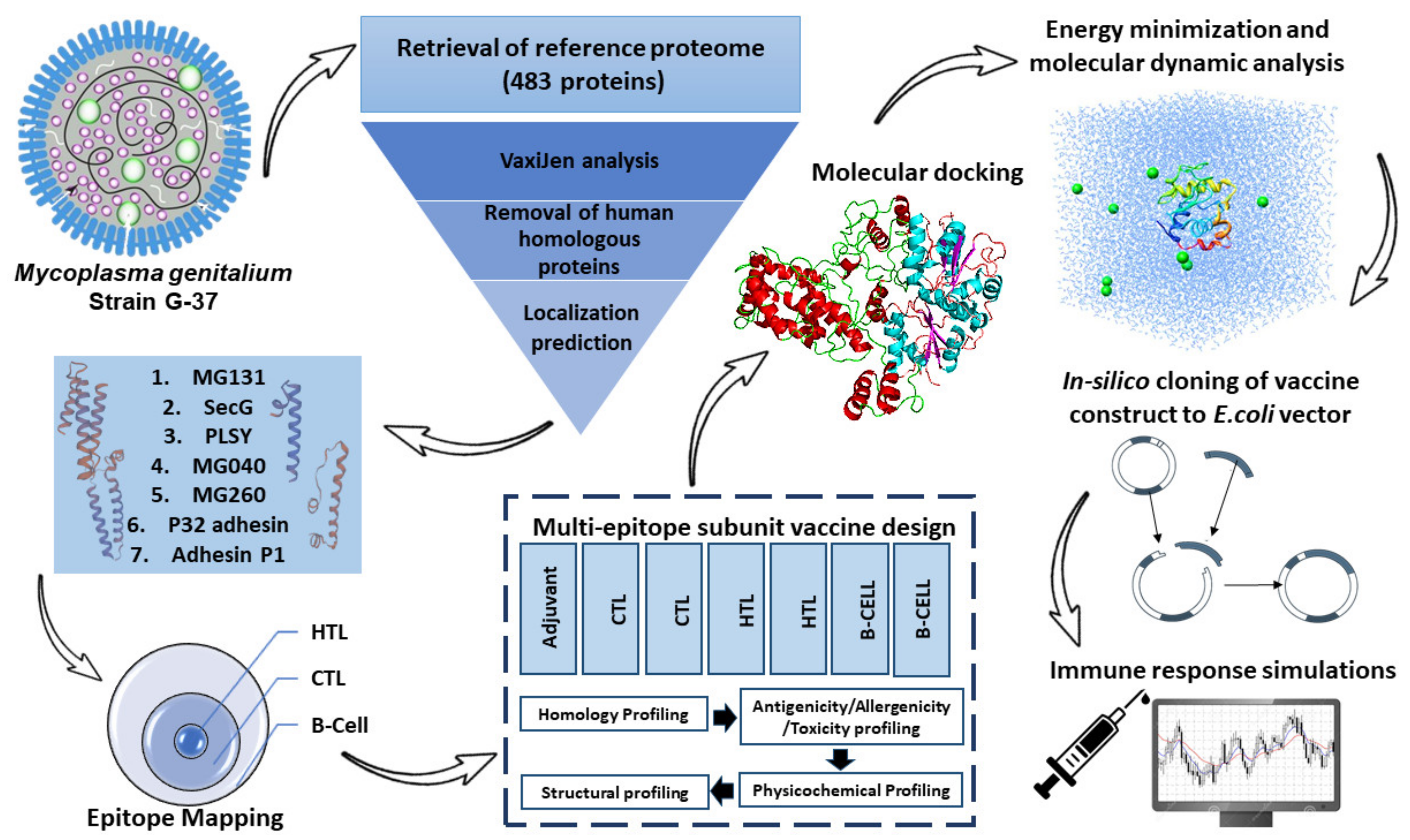
2. Materials and Methods
2.1. Retrieval of M. genitalium Reference Proteome
2.2. Epitope Mapping
2.3. Conserved Epitope Analysis
2.4. Epitopes Assembly
2.5. Profiling of Immunogenicity, Allergenicity, and Physicochemical Characteristics
2.6. Solubility Analysis
2.7. Computation of Secondary Structure
2.8. Computation of Tertiary Structure (3D)
2.9. 3D Model Validation and Refinement
2.10. Probing of Discontinuous BCEs
2.11. Docking Studies of the Modeled Vaccine with Toll-like Receptors
2.12. Energy Optimization and Simulation of Molecular Dynamics
2.13. Simulated Immune Responses
2.14. Reverse Transcription and Computational Cloning into Vector Backbone
3. Results
3.1. Finalization of Protein Sequences for Development of Vaccine Based on Immunogenicity Analysis
3.2. Finalization of Cytotoxic T-Lymphocyte Epitopes
3.3. Identification of Helper T-Lymphocyte Epitopes
3.4. Probing the Linear BCEs
3.5. Fusion of Final Candidates
3.6. Physicochemical and Antigenicity Profiling of Chimeric Construct
3.7. Solubility Analysis
3.8. Projection of Vaccine’s 2-D Structure
3.9. 3D Modeling of the Proposed Construct
3.10. 3D Model Enhancement and Quality Inspection
3.11. Probing Non-Linear BCEs
3.12. Docking Studies of the Modeled Vaccine with Toll-like Receptors
3.13. Binding Energy Analysis
3.14. MD Simulations
3.15. Immune Simulations
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Unemo, M.; Jensen, J.S. Antimicrobial-Resistant Sexually Transmitted Infections: Gonorrhoea and Mycoplasma genitalium. Nat. Rev. Urol. 2017, 14, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Edouard, S.; Tissot-Dupont, H.; Dubourg, G.; Bernard, A.; Fournier, P.-E.; Ravaux, I.; Stein, A.; Raoult, D. Mycoplasma genitalium, an Agent of Reemerging Sexually Transmitted Infections. APMIS 2017, 125, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, E.L.; Lowry, K.; Bletchly, C.; Nimmo, G.R.; Whiley, D.M. Mycoplasma genitalium Infections Can Comprise a Mixture of Both Fluoroquinolone-Susceptible and Fluoroquinolone-Resistant Strains. J. Antimicrob. Chemother. 2021, 76, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Biggest Threats and Data|Antibiotic/Antimicrobial Resistance|CDC. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html (accessed on 30 August 2021).
- Ona, S.; Molina, R.L.; Diouf, K. Mycoplasma genitalium: An Overlooked Sexually Transmitted Pathogen in Women? Infect. Dis. Obstet. Gynecol. 2016, 2016, 4513089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, J.S. Mycoplasma and Ureaplasma. In Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1660–1665.e2. [Google Scholar] [CrossRef]
- Taylor-Robinson, D.; Jensen, J.S. Mycoplasma genitalium: From Chrysalis to Multicolored Butterfly. Clin. Microbiol. Rev. 2011, 24, 498–514. [Google Scholar] [CrossRef] [Green Version]
- Tully, J.; Cole, R.; Taylor-Robinson, D.; Rose, D. A Newly Discovered Mycoplasma in The Human Urogenital Tract. Lancet 1981, 317, 1288–1291. [Google Scholar] [CrossRef]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The Minimal Gene Complement of Mycoplasma genitalium. Science 1995, 270, 397–403. [Google Scholar] [CrossRef]
- Sethi, S.; Zaman, K.; Jain, N. Mycoplasma genitalium Infections: Current Treatment Options and Resistance Issues. Infect. Drug Resist. 2017, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.L.; Goje, O.J. Mycoplasma genitalium: An Emerging Sexually Transmitted Infection. Scientifica 2016, 2016, 7537318. [Google Scholar] [CrossRef]
- Sweeney, E.L.; Trembizki, E.; Bletchly, C.; Bradshaw, C.S.; Menon, A.; Francis, F.; Langton-Lockton, J.; Nimmo, G.R.; Whiley, D.M. Levels of Mycoplasma genitalium Antimicrobial Resistance Differ by Both Region and Gender in the State of Queensland, Australia: Implications for Treatment Guidelines. J. Clin. Microbiol. 2019, 57, e01555-18. [Google Scholar] [CrossRef] [Green Version]
- Pitt, R.; Unemo, M.; Sonnenberg, P.; Alexander, S.; Beddows, S.; Cole, M.J.; Clifton, S.; Mercer, C.H.; Johnson, A.M.; Ison, C.A.; et al. Antimicrobial Resistance in Mycoplasma genitalium Sampled from the British General Population. Sex. Transm. Infect. 2020, 96, 464–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Schalk, T.E.; Braam, J.F.; Kusters, J.G. Molecular Basis of Antimicrobial Resistance in Mycoplasma genitalium. Int. J. Antimicrob. Agents 2020, 55, 105911. [Google Scholar] [CrossRef] [PubMed]
- Workowski, K.A. Centers for Disease Control and Prevention Sexually Transmitted Diseases Treatment Guidelines. Clin. Infect. Dis. 2015, 61 (Suppl. 8), S759–S762. [Google Scholar] [CrossRef] [PubMed]
- McGowin, C.L.; Anderson-Smits, C. Mycoplasma genitalium: An Emerging Cause of Sexually Transmitted Disease in Women. PLOS Pathog. 2011, 7, e1001324. [Google Scholar] [CrossRef] [Green Version]
- Vesty, A.; McAuliffe, G.; Roberts, S.; Henderson, G.; Basu, I. Mycoplasma genitalium Antimicrobial Resistance in Community and Sexual Health Clinic Patients, Auckland, New Zealand—Volume 26, Number 2—February 2020—Emerging Infectious Diseases Journal—CDC. Emerg. Infect. Dis. 2020, 26, 332–335. [Google Scholar] [CrossRef]
- Nye, M.B.; Harris, A.B.; Pherson, A.J.; Cartwright, C.P. Prevalence of Mycoplasma genitalium Infection in Women with Bacterial Vaginosis. BMC Women’s Health 2020, 20, 62. [Google Scholar] [CrossRef]
- Parmar, N.R.; Mushanski, L.; Wanlin, T.; Lepe, A.; Lang, A.; Minion, J.; Dillon, J.A.R. High Prevalence of Macrolide and Fluoroquinolone Resistance-Mediating Mutations in Mycoplasma genitalium -Positive Urine Specimens from Saskatchewan. Sex. Transm. Dis. 2021, 48, 680–684. [Google Scholar] [CrossRef]
- Horseman, T.S.; Crecelius, E.M.; Miller, M.A.; Lustik, M.B.; Lee, B.C.; Brazer, M.L.; O’Neal, L.L.; Kim, D.M.; Fong, K.S.K.; Chang, T.W. Prevalence and Epidemiology of Mycoplasma genitalium in a Pacific-Region Military Population. Sex. Transm. Dis. 2021, 48, 578–582. [Google Scholar] [CrossRef]
- Munson, E.; Morgan, E.; Sienkiewicz, L.; Thomas, Y.; Buehler, K.; Ryan, D.; Clifford, A.; Mustanski, B. Molecular Screening in a Longitudinal Cohort of Young Men Who Have Sex with Men and Young Transgender Women: Associations with Focus on the Emerging Sexually Transmitted Pathogen Mycoplasma genitalium. Sex. Transm. Infect. 2021, 97, 434–440. [Google Scholar] [CrossRef]
- Getman, D.; Jiang, A.; O’Donnell, M.; Cohen, S. Mycoplasma genitalium Prevalence, Coinfection, and Macrolide Antibiotic Resistance Frequency in a Multicenter Clinical Study Cohort in the United States. J. Clin. Microbiol. 2016, 54, 2278–2283. [Google Scholar] [CrossRef] [Green Version]
- Vandepitte, J.; Weiss, H.A.; Bukenya, J.; Kyakuwa, N.; Muller, E.; Buvé, A.; Van der Stuyft, P.; Hayes, R.J.; Grosskurth, H. Association between Mycoplasma genitalium Infection and HIV Acquisition among Female Sex Workers in Uganda: Evidence from a Nested Case–Control Study. Sex. Transm. Infect. 2014, 90, 545–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Robinson, D. Diagnosis and Antimicrobial Treatment of Mycoplasma genitalium Infection: Sobering Thoughts. Expert Rev. Anti-Infect. Ther. 2014, 12, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Chen, Y.; You, X. Subversion of the Immune Response by Human Pathogenic Mycoplasmas. Front. Microbiol. 2019, 10, 1934. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, E.C.; Ward, R.W. Mucosal Vaccines—Fortifying the Frontiers. Nat. Rev. Immunol. 2021, 22, 236–250. [Google Scholar] [CrossRef]
- Mahmud, S.; Rafi, M.O.; Paul, G.K.; Promi, M.M.; Shimu, M.S.S.; Biswas, S.; Emran, T.B.; Dhama, K.; Alyami, S.A.; Moni, M.A.; et al. Designing a Multi-Epitope Vaccine Candidate to Combat MERS-CoV by Employing an Immunoinformatics Approach. Sci. Rep. 2021, 11, 15431. [Google Scholar] [CrossRef]
- Zhang, L. Multi-Epitope Vaccines: A Promising Strategy against Tumors and Viral Infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [Green Version]
- Fookes, M.C.; Hadfield, J.; Harris, S.; Parmar, S.; Unemo, M.; Jensen, J.S.; Thomson, N.R. Mycoplasma genitalium: Whole Genome Sequence Analysis, Recombination and Population Structure. BMC Genom. 2017, 18, 993. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Yu, C.-S.; Cheng, C.-W.; Su, W.-C.; Chang, S.-C.; Huang, S.-W.; Hwang, J.-K.; Lu, C.-H. CELLO2GO: A Web Server for Protein SubCELlular LOcalization Prediction with Functional Gene Ontology Annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-Scale Validation of Methods for Cytotoxic T-Lymphocyte Epitope Prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune Epitope Database Analysis Resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction Methods for B-Cell Epitopes. Methods Mol. Biol. 2007, 409, 387–394. [Google Scholar] [CrossRef] [PubMed]
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting Linear B-Cell Epitopes Using String Kernels. J. Mol. Recognit. 2008, 21, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Yao, B.; Zheng, D.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict B-Cell Linear Antigenic Epitopes. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2020; Volume 2131, pp. 299–307. [Google Scholar]
- Chen, X.; Zaro, J.; Shen, W.-C. Fusion Protein Linkers: Property, Design and Functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357. [Google Scholar] [CrossRef] [Green Version]
- McGowin, C.L.; Ma, L.; Martin, D.H.; Pyles, R.B. Mycoplasma genitalium-Encoded MG309 Activates NF-KappaB via Toll-like Receptors 2 and 6 to Elicit Proinflammatory Cytokine Secretion from Human Genital Epithelial Cells. Infect. Immun. 2009, 77, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- He, J.; You, X.; Zeng, Y.; Yu, M.; Zuo, L.; Wu, Y. Mycoplasma genitalium-Derived Lipid-Associated Membrane Proteins Activate NF-ΚB through Toll-Like Receptors 1, 2, and 6 and CD14 in a MyD88-Dependent Pathway. Clin. Vaccine Immunol. 2009, 16, 1750. [Google Scholar] [CrossRef] [Green Version]
- Campos, M.A.S.; Almeida, I.C.; Takeuchi, O.; Akira, S.; Valente, E.P.; Procópio, D.O.; Travassos, L.R.; Smith, J.A.; Golenbock, D.T.; Gazzinelli, R.T. Activation of Toll-Like Receptor-2 by Glycosylphosphatidylinositol Anchors from a Protozoan Parasite. J. Immunol. 2001, 167, 416–423. [Google Scholar] [CrossRef]
- Gupta, N.; Regar, H.; Verma, V.K.; Prusty, D.; Mishra, A.; Prajapati, V.K. Receptor-Ligand Based Molecular Interaction to Discover Adjuvant for Immune Cell TLRs to Develop next-Generation Vaccine. Int. J. Biol. Macromol. 2020, 152, 535–545. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-Throughput Prediction of Protein Antigenicity Using Protein Microarray Data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB Bioinformatics Resource Portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate Sequence-Based Prediction of Protein Solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [Green Version]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein-Sol: A Web Tool for Predicting Protein Solubility from Sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004, 32, W526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, M. Computational Protein Structure Refinement: Almost There, yet Still so Far to Go. WIREs Comput. Mol. Sci. 2017, 7, e1307. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure Validation by Cα Geometry: ϕ,ψ and Cβ Deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Ansari, H.R.; Raghava, G.P. Identification of Conformational B-Cell Epitopes in an Antigen from Its Primary Sequence. Immunome Res. 2010, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A New Structure-Based Tool for the Prediction of Antibody Epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, V.; Tiwari, V. Subtractive Proteomics to Identify Novel Drug Targets and Reverse Vaccinology for the Development of Chimeric Vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 9044. [Google Scholar] [CrossRef] [Green Version]
- Ramos, E.I.; Das, K.; Harrison, A.L.; Garcia, A.; Gadad, S.S.; Dhandayuthapani, S. Mycoplasma genitalium and M. Pneumoniae Regulate a Distinct Set of Protein-Coding Genes in Epithelial Cells. Front. Immunol. 2021, 12, 4072. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein-Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK Server for Integrated Protein–Protein Docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein-Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J. PDBsum: Structural Summaries of PDB Entries. Protein Sci. 2017, 27, 129–134. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Turner: XMGRACE, Version 5.1. 19—Google Scholar. Available online: https://scholar.google.com/scholar_lookup?title=XMGRACE,+Version+5.1.19&author=PJ+Turner&publication_year=2005& (accessed on 28 April 2021).
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, 9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, K.; Irum, S.; Ullah, S.R.; Andleeb, S. In-Silico Vaccine Design Based on a Novel Vaccine Candidate Against Infections Caused by Acinetobacter baumannii. Int. J. Pept. Res. Ther. 2022, 28, 16. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. Identifying Candidate Subunit Vaccines Using an Alignment-Independent Method Based on Principal Amino Acid Properties. Vaccine 2007, 25, 856–866. [Google Scholar] [CrossRef]
- de Kraker, M.E.A.; Stewardson, A.J.; Harbarth, S. Will 10 Million People Die a Year Due to Antimicrobial Resistance by 2050? PLoS Med. 2016, 13, 1002184. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Sarma, D.K.; Shubham, S.; Kumawat, M.; Verma, V.; Nina, P.B.; JP, D.; Kumar, S.; Singh, B.; Tiwari, R.R. Futuristic Non-Antibiotic Therapies to Combat Antibiotic Resistance: A Review. Front. Microbiol. 2021, 12, 16. [Google Scholar] [CrossRef]
- Arnon, R.; Ben-Yedidia, T. Old and New Vaccine Approaches. Int. Immunopharmacol. 2003, 3, 1195–1204. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of Vaccine Candidates against Serogroup B Meningococcus by Whole-Genome Sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A Candidate Multi-Epitope Vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar] [CrossRef] [PubMed]
- Nain, Z.; Abdulla, F.; Rahman, M.M.; Karim, M.M.; Khan, M.S.A.; Sayed, S.B.; Mahmud, S.; Rahman, S.M.R.; Sheam, M.M.; Haque, Z.; et al. Proteome-Wide Screening for Designing a Multi-Epitope Vaccine against Emerging Pathogen Elizabethkingia anophelis Using Immunoinformatic Approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Y.; Shi, Y.; Wu, C.; Zhang, W.J.; Mao, X.H.; Guo, G.; Li, H.X.; Zou, Q.M. Therapeutic Efficacy of a Multi-Epitope Vaccine against Helicobacter pylori Infection in BALB/c Mice Model. Vaccine 2009, 27, 5013–5019. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Tarang, S.; Kesherwani, V.; LaTendresse, B.; Lindgren, L.; Rocha-Sanchez, S.M.; Weston, M.D. In Silico Design of a Multivalent Vaccine Against Candida albicans. Sci. Rep. 2020, 10, 1066. [Google Scholar] [CrossRef] [Green Version]
- Aldakheel, F.M.; Abrar, A.; Munir, S.; Aslam, S.; Allemailem, K.S.; Khurshid, M.; Ashfaq, U.A. Proteome-Wide Mapping and Reverse Vaccinology Approaches to Design a Multi-Epitope Vaccine against Clostridium perfringens. Vaccines 2021, 9, 1079. [Google Scholar] [CrossRef]
- Ali, S.; Ali, S.; Javed, S.O.; Shoukat, S.; Ahmad, S.; Ali, S.S.; Hussain, Z.; Waseem, M.; Rizwan, M.; Suleman, M.; et al. Proteome Wide Vaccine Targets Prioritization and Designing of Antigenic Vaccine Candidate to Trigger the Host Immune Response against the Mycoplasma genitalium Infection. Microb. Pathog. 2021, 152, 104771. [Google Scholar] [CrossRef]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of Putative Vaccine Candidates against Helicobacter pylori Exploiting Exoproteome and Secretome: A Reverse Vaccinology Based Approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Pulendran, B.; Arunachalam, P.S.; O’Hagan, D.T. Emerging Concepts in the Science of Vaccine Adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Adeleke, V.T.; Adeniyi, A.A.; Adeleke, M.A.; Okpeku, M.; Lokhat, D. The Design of Multiepitope Vaccines from Plasmids of Diarrheagenic Escherichia coli against Diarrhoea Infection: Immunoinformatics Approach. Infect. Genet. Evol. 2021, 91, 104803. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot Entry Identifier | Protein | Antigenicity (VaxiJen) | Non-Allergen | Subcellular Localization | |
|---|---|---|---|---|---|
| 1 | P47377 | MG131 (Hypothetical protein) | 1.16 | Yes | Cytoplasmic membrane |
| 2 | P58061 | SECG (Probable protein-export membrane protein SecG) | 0.93 | Yes | Cytoplasmic membrane |
| 3 | P47489 | PLSY (Glycerol-3-phosphate acyltransferase) | 0.92 | Yes | Cytoplasmic membrane |
| 4 | P47286 | MG040 (ABC transporter substrate binding protein pnrA-like) | 0.76 | Yes | Cytoplasmic membrane |
| 5 | P47502 | MG260 (Mycoplasma lipoprotein) | 0.76 | Yes | Cytoplasmic Membrane |
| 6 | Q49417 | P32 adhesin | 0.69 | Yes | Cytoplasmic Membrane |
| 7 | P20796 | Adhesin P1 | 0.52 | Yes | Cytoplasmic Membrane |
| Protein ID | CTL Epitopes (9-mer) | MHC Class(I) Supertypes | Binding Affinity | VaxiJen Score | Non-Allergen | Toxicity | Conservation |
|---|---|---|---|---|---|---|---|
| P47377 | YSALIPLFI | A1, A24 | 0.24 | 1.8319 | Yes | No | 100% |
| P58061 | AVICLIIGL | A1, A2 | 0.57 | 0.82 | Yes | No | 100% |
| P47489 | YQSTYFLSY | A3, A24 | 0.17 | 1.22 | Yes | No | 100% |
| P47286 | YIIWELIPF | A1, A24 | 0.11 | 2.1807 | Yes | No | 100% |
| P47502 | SVTLEQGWY | A2, A3, A24 | 0.46 | 0.82 | Yes | No | 100% |
| Protein ID | HTL Epitopes (15-mer) | Percentile Rank | IC50 | VaxiJen Score | Non-Allergen | Toxicity | Conser- Vation |
|---|---|---|---|---|---|---|---|
| P47377 | ALIPLFILLISLVLF | 0.01 | 0.21 | 2.11 | Yes | No | 100% |
| P58061 | GFVKILQIIMFILVV | 0.02 | 0.32 | 0.82 | Yes | No | 100% |
| P47489 | EKVYQSTYFLSYLSC | 0.04 | 0.41 | 0.59 | Yes | No | 100% |
| P47286 | FDLVLLWFLFVPLLI | 0.01 | 0.69 | 3.53 | Yes | No | 100% |
| P47502 | AKKAFRLYKKKISTS | 0.03 | 0.81 | 0.51 | Yes | No | 100% |
| P20796 | KLVLFLLAIVFLMLG | 0.15 | 0.95 | 1.29 | Yes | No | 100% |
| No. | Residues | Total Residues | Score |
|---|---|---|---|
| 1 | A:M1, A:A2, A:K3, A:L4, A:S5, A:T6, A:D7, A:E8, A:L9, A:L10, A:D11, A:A12, A:F13, A:K14, A:E15, A:M16, A:T17, A:L18, A:L19, A:E20, A:L21, A:S22, A:D23, A:F24, A:V25, A:K26, A:K27, A:F28, A:E29, A:E30, A:T31, A:F32, A:E33, A:V34, A:T35, A:A36, A:A37, A:A38, A:P39, A:V40, A:A41, A:V42, A:A43, A:A44, A:A45, A:G46, A:A47, A:A48, A:P49, A:P381 | 50 | 0.841 |
| 2 | A:E68, A:A69, A:G71, A:D72, A:K73, A:I75, A:G76, A:V77, A:I78, A:K79, A:V80, A:V81, A:R82, A:E83, A:I84, A:V85, A:S86, A:G87, A:L88, A:G89, A:L90, A:K91, A:E92, A:A93, A:K94, A:D95, A:L96, A:V97, A:D98, A:G99, A:A100, A:P101, A:K102, A:P103, A:L104, A:L105, A:E106, A:K107, A:V108, A:A109, A:K110, A:E111, A:A112, A:A113, A:D114, A:E115, A:A116, A:K117, A:A118, A:L120, A:E121, A:A122, A:G124, A:A125, A:T126, A:T128, A:K290, A:K291, A:I292, A:S293, A:T294, A:S295, A:G296, A:P297, A:G298, A:P299, A:G300, A:K301, A:L302, A:V303, A:L304, A:F305, A:L306, A:A308, A:I309, A:V310, A:L312, A:M313, A:L314, A:G315, A:F316, A:S317 | 82 | 0.699 |
| 3 | A:W178, A:E179, A:L180, A:I181, A:P182, A:F183, A:A184, A:A185, A:K186, A:S187, A:V188, A:T189, A:L190, A:E191, A:Q192, A:G193, A:W194, A:Y195, A:G196, A:P197, A:G198, A:P199, A:G200, A:A201, A:I207, A:S211, A:L212, A:V213, A:L214, A:F215, A:G216, A:P217, A:G218, A:P219, A:F222, A:V223, A:K224, A:I225, A:L226, A:Q227, A:I228, A:I229, A:M230, A:F231, A:I232, A:L233, A:V234, A:V235, A:G236, A:P237, A:G238, A:P239, A:G240, A:E241, A:K242, A:V243, A:Y244, A:Q245, A:S246, A:T247, A:Y248, A:F249, A:L250, A:S251, A:Y252, A:L253, A:S254, A:C255, A:G256, A:P257, A:G258, A:P259, A:G260, A:F261, A:D262, A:L263, A:L266, A:W267 | 78 | 0.687 |
| 4 | A:D64, A:V65, A:I66, A:E334 | 4 | 0.576 |
| 5 | A:K130, A:A132, A:A133, A:A134, A:K135, A:Y139 | 6 | 0.56 |
| Vaccine Construct | TLRs | Solution | a GBE | b aVdW | c HBE | d ACE | e Score | f Area |
| TLR 1 | 1 | −37.53 | −40.86 | −2.37 | 4.44 | 20,226 | 3124.50 | |
| TLR 2 | 7 | −12.41 | −52.34 | −3.94 | −0.55 | 17,078 | 2019.00 | |
| TLR 6 | 4 | −7.79 | −24.19 | −5.19 | 8.37 | 15,410 | 2584.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, K.; Hussain, T.; Jamil, Z.; Alrokayan, K.S.; Ahmad, B.; Waheed, Y. Vaccinomics-Aided Development of a Next-Generation Chimeric Vaccine against an Emerging Threat: Mycoplasma genitalium. Vaccines 2022, 10, 1720. https://doi.org/10.3390/vaccines10101720
Khalid K, Hussain T, Jamil Z, Alrokayan KS, Ahmad B, Waheed Y. Vaccinomics-Aided Development of a Next-Generation Chimeric Vaccine against an Emerging Threat: Mycoplasma genitalium. Vaccines. 2022; 10(10):1720. https://doi.org/10.3390/vaccines10101720
Chicago/Turabian StyleKhalid, Kashaf, Tajamul Hussain, Zubia Jamil, Khalid Salman Alrokayan, Bashir Ahmad, and Yasir Waheed. 2022. "Vaccinomics-Aided Development of a Next-Generation Chimeric Vaccine against an Emerging Threat: Mycoplasma genitalium" Vaccines 10, no. 10: 1720. https://doi.org/10.3390/vaccines10101720
APA StyleKhalid, K., Hussain, T., Jamil, Z., Alrokayan, K. S., Ahmad, B., & Waheed, Y. (2022). Vaccinomics-Aided Development of a Next-Generation Chimeric Vaccine against an Emerging Threat: Mycoplasma genitalium. Vaccines, 10(10), 1720. https://doi.org/10.3390/vaccines10101720