1. Introduction
Viticulture (
Vitis vinifera L.) is one of the world’s most important agricultural activities, producing up to 77 million tons of grapes every year. Around 75–80% of this crop is used to produce wine, which, with a demand of 25 billion liters per year, has earned the title of the most important alcoholic beverage in the world [
1]. The production process, mainly located in Europe and America, followed by Australia, China, and South Africa [
2], starts from the seasonal grape harvest and then proceeds to operations that produce the finished product, which can be diversified according to the various characteristics requested. However, winemaking also involves the parallel production of large quantities of processing waste, from the destemming, pressing, settling, and washing operations, and thus has a significant environmental impact and disposal costs. On the one hand, it has been estimated that for each liter of wine produced, one to several liters of wastewater are derived. On the other hand, the amount of solid residue resulting from this supply chain, which includes grape marc and stalks, is equally impressive, and reaches 5 tons per year for each hectare cultivated [
3,
4]. Stalks make up between 2.5% and 7.5% of the grape weight, depending on the grape cultivar and pressing methods, and about 13% and 16% of white- and red-wine solid waste, respectively. Besides being essentially composed of cellulose and lignin [
5,
6], the grape-bunch skeleton also contains lipophilic compounds (e.g., sterols and triterpenes) [
7] and polyphenols, which can be divided into flavonoids, e.g., flavonols, anthocyanins, and flavan-3-ols (monomer, dimers, oligomers, and polymers), and non-flavonoids, mainly phenolic acids and stilbenes [
8,
9]. All of these compounds can have a beneficial influence on human health, including anti-carcinogenic, anti-atherogenic, and anti-mutagenic effects; protection against cardiovascular diseases; and anti-inflammatory activity [
10]. Moreover, it has been shown that winery by-products can be a good source of natural preservatives, because they contain molecules with antibacterial and antioxidant properties, such as hydroxycinnamic acid derivatives, tannins, catechins, and the other above-mentioned phenols [
11]. The shelf-life of food products, especially when they contain oils and fats, can therefore be prolonged by the addition of natural and safe secondary metabolites of wine [
12].
Although the stalk fingerprints for the polyphenols composition and concentration depend on several factors, including grape variety, climate, time of harvest, and geographic origin, there is important commercial value in this type of purified bioactive molecule [
13]. In fact, we are currently witnessing a constant increase in the number of drugs, cosmetics, and nutraceutical-product formulations that are making a high antioxidant power, and therefore high polyphenolic content, one of their main strengths. The exploitation of this common waste as a negative cost feedstock may provide a significant economic advantage. Industrial waste valorization is now, therefore, a mandatory objective as a means to decrease pollution and simultaneously recover compounds with a high added-value, such as polyphenols, which would otherwise be lost, often with detrimental effects on the flora and fauna in the affected areas.
Agro-food industry waste treatment often involves the extraction and purification of natural compounds that can be employed in functional foods, nutraceuticals, bio-fuels, cosmetics, drugs, and other bio-products [
14,
15]. The management of winery and grape-juice residues is related to the high content of organic matter as well as the large amount generated over a short period of the year, with the consequent environmental problems of biological and chemical oxygen demand [
16]. The “green chemistry” concept is strictly connected to the prospect of a cleaner and more sustainable world, and includes any practice that aims to reduce the use of hazardous and polluting substances and any waste released into the environment, and thus supports any action that enables the reuse, treatment, and disposal of such materials. Synergistically, “green engineering” bases its principles on ensuring the design of plants with reduced risks, taking into account not only the reliability of the control systems, but also ensuring maximum safety. The 12 principles of green chemistry and the 12 principles of green engineering, which can be summarized by the acronyms “IMPROVEMENTS” and “PRODUCTIVELY”, respectively, can be combined in order to allow for improvements in production together with reliable scale-up criteria [
17].
The idea of optimizing extraction techniques, also when used to valorize agro-food wastes, fits perfectly with these concepts. It is important to revolutionize conventional procedures with the purpose of intensifying processes, while also enhancing their sustainability. Chemat et al. [
18] have indicated what needs to be done in designing a competitive extraction process, namely: innovation, flexibility, the use of alternative solvents (mainly water), reduction in energy consumption, co-product production instead of waste, process safety, limited operative units, and the avoidance of contaminants. Extreme attention must also be paid to the downstream separation and purification steps. Improving existing processes involves the use of enabling technologies, which can provide the two-fold effects of increasing efficiency, while greatly reducing the environmental and economic impact [
19]. As shown in the high number of recently published works (about 5000 only in the last 5 years), ultrasound-assisted extraction (UAE) is now fully recognized as a green technique by the pharmaceutical, cosmetic, and food industries [
20]. Only physical effects are exerted on an immersed/dispersed matrix when ultrasound (US) waves cross a medium at a frequency of 20–100 kHz, because they only exert a mechanical action at a low frequency and high intensity. In addition to the sonotrode used in the extraction processes, either immersion and cup-horn systems, a transducer is used to transform the signal of a generator into mechanical waves, with compression/rarefaction cycles in the US frequencies, giving rise to the well-known cavitation phenomenon. Several physical phenomena develop as a consequence of this effect, and macro-turbulence and micro-mixing are generated in the case of a liquid–solid system. The synergy of several independent factors, including fragmentation, erosion, capillarity, destructuring, and sonoporation, lead to the disruption of vegetal matrices and to a higher solvent-penetration coefficient, thus significantly improving mass transfer kinetics during an extraction procedure [
20,
21]. Operating times are reduced, the purity of the final product is increased, and, furthermore, the plant can be simplified by reducing the (L/S) ratio, leading to reductions in the solvent amount and the minimization of wastewater post treatment. It is worth noting that all of these goals can also be achieved when working at room temperature and using a fraction of the energy required for a conventional extraction process, which, in the case of solid waste from wine making, commonly involves alcohol or hydro alcoholic mixtures and heating above 90 °C [
22].
The grape stalks are essentially recycled as fertilizer [
23], and the possibility of their valorisation to produce health-promoting compounds on an industrial scale must still be investigated.
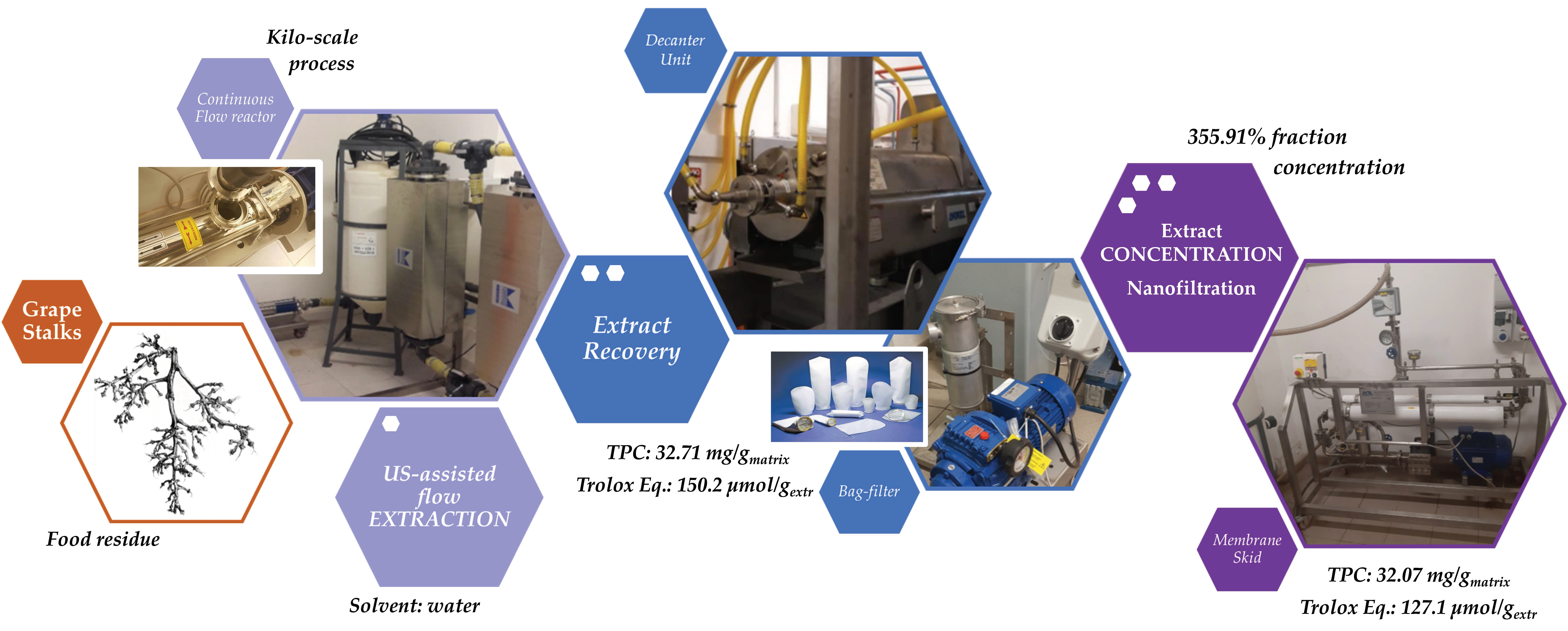
The present work has screened polyphenols UAE from grape stalks in water and has focused on extraction kinetics and scale-up factors. The total phenolic content (TPC) was chosen as the parameter to rapidly assess process efficiency. The results have been compared with those of the conventional extraction and of runs with different feedstock-size distributions. Peleg’s hyperbolic model was used to describe the UAE kinetic curves in lab-scale apparatus, and, consequently, to determine the best trade-off between the extraction rate and process time. The related volumetric mass transfer coefficient (kLa) was crucial for the investigation of the process scale-up design, up to an industrial-plant scale, with a multistage UAE section. A flow approach, in a 15 L reactor and using a 120 L tank in recirculating extraction, processed between 2 to 6 kg of grape stalks. The scaled-up protocol required dedicated facilities for the post-extraction treatment, such as a decanter unit and a bag-filter (150 µm mesh), and the resulting particle-free solution underwent treatment on a nanofiltration (NF) membrane pilot skid in order to give a concentrated polyphenols-rich fraction. Lab-scale extracts were characterized using HPLC-DAD, UPLC-ESI-MS/MS, DPPH• scavenging activity (2,2-diphenyl-1-picrylhydrazyl assay), TEAC assay (Trolox equivalent antioxidant capacity), and proanthocyanidin content determination. The antioxidant activity and TPC were used to follow the efficiency of the UAE scale-up from the lab- to pilot-scale.
2. Materials and Methods
2.1. Chemicals
EtOH (ACS grade, ≥99%), methanol (MeOH, HPLC, ≥99.9%), and acetone (ACS grade, ≥99%), used for the extractions and micro-assays, and acetonitrile, used for the HPLC analyses (MeCN, HPLC Plus, ≥99.9%), were purchased from Sigma-Aldrich (Sigma-Aldrich, Milan, Italy), while Milli-Q H2O was obtained in the laboratory using a Milli-Q Reference A + System (Merck Millipore, Darmstadt, Germany). Glacial acetic acid (AcOH, ≥96%) was purchased from Merck (Darmstadt, Germany).
Analytical standards of gallic; gentisic; o-hydroxybenzoic (salicylic); p-hydroxybenzoic; protocatechuic; vanillic; 3,4-dimethoxybenzoic (veratric); syringic, ellagic; cinnamic; o-, m-, and p-coumaric; caffeic; chlorogenic; ferulic; 3-hydroxy-4-methoxycinnamic; 3,4-dimethoxycinnamic; 3-methoxycinnamic; 4-methoxycinnamic; and sinapic acids, as well as (+)-ε-viniferin, (−)-epicatechin, quercetin-3-glucoside, apigenin-3-glucoside, and luteolin-3-glucoside for the HPLC analyses were purchased from Sigma-Aldrich. The Green Tea Catechin Mix (100 μg/mL caffeine, (+)-catechin, (−)-catechin 3-gallate, (−)-epicatechin, (−)-epicatechin-3-gallate, (−)-epigallocatechin 3-gallate, (−)-gallocatechin, and (−)-gallocatechin 3-gallate in acetonitrile:water (8:2) with 5% 1M HCl, Cerilliant®, Sigma Aldrich) was used for the HPLC analyses of catechins.
A procyanidin B2 standard, sodium metabisulfite, n-butanol, and hydrochloric acid for the proanthocyanidin content determination; Trolox® and the 2,2-diphenyl-1-picrylhydrazyl radical (DPPH•) for antioxidant activity determination; and the Folin–Ciocalteu reagent, sodium carbonate, and DMSO for the total polyphenol content (TPC) determination were all purchased from Sigma Aldrich.
2.2. Matrix Analysis
Dried and milled grape stalks were kindly provided by Cantine Ascheri Giacomo (Bra-CN, Italy), obtained by mechanical separation from grapes. The resulting material was not submitted to any dedicated drying process. The matrix was stored at room temperature until the extraction procedures.
The water content and the organic and ashes fractions were obtained by means of thermogravimetric analysis. First, 200 mg of raw grape stalks were maintained at 100 °C over night, in order to register the biomass water content as a mass loss, and then for 4 h at 650 °C so as to allow for discriminating between the organic and inorganic residual content.
The matrix granulometry was analyzed by means of physical separation. An average quantity of 7.5 g of grape stalks was passed through sieves with decreasing mesh sizes (Giuliani, Italy 1000, 500 and 212 µm). The different weights were registered and applied for percentage compositions.
2.3. Lab-Scale Ultrasound-Assisted Extraction (l-UAE)
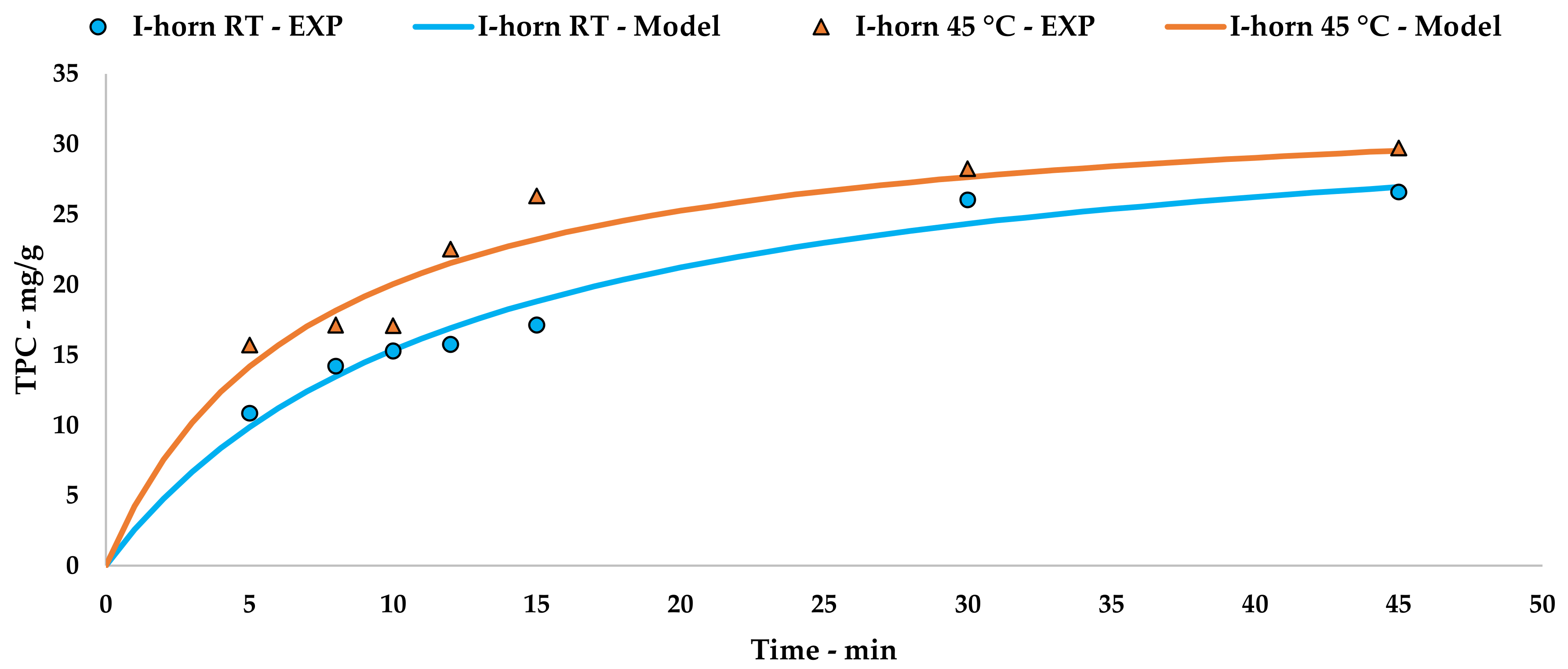
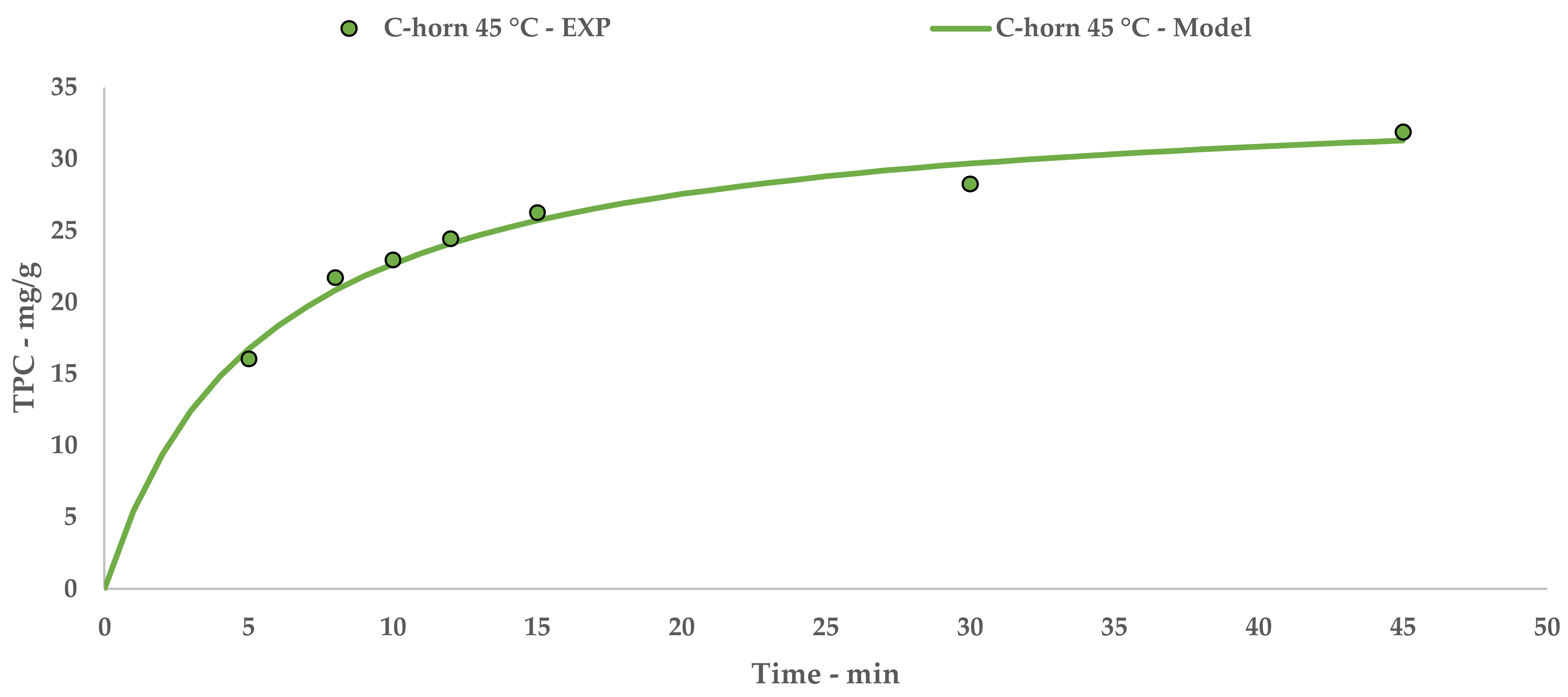
l-UAE was performed using two different US devices, an immersion horn (I-horn, HNG-20500-SP, Hainertec Suzhou, China) and a cup-horn (C-horn, PEX1, R.E.U.S., France). The two systems work under different conditions, namely: 500W of power at a frequency of 21 kHz and 200 W of power at a frequency of 25 kHz, respectively.
In the function of the extraction vessel volume, the experiments were performed by mixing 7.5 g of grape stalks (<1000 µm and 212 µm) with the correct amount of deionised water in order to achieve an S/L ratio of 1:20. For the I-horn, the solution was placed in a Pyrex® thimble (r: 4 cm and h: 19 cm), while the C-horn did not require other glassware. The temperature was registered throughout the UAE, and heat exchange was provided using two different approaches—an ice bath for the I-horn and a cooling jacket with a continuous flow of tap water for the C-horn. Where required, an extraction temperature of 45 °C was achieved via US-derived heating for a few minutes. For this purpose, the cooling system was removed until the fixed limit was reached.
After extraction, the solutions were vacuum filtered, and the matrices were thoroughly washed with deionized water. The crude extracts were then freeze dried (LyoQuest–85, Telstar, Spain), and the dry material was weighted and analysed for the total polyphenol content (TPC) determination. The optimized extract (C-horn, 45 °C, 45 min) was also characterized using HPLC analyses, antioxidant activity, and oligomeric proanthocyanidin (OPC) content assays.
2.4. Conventional Extractions
For the sake of comparison, three types of conventional approaches were also carried out, namely: (1) silent extraction, (2) water reflux multi-step extraction, and (3) a reflux hydroalcoholic benchmark. For (1), the optimized parameters for l-UAE were exactly reproduced without US irradiation. In approach (2), 7.5 g of stalks were mixed with 150 mL of deionised water (S/L 1:20), and were extracted for 180 min at a reflux temperature. The matrix was then recovered, washed, and extracted twice under the same conditions. The crude extracts derived from procedures (1) and (2) were freeze dried and analysed separately for the TPC. Finally, extraction (3) was conducted by reproducing the hydroalcoholic extraction as found in the literature [
22] for <1000 µm and 212 µm granulometries. Then, 7.5 g of the selected matrix was mixed with 150 mL of a hydroalcoholic solution (S/L ratio of 1:20, EtOH/H
2O 60:40), and heated for 23 min at 95 °C under stirring. The matrix was then filtered and washed three times with a fresh extraction solvent. The crude extracts underwent the sequential removal of EtOH and water by means of rotary evaporation and lyophilisation, respectively. The dry product was analyzed for TPC determination, and for the reference sample (hydroalcoholic mixture), HPLC analyses, antioxidant activity, and OPC assays were performed.
2.5. Scale-Up Ultrasound-Assisted Extraction (S-UAE)
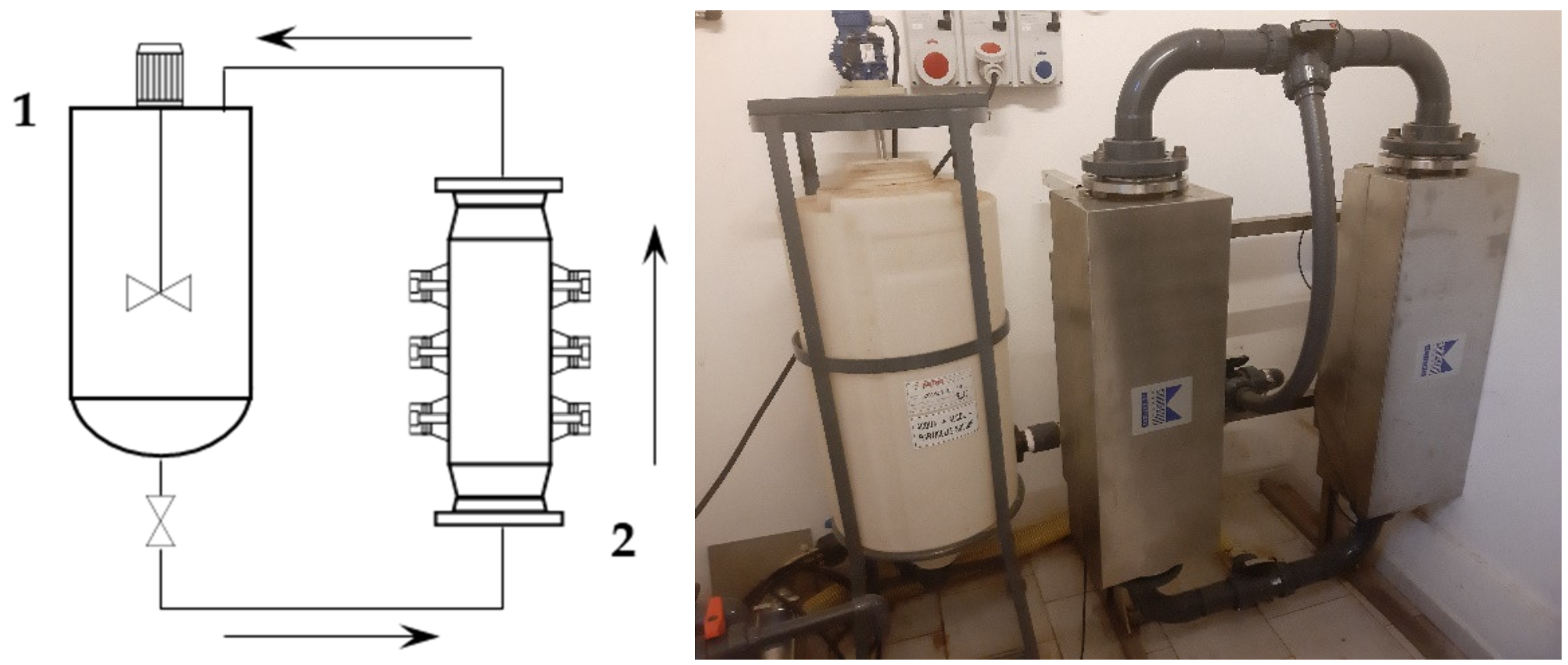
S-UAE was performed using a Biopush flow-through cell reactor from Weber Ultrasonics GA (Germany), which is able to simultaneously process 15 L of solution under irradiation at up to 2000 W, at a frequency of 29 kHz. In order to reflect the information obtained under the l-UAE optimized conditions as closely as possible, the system was used in a loop configuration. The loop also included a squared section (460 × 460 mm) with a 45° pyramidal bottom 120 L tank, equipped with an impeller with four inclined blades (blade inclination: 45, impeller diameter: 200 mm, impeller position: 110 mm above pyramidal cone starting plane, installed power: 0.18 kW, rotational speed: 1340 rpm) to form the solid–liquid suspension and to maintain it in a homogeneous state, so that it could be pumped by the dedicated progressing cavity pump (MAE 50-1/AA. T33A, CSF, Italy).
First, 2 kg of sieved grape stalks (<1000 µm) were mixed with water at 45 °C to reach the desired S/L ratio of 1:30, for a total addition of 60 L. The pump speed was set to 300 rpm, resulting in a flow rate of 30 L/min. The recirculated solution entered the middle section of the tank, and thus provided additional suspension mixing together with the blade mixer. The tank acted as a 45 L buffer while 15 L were processed. The ultrasonic reactor BioPush Weber, with a generator input power of 2000 W, was used for all of the S-UAE, thus allowing for an automatic time control to be set. Then, 50–100 mL of samples were collected at fixed times. A schematic representation and relative picture can be found in
Figure 1.
At the end of the sonication, the suspension was pumped into a dedicated 250 L tank and the extraction system was thoroughly washed. The resulting solution was separated by means of an F2000 decanter (Andritz, The Netherlands), running at a differential speed of 7 rpm and a pipette position of 10 cm. The system was fed with a 500 kg/h flow, and was finally washed with water to complete the extract recovery.
The solid stalk residues were collected below the decanter, while the liquid fraction was recovered in a separate tank and sent to concentration through NF. The samples were collected and freeze dried according to the l-UAE protocol. All of the collected samples were dried and then analyzed for TPC determination.
2.6. Nanofiltration Concentration (NF Concentration)
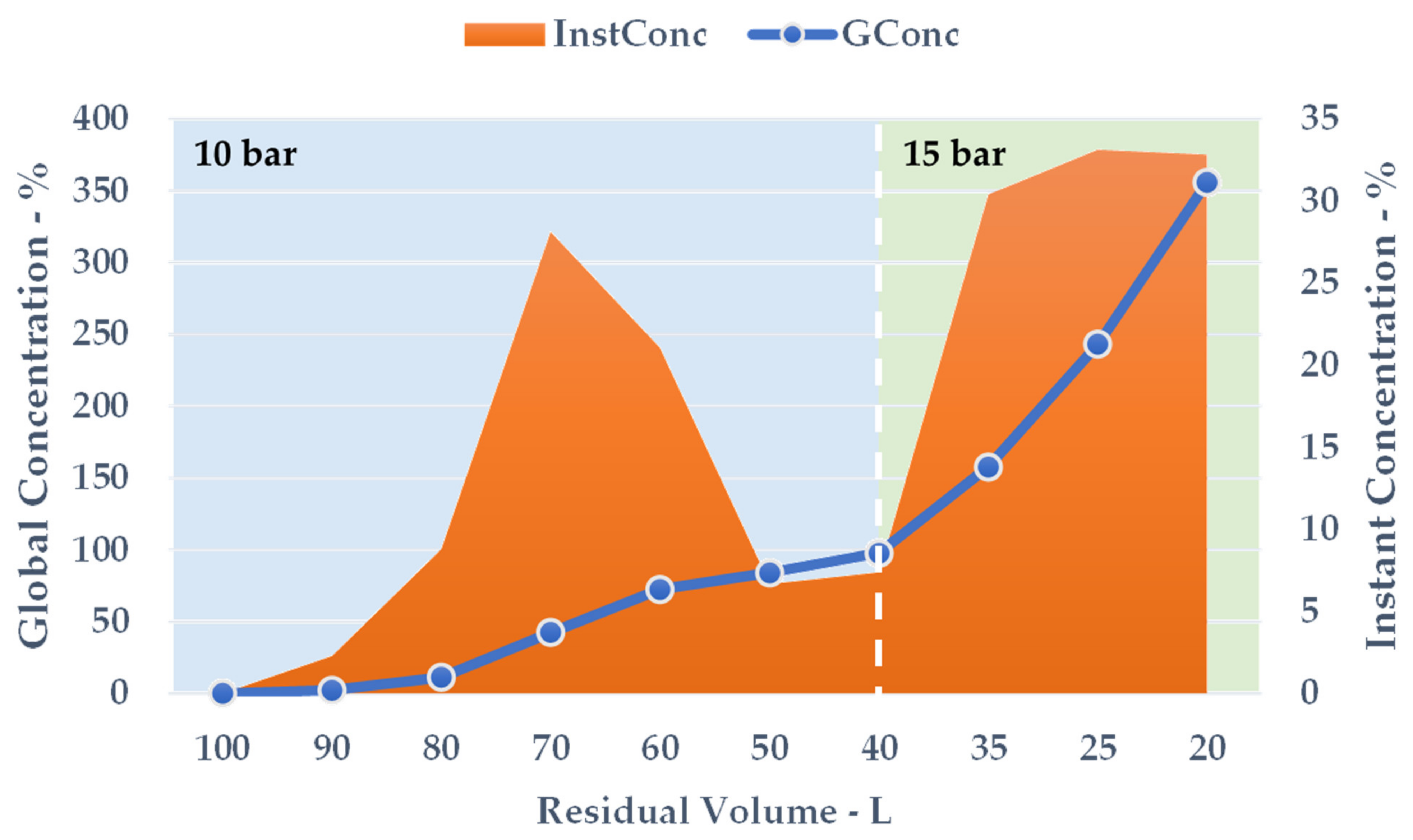
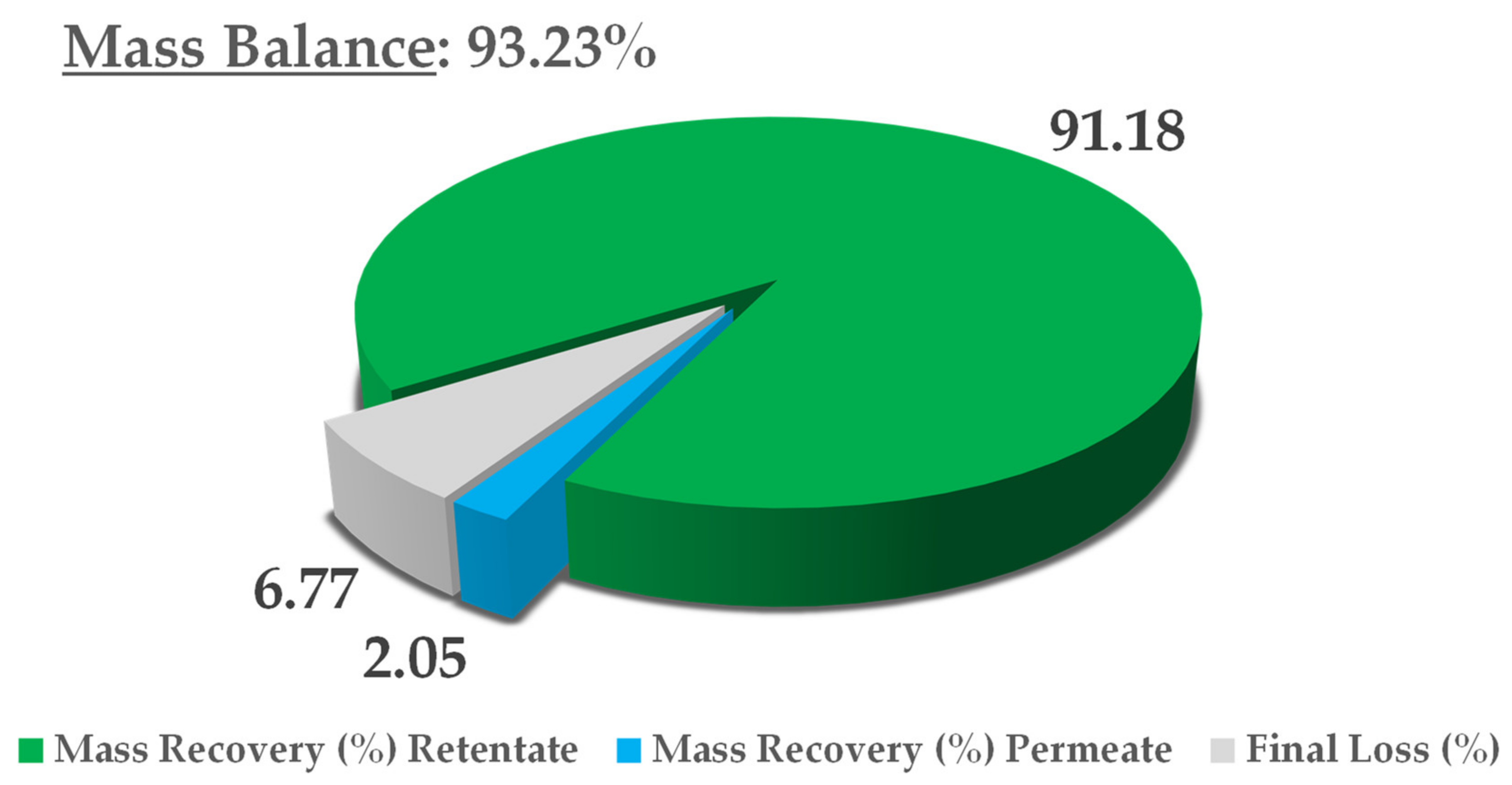
The crude extract recovered from S-UAE amounted to approximately 100 L, and was processed by NF performed on a HARI P2B200-400 membrane skid (HydroAir Research, Lodi, Italy) that was equipped with a DLU 19 F cartridge (2.5 m2) with an approximative MWCO (molecular weight cut-off) span of 150–300 Da. The flow rate was set to 1000 L/h by means of a by-pass and counter-pressure regulation. The latter was raised to 10 bar and maintained until a constant output was observed. Once the residual volume reached 40 L, the pressure was increased to 15 bar for the remaining time. The temperature was monitored, and ranged from 25 to 30 °C. The process was concluded when precipitation due to solvent removal was incipient, at a residual volume of roughly 20 L. The concentrated fraction corresponded to the retentate stream, continuously recycled into the feed tank, while the removed aqueous solution converged into the permeate output. The retentate was collected during the process and the dry extract (DE) yield was derived by freeze drying. The NF trend was defined by the percentage global concentration (GConc) and instant concentration (InstConc). GConc considered t = 0 dry yield as a reference, while InstConc provided a ratio between two adjacent samples. An aliquot of the permeate was also freeze dried so as to determine the mass balance.
2.7. Total Phenolic Content (TPC)
TPC was determined according to the method developed by Cicco et al. [
24]. Quantification was carried out using a standard curve of gallic acid as the reference phenolic compound, at dilutions of between 5 and 250 µg/mL in a H
2O/DMSO 1:1 mixture. Dried samples were dissolved in a H
2O/DMSO 1:1 mixture at a concentration of nearly 0.5 mg/mL. The gallic acid and extract solutions (250 µL) were placed into test tubes. The following solutions were added sequentially to each tube: 250 µL of Folin–Ciocalteu (diluted 1:1 with distilled H
2O), 500 µL of a 10%
p/v Na
2CO
3 solution, and 4 mL of distilled H
2O. The resulting solution was vigorously shaken and left at room temperature for 25 min prior to the analysis. The absorption of the final mixtures was measured at 740 nm, in a 1 cm cuvette using a Cary 60 UV-VIS spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). These conditions provided the assay with a high accuracy and reproducibility. The TPC was expressed as gallic acid equivalents (GAE; mg/g) over the dried matrix (DM). All of the analyses were performed in triplicate and were expressed as averages. The standard errors (SE) are specified in the tables at confidence level of 0.95.
2.8. Peleg Method
Peleg’s hyperbolic model (see Equation (1)) [
25] was applied to evaluate the extraction kinetics and to determine the point of maximum extraction rate by means of the related constants.
C(t) is the concentration of the extract after extraction time t, while C
0 is equal to 0 at the beginning of the process. The Peleg initial extraction rate (k
1) is correlated to the starting extraction rate (B
0, Equation (2)), and can be used to calculate the relative extraction rate at each moment of the extraction (B
t).
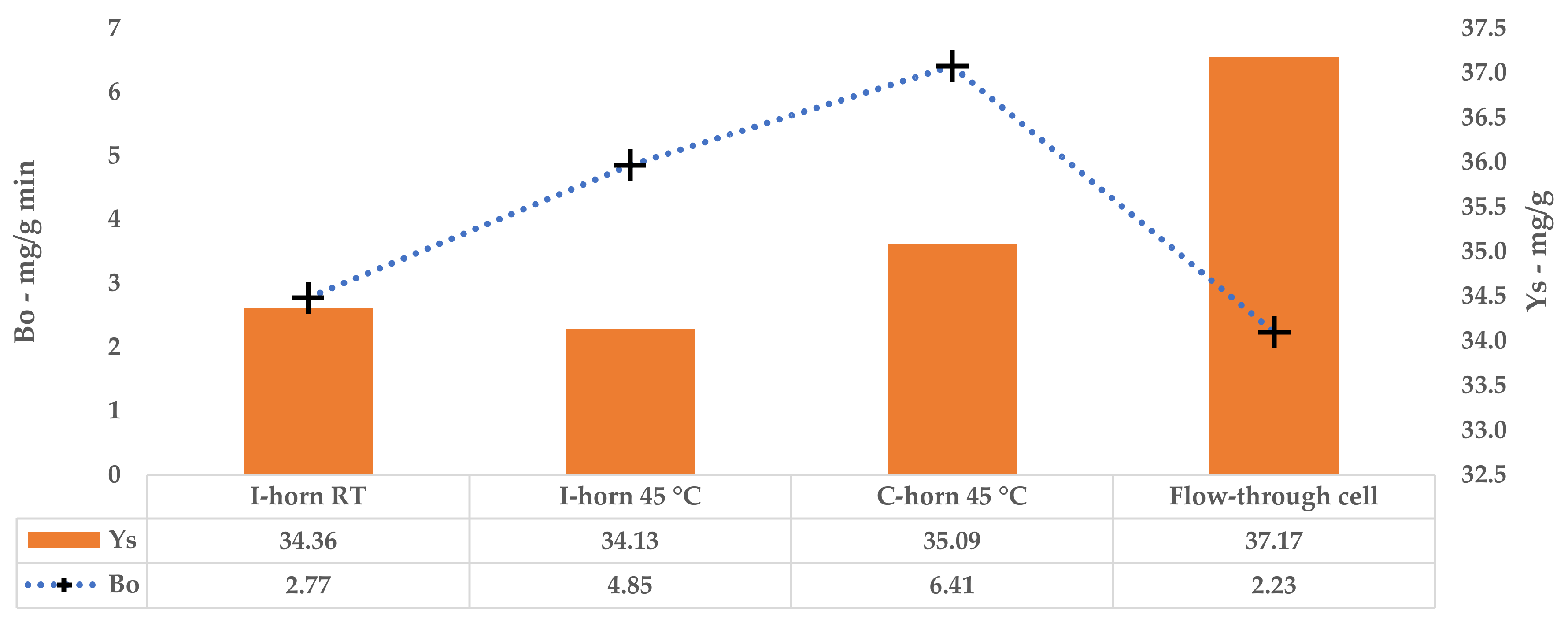
Bo allows the maximum extraction rate achievable when the mass-transfer driving force is at its maximum to be calculated (i.e., when the difference in the solute concentration in the solid matrix and in the liquid extractant is at its maximum), which is an important factor for cross-flow and counter-current industrial-scale extraction processes.
The Peleg capacity constant (k
2) is correlated to the highest extraction yield achievable at the steady state with a single equilibrium stage (Y
s, Equation (3)). It is also important data that are needed to define the number of extraction stages that the industrial process must have in order to achieve the required overall extraction yield. From a graphic point of view, this parameter represents a horizontal asymptote.
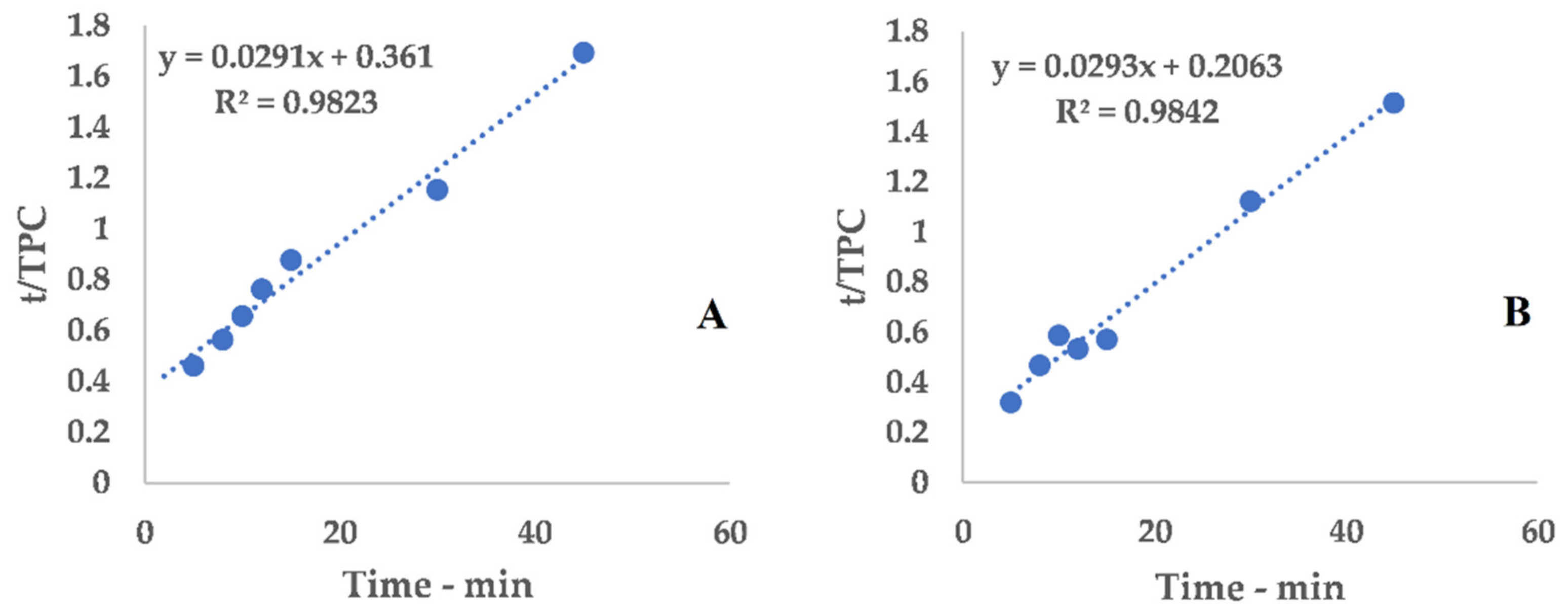
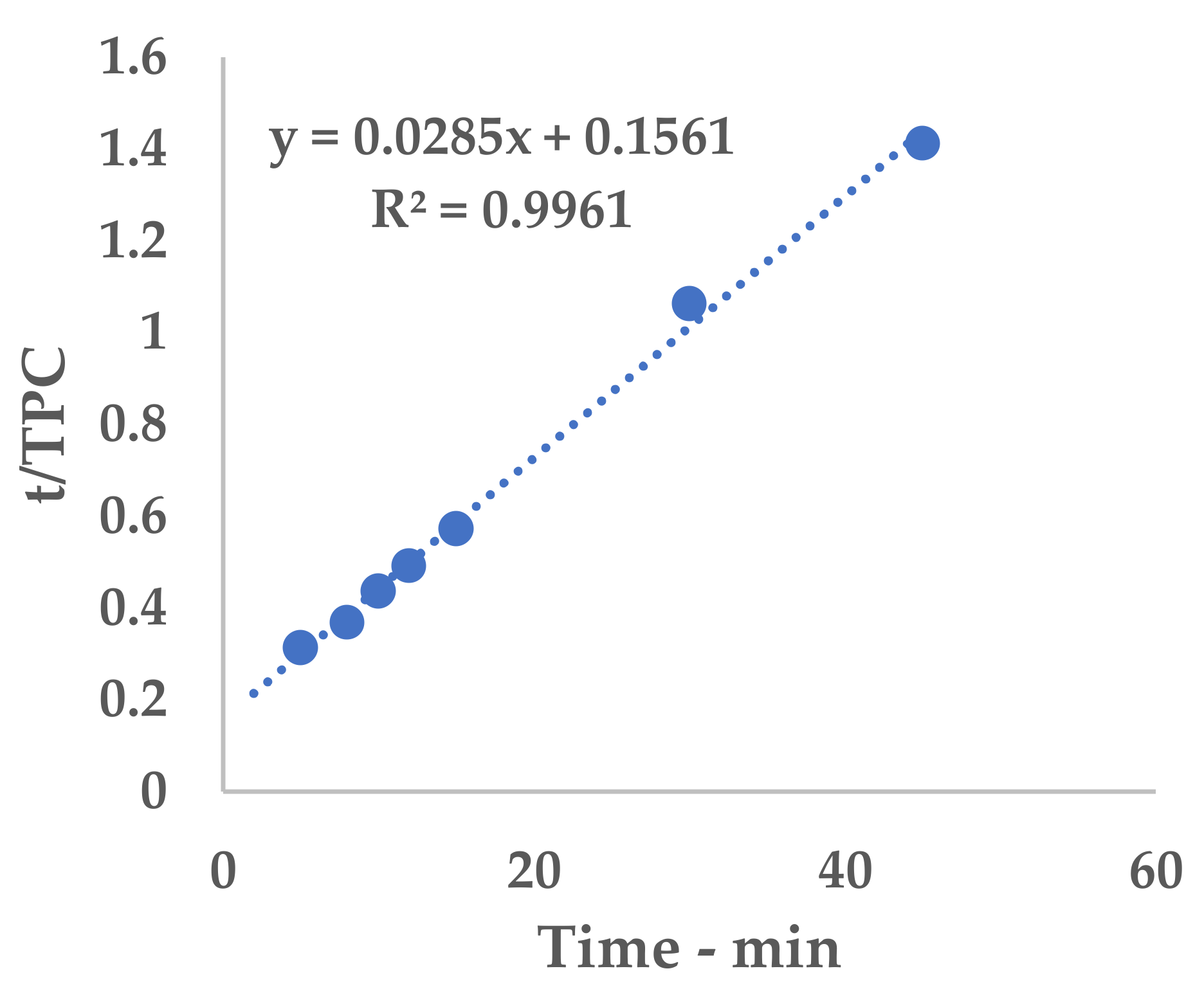
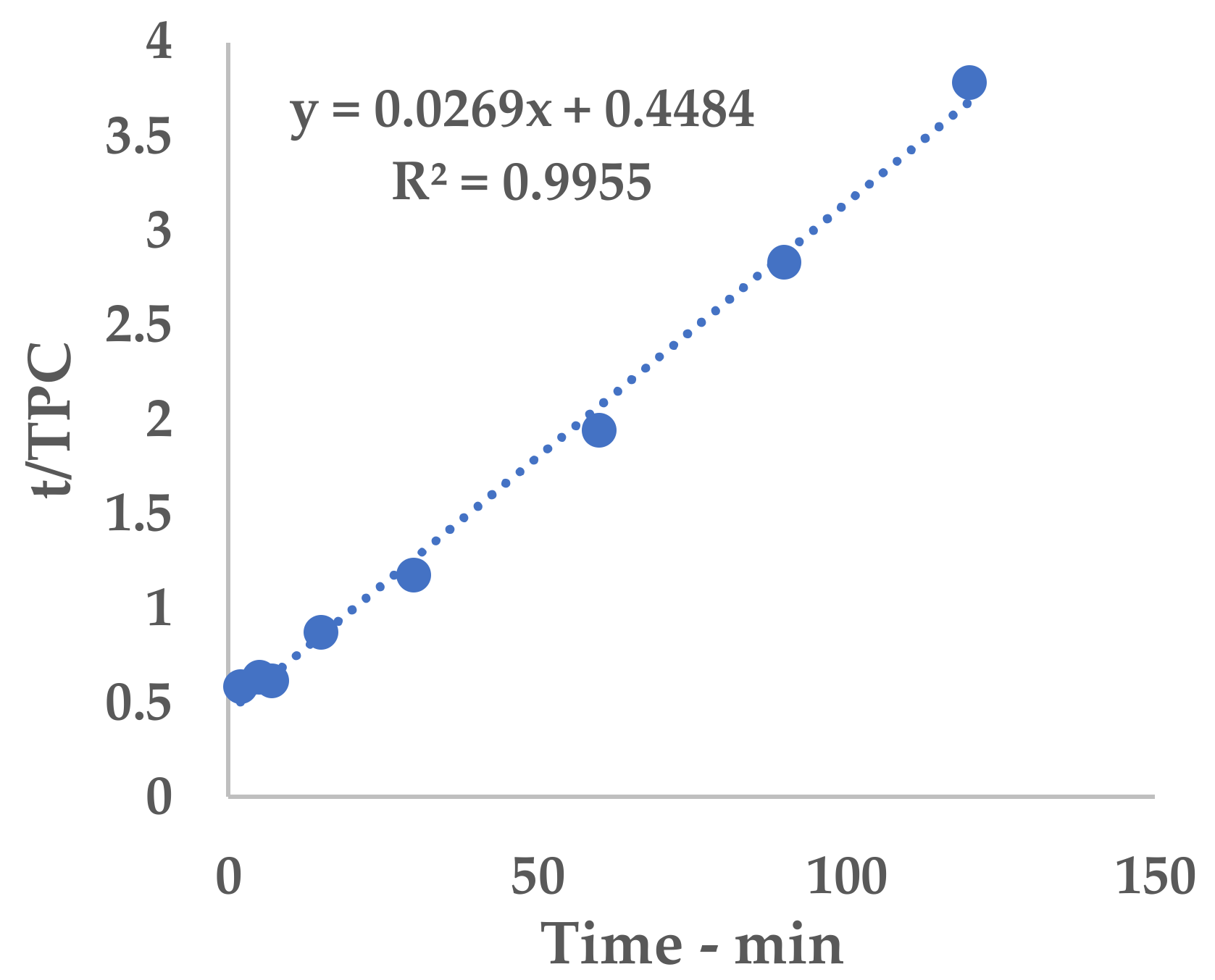
Equation (1) can be conveniently linearized into Equation (4), thus providing a fast and easy way to extrapolate k
1 and k
2 as the intercept and slope, respectively. The kinetic constants can be calculated via the linear interpolation of the experimental yields at different extraction times (see
Table A1 and
Figure A1 for 6 mm, and
Table A2 and
Figure A2 for 2 mm), and then filled into Equation (1).
The obtained hyperbolic curve describes a time-dependent extraction trend. This model is useful to display the horizontal asymptote of Y
s and the extraction rates (slope of the curve). Furthermore, the so-called “knee-point” can be exploited to determine the best trade-off between productivity and the process extent. This graphical feature can be identified where the extraction efficiency starts to steeply increase and it no longer worth the time consumption. In the present work, the abovementioned parameter could not be directly applied because of the necessary technological transposition (see
Section 3.5).
2.9. Antioxidant Activity (DPPH• Radical Scavenging Method and TEAC)
The radical scavenging ability of the extracts was evaluated using the stable free radical DPPH
•, according to the method described by Brand-Williams, Cuvelier, and Berset [
26]. Details of the procedure and calculations have been reported by Boffa et al. [
27]. The bleaching rate of the DPPH
• radical was monitored in the presence of solutions of grape-stalks extracts, as well as in that of Trolox
® (antioxidant standard for TEAC determination), in order to calculate the EC
50 (half maximal effective concentration or amount of compound/extract necessary to decrease the initial concentration of DPPH
• to 50% at equilibrium). The radical scavenging activity of the Trolox
® solutions was measured in concentrations from 1 to 18 µg/mL, corresponding to 0.004, 0.008, 0.016, 0.020, 0.028, 0.040, 0.048, and 0.072 µmol/mL. Various concentrations of grape-stalk extracts, between 5 and 500 µg/mL, were also tested. All of the samples were prepared in triplicate, and the DPPH
• radical scavenging activity was expressed as µg compound/dried extract per mL solution ± standard deviation. The Trolox
® equivalents (TE) µmol/g of the extract were calculated according to the EC
50 values.
2.10. Oligomeric Proanthocyanidin Content (OPC)
The OPC in the extracts obtained using l-UAE (C-horn, 45 °C, 45 min) and conventional hydroalcoholic extraction (23 min, 95 °C under stirring) was quantified using the butanol-HCl method with procyanidