
Rapid In-Process Measurement of Live Virus Vaccine Potency Using Laser Force Cytology: Paving the Way for Rapid Vaccine Development

 , ,
, ,
Abstract
:
1. Introduction
2. Results
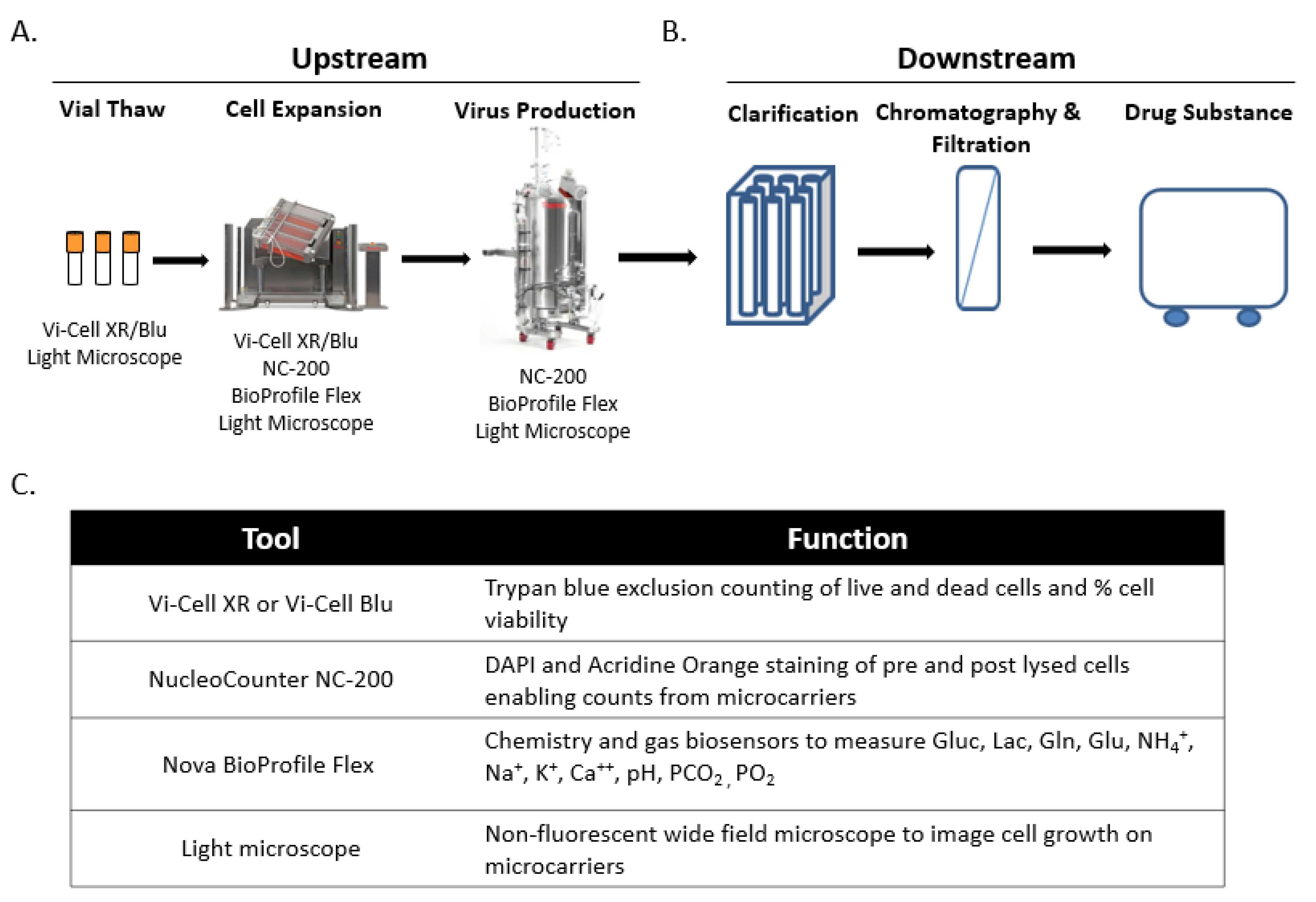
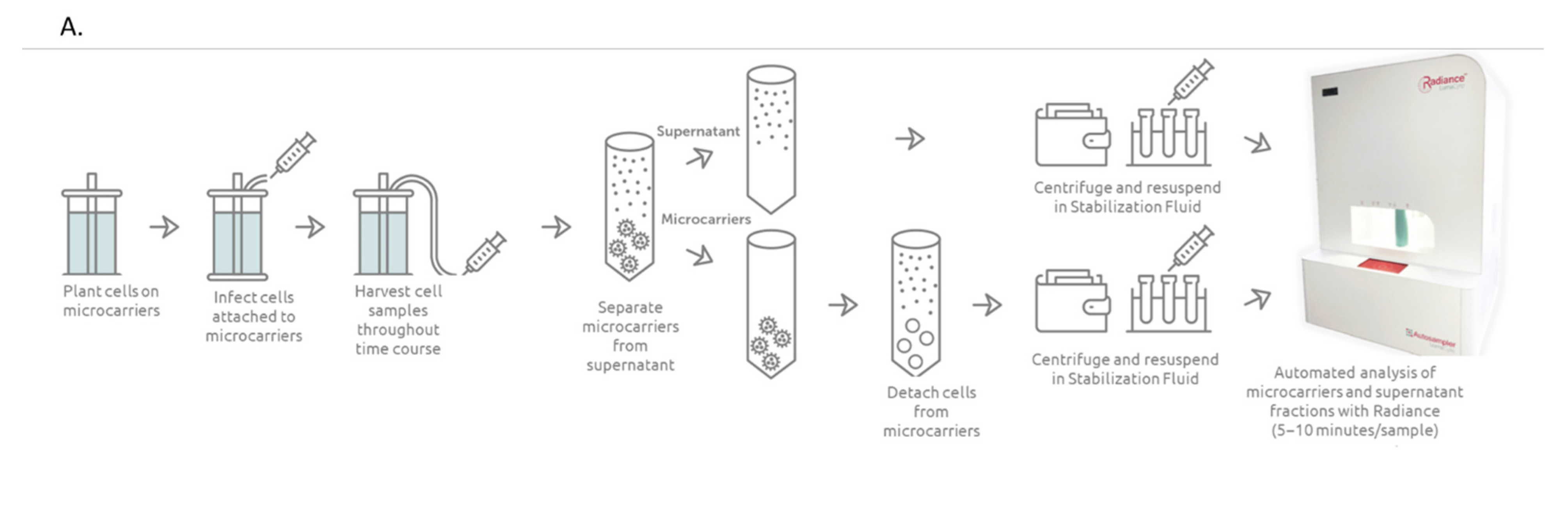
2.1. Live Virus Microcarrier Culture Bioreactor Monitoring Current Practice
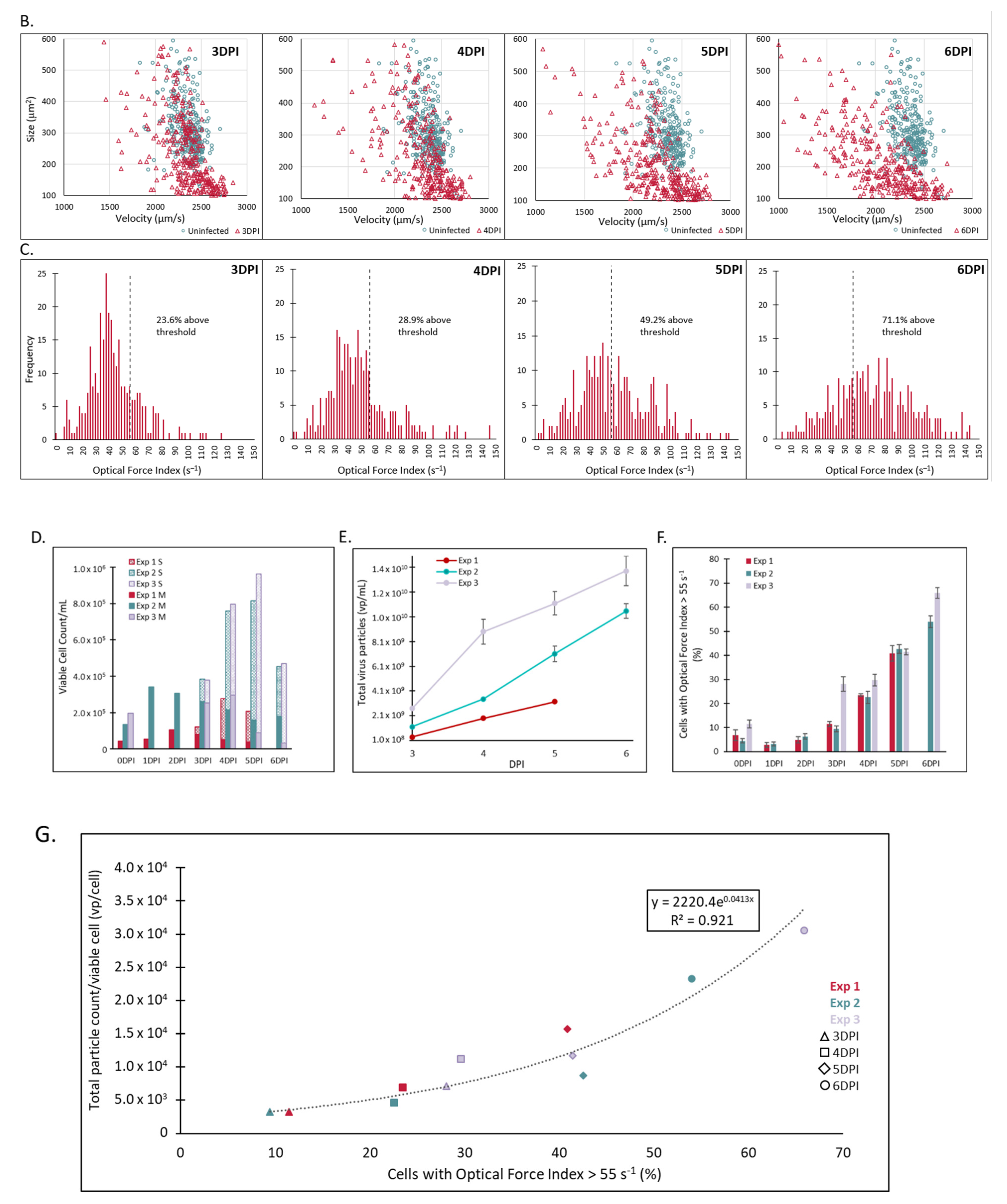
2.2. Laser Force Cytology Cellular Fingerprinting of Measles Virus Infection Correlates with Total Virus Particle Counts and Acts as a Surrogate to Potency
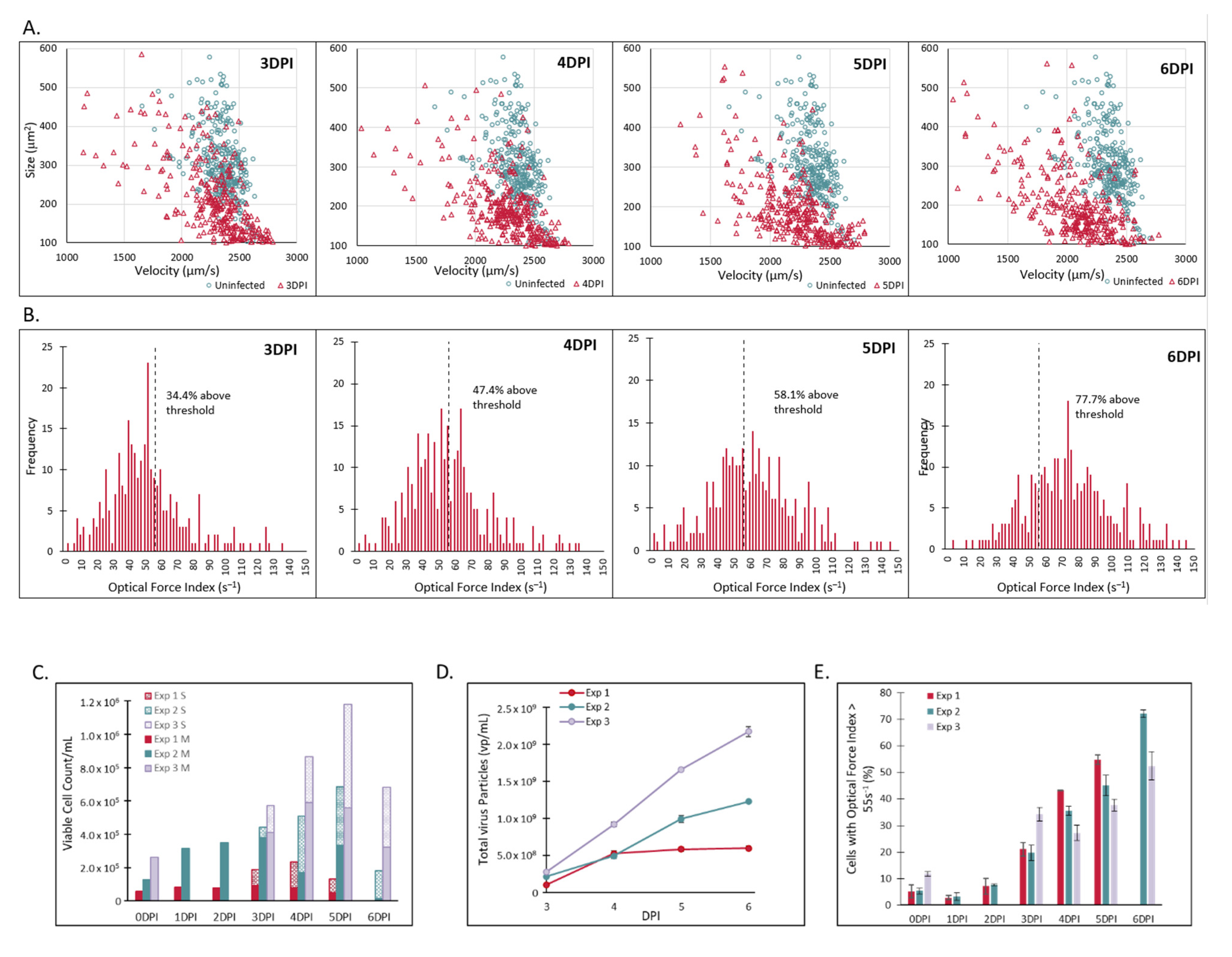
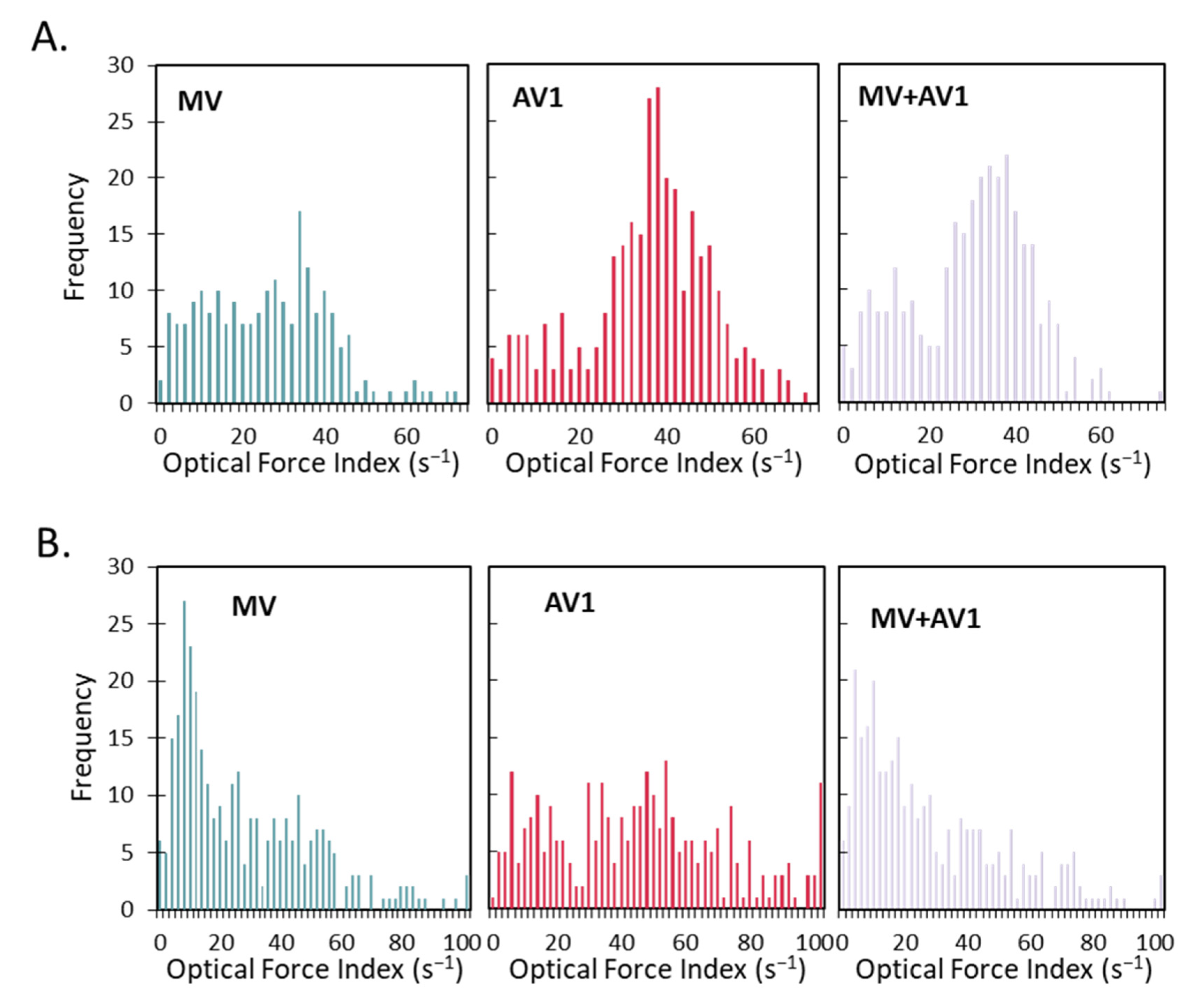
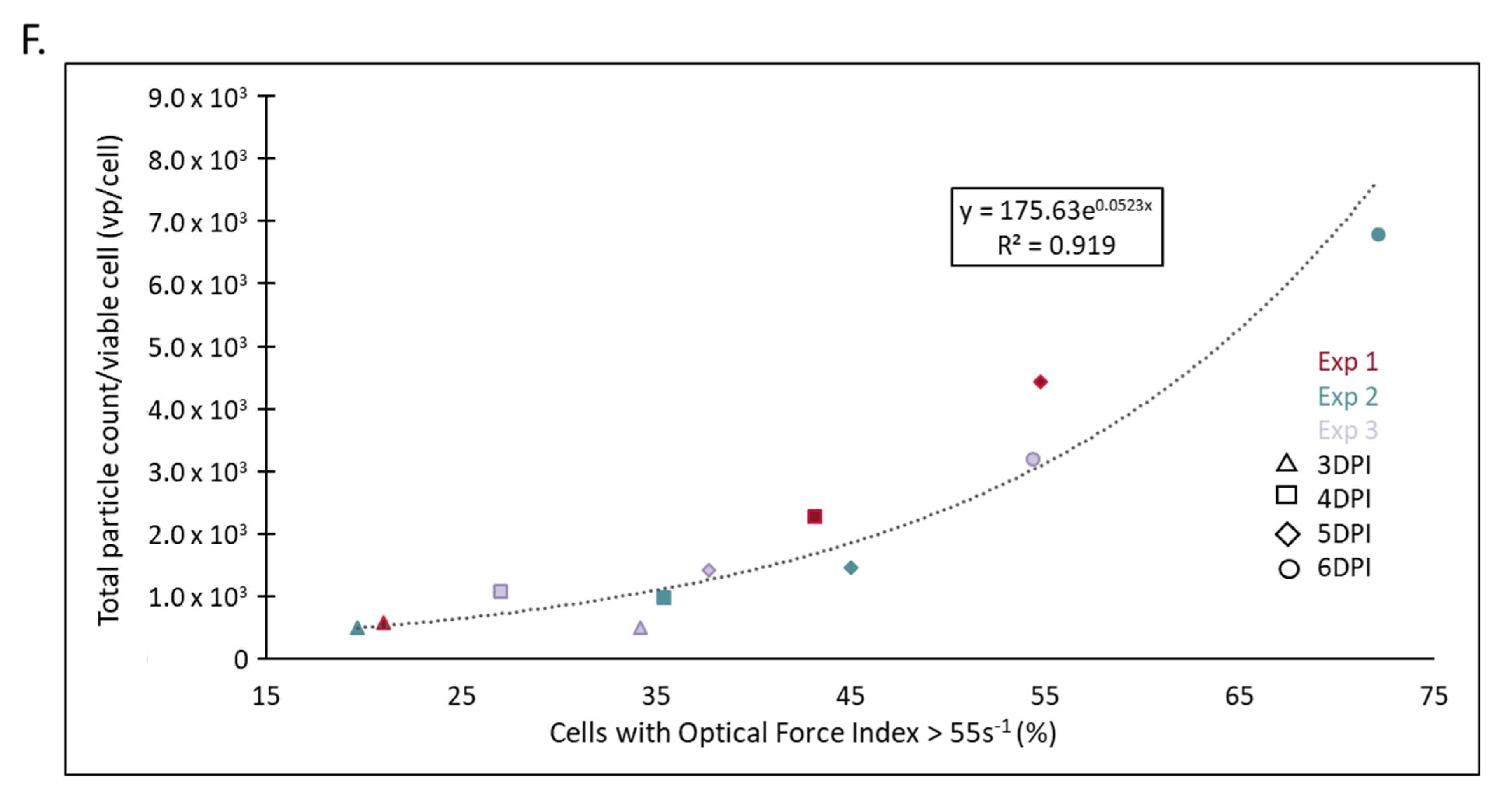
2.3. A Unique and Non-Overlapping Fingerprint of the Adventitious Virus Is Established in a Microcarrier Bioreactor Culture and Correlated to Total Virus Particle Counts
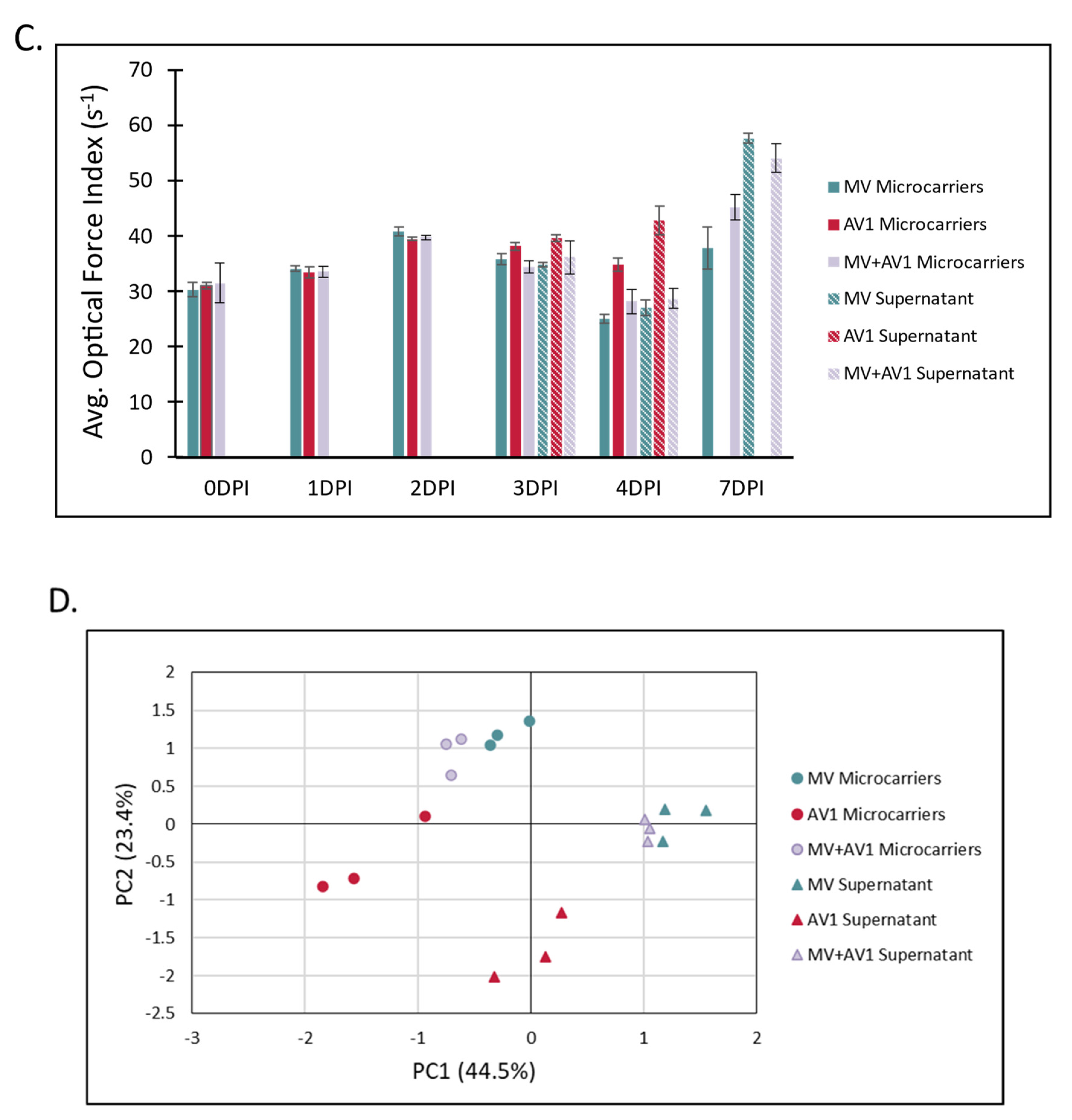
2.4. The Unique Cellular Responses Measured by Laser Force Cytology Are Used to Identify and Distinguish between Measles and an Adventitious Virus in a Co-Infected Production Bioreactor
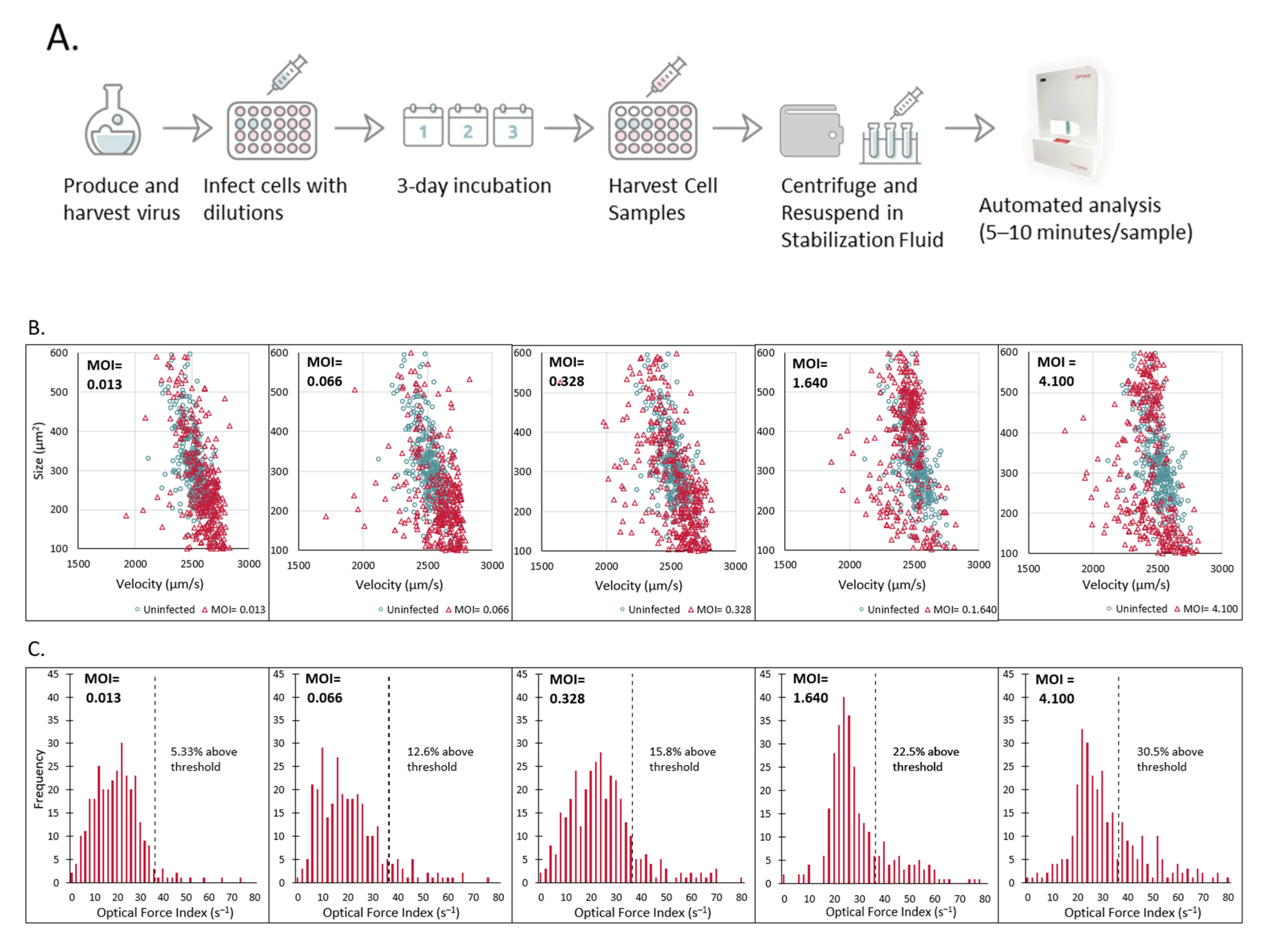
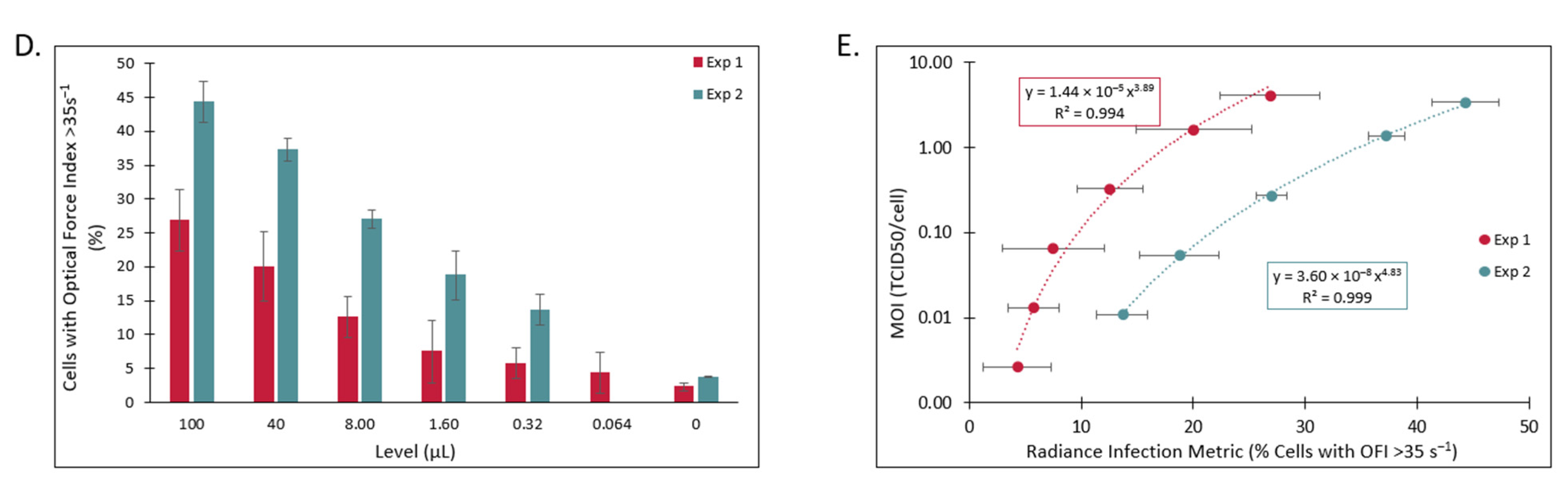
2.5. Measurement of Measles Virus Potency in a Plate-Based Assay by Laser Force Cytology Shows Robust Correlation to the Traditional TCID50 Assay
3. Methods
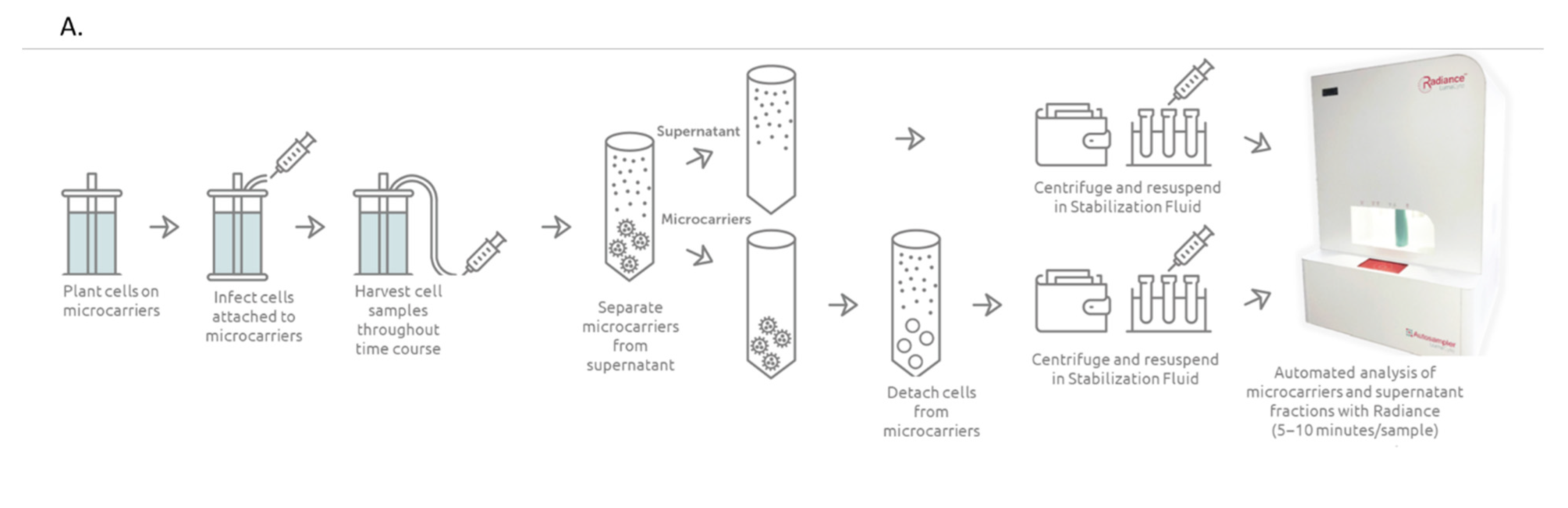
3.1. Bioreactor Sampling
3.2. Cell Culture
3.3. Viral Infectivity Assay
3.4. Sample Collection for Infectivity Assay
3.5. CytoFLEX Assay
3.6. Data Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van de Berg, D.; Kis, Z.; Behmer, C.F.; Samnuan, K.; Blakney, A.K.; Kontoravdi, C.; Shattock, R.; Shah, N. Quality by design modelling to support rapid RNA vaccine production against emerging infectious diseases. NPJ Vaccines 2021, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, R.; Reichl, U. Editorial: Can modern vaccine technology pursue the success of traditional vaccine manufacturing? Biotechnol. J. 2015, 10, 657–658. [Google Scholar] [CrossRef] [PubMed]
- Largoni, M.; Facco, P.; Bernini, D.; Bezzo, F.; Barolo, M. Quality-by-Design approach to monitor the operation of a batch bioreactor in an industrial avian vaccine manufacturing process. J. Biotechnol. 2015, 211, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C. Coping with vaccine manufacturing delays. Clevel. Clin. J. Med. 2000, 67, 898–900. [Google Scholar] [CrossRef]
- Plotkin, S.; Robinson, J.M.; Cunningham, G.; Iqbal, R.; Larsen, S. The complexity and cost of vaccine manufacturing–An overview. Vaccine 2017, 35, 4064–4071. [Google Scholar] [CrossRef]
- Ulmer, J.B.; Valley, U.; Rappuoli, R. Vaccine manufacturing: Challenges and solutions. Nat. Biotechnol. 2006, 24, 1377–1383. [Google Scholar] [CrossRef]
- Sashihara, J.; Burbelo, P.D.; Savoldo, B.; Pierson, T.C.; Cohen, J.I. Human antibody titers to Epstein–Barr Virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology 2009, 391, 249–256. [Google Scholar] [CrossRef]
- Heppner, D.G., Jr.; Kemp, T.L.; Martin, B.K.; Ramsey, W.J.; Nichols, R.; Dasen, E.J.; Link, C.J.; Das, R.; Xu, Z.J.; Sheldon, E.A.; et al. Safety and immunogenicity of the rVSVG-ZEBOV-GP Ebola virus vaccine candidate in healthy adults: A phase 1b randomised, multicentre, double-blind, placebo-controlled, dose-response study. Lancet Infect. Dis. 2017, 17, 854–866. [Google Scholar] [CrossRef]
- Salje, H.; Rodríguez-Barraquer, I.; Rainwater-Lovett, K.; Nisalak, A.; Thaisomboonsuk, B.; Thomas, S.J.; Fernandez, S.; Jarman, R.G.; Yoon, I.-K.; Cummings, D.A.T. Variability in Dengue Titer Estimates from Plaque Reduction Neutralization Tests Poses a Challenge to Epidemiological Studies and Vaccine Development. PLoS Negl. Trop. Dis. 2014, 8, e2952. [Google Scholar]
- Wang, J.; Peng, Y.; Xu, H.; Cui, Z.; Williams, R.O., 3rd. The COVID-19 Vaccine Race: Challenges and Opportunities in Vaccine Formulation. AAPS PharmSciTech 2020, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Azizi, A.; Mironov, G.G.; Muharemagic, D.; Wehbe, M.; Bell, J.C.; Berezovski, M.V. Viral Quantitative Capillary Electrophoresis for Counting and Quality Control of RNA Viruses. Anal. Chem. 2012, 84, 9585–9591. [Google Scholar] [CrossRef]
- Mironov, G.G.; Chechik, A.V.; Ozer, R.; Bell, J.C.; Berezovski, M.V. Viral Quantitative Capillary Electrophoresis for Counting Intact Viruses. Anal. Chem. 2011, 83, 5431–5435. [Google Scholar] [CrossRef] [PubMed]
- Hebert, C.G.; DiNardo, N.; Evans, Z.L.; Hart, S.J.; Hachmann, A.-B. Rapid quantification of vesicular stomatitis virus in Vero cells using Laser Force Cytology. Vaccine 2018, 36, 6061–6069. [Google Scholar] [CrossRef]
- Hebert, C.G.; Hart, S.J.; Terray, A. Label free detection of pseudorabies virus infection in Vero cells using laser force analysis. Analyst 2014, 139, 1472–1481. [Google Scholar] [CrossRef]
- Hebert, C.G.; Hart, S.; Leski, T.A.; Terray, A.; Lu, Q. Label-Free Detection of Bacillus anthracis Spore Uptake in Macrophage Cells Using Analytical Optical Force Measurements. Anal. Chem. 2017, 89, 10296–10302. [Google Scholar] [CrossRef]
- Kaneta, T.; Makihara, J.; Imasaka, T. An “Optical Channel”: A Technique for the Evaluation of Biological Cell Elasticity. Anal. Chem. 2001, 73, 5791–5795. [Google Scholar] [CrossRef] [PubMed]
- Hebert, C.G.; Terray, A.; Hart, S.J. Toward Label-Free Optical Fractionation of Blood-Optical Force Measurements of Blood Cells. Anal. Chem. 2011, 83, 5666–5672. [Google Scholar] [CrossRef] [PubMed]
- Hebert, C.G.; Rodrigues, K.L.; DiNardo, N.; Hachmann, A.B. Viral Infectivity Quantification and Neutralization Assays Using Laser Force Cytology. In Vaccine Delivery Technology: Methods and Protocols; Pfeifer, B.A., Hill, A., Eds.; Springer: New York, NY, USA, 2021; pp. 575–585. [Google Scholar]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Leroux-Roels, G.; Bonanni, P.; Tantawichien, T.; Zepp, F. Vaccine Development, in Understanding Modern Vaccines. Perspect. Vaccinol. 2011, 1, 115–150. [Google Scholar] [CrossRef]
- Struck, M.M. Vaccine R&D success rates and development times. Nat. Biotechnol. 1996, 14, 591–593. [Google Scholar]
- World Health Organization. Manual of Laboratory Methods for Testing of Vaccines Used in the WHO Expanded Programme on Immunization; World Health Organization: Geneva, Switzerland, 1997.
- Food and Drug Administration, Center for Biologics Evaluation and Research. Guidance for Industry on Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications; United States Department of Health and Human Services, Ed.; Office of Communication, Outreach and Development (OCOD) (HFM-40): Rockville, MD, USA, 2010.
- Petrova, A.B.; Herold, C.; Petrov, E.P. Conformations and membrane-driven self-organization of rodlike fd virus particles on freestanding lipid membranes. Soft Matter 2017, 13, 7172–7187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherstvy, A.G.; Petrov, E.P. Petrov, Modeling DNA condensation on freestanding cationic lipid membranes. Phys. Chem. Chem. Phys. 2014, 16, 2020–2037. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | DPI | Radiance Infection Metric (% OFI > 55 s−1) | CytoFLEX Surrogate Potency (vp/Viable Cell) | Calculated Potency (vp/Viable Cell) | Log10 Difference |
|---|---|---|---|---|---|
| Exp 1 | 3 | 11.53 | 3.15 × 103 | 3.57 × 103 | 0.055 |
| 4 | 23.52 | 6.86 × 103 | 5.87 × 103 | −0.068 | |
| 5 | 40.92 | 1.57 × 104 | 1.20 × 104 | −0.115 | |
| Exp 2 | 3 | 9.49 | 3.17 × 103 | 3.29 × 103 | 0.015 |
| 4 | 22.60 | 4.56 × 103 | 5.65 × 103 | 0.093 | |
| 5 | 42.57 | 8.73 × 103 | 1.29 × 104 | 0.169 | |
| 6 | 54.02 | 2.33 × 104 | 2.07 × 104 | −0.052 | |
| Exp 3 | 3 | 28.12 | 7.05 × 103 | 7.10 × 103 | 0.003 |
| 4 | 29.69 | 1.12 × 104 | 7.57 × 103 | −0.169 | |
| 5 | 41.48 | 1.16 × 104 | 1.23 × 104 | 0.025 | |
| 6 | 65.92 | 3.05 × 104 | 3.38 × 104 | 0.045 | |
| Absolute Average | 0.074 | ||||
| Experiment | DPI | Radiance Infection Metric (% OFI > 55 s−1) | CytoFLEX Surrogate Potency (vp/Viable Cell) | Calculated Potency (vp/Viable Cell) | Log10 Difference |
|---|---|---|---|---|---|
| Exp 1 | 3 | 21.06 | 5.72 × 102 | 5.29 × 102 | −0.034 |
| 4 | 43.19 | 2.26 × 103 | 1.68 × 103 | −0.129 | |
| 5 | 54.78 | 4.41 × 103 | 3.09 × 103 | −0.155 | |
| Exp 2 | 3 | 19.72 | 4.95 × 102 | 4.93 × 102 | −0.002 |
| 4 | 35.46 | 9.71 × 102 | 1.12 × 103 | 0.064 | |
| 5 | 45.07 | 1.45 × 103 | 1.86 × 103 | 0.108 | |
| 6 | 72.13 | 6.80 × 103 | 7.66 × 103 | 0.052 | |
| Exp 3 | 3 | 34.24 | 4.96 × 102 | 1.05 × 103 | 0.328 |
| 4 | 27.07 | 1.06 × 103 | 7.25 × 102 | −0.166 | |
| 5 | 37.75 | 1.41 × 103 | 1.27 × 103 | −0.046 | |
| 6 | 54.44 | 3.18 × 103 | 3.04 × 103 | −0.020 | |
| Absolute Average | 0.100 | ||||
| Experiment 1 | |||||
|---|---|---|---|---|---|
| Input MOI (TCID50/Cell) | Level (µL) | Radiance® Infection Metric (% Cells with OFI > 35 s−1) | Calculated MOI (TCID50/Cell) | Calculated Titer (TCID50/mL) | Log10 Difference from Stock |
| Stock | N/A | N/A | N/A | 8.20 × 106 | N/A |
| 4.100 | 100 | 26.9 | 5.299 | 1.06 × 107 | 0.111 |
| 1.640 | 40 | 20.1 | 1.694 | 8.47 × 106 | 0.014 |
| 0.328 | 8.0 | 12.6 | 0.276 | 6.90 × 106 | −0.075 |
| 0.066 | 1.6 | 7.5 | 0.037 | 4.57 × 106 | −0.253 |
| 0.013 | 0.32 | 5.7 | 0.013 | 8.15 × 106 | −0.003 |
| 0.003 | 0.064 | 4.3 | 0.004 | 1.33 × 107 | 0.211 |
| Average | 8.67 × 106 | 0.024 | |||
| Experiment 2 | |||||
| Stock | N/A | N/A | N/A | 6.81 × 106 | N/A |
| 3.407 | 100 | 44.3 | 3.227 | 6.45 × 106 | −0.024 |
| 1.363 | 40 | 37.3 | 1.405 | 7.03 × 106 | 0.013 |
| 0.273 | 8.0 | 27.0 | 0.295 | 7.39 × 106 | 0.035 |
| 0.055 | 1.6 | 18.7 | 0.051 | 6.33 × 106 | −0.032 |
| 0.011 | 0.32 | 13.7 | 0.011 | 6.91 × 106 | 0.006 |
| Average | 6.82 × 106 | 0.001 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCracken, R.; Al-Nazal, N.; Whitmer, T.; Yi, S.; Wagner, J.M.; Hebert, C.G.; Lowry, M.J.; Hayes, P.R.; Schneider, J.W.; Przybycien, T.M.; et al. Rapid In-Process Measurement of Live Virus Vaccine Potency Using Laser Force Cytology: Paving the Way for Rapid Vaccine Development. Vaccines 2022, 10, 1589. https://doi.org/10.3390/vaccines10101589
McCracken R, Al-Nazal N, Whitmer T, Yi S, Wagner JM, Hebert CG, Lowry MJ, Hayes PR, Schneider JW, Przybycien TM, et al. Rapid In-Process Measurement of Live Virus Vaccine Potency Using Laser Force Cytology: Paving the Way for Rapid Vaccine Development. Vaccines. 2022; 10(10):1589. https://doi.org/10.3390/vaccines10101589
Chicago/Turabian StyleMcCracken, Reilly, Noor Al-Nazal, Travis Whitmer, Sijia Yi, James M. Wagner, Colin G. Hebert, Matthew J. Lowry, Peter R. Hayes, James W. Schneider, Todd M. Przybycien, and et al. 2022. "Rapid In-Process Measurement of Live Virus Vaccine Potency Using Laser Force Cytology: Paving the Way for Rapid Vaccine Development" Vaccines 10, no. 10: 1589. https://doi.org/10.3390/vaccines10101589
APA StyleMcCracken, R., Al-Nazal, N., Whitmer, T., Yi, S., Wagner, J. M., Hebert, C. G., Lowry, M. J., Hayes, P. R., Schneider, J. W., Przybycien, T. M., & Mukherjee, M. (2022). Rapid In-Process Measurement of Live Virus Vaccine Potency Using Laser Force Cytology: Paving the Way for Rapid Vaccine Development. Vaccines, 10(10), 1589. https://doi.org/10.3390/vaccines10101589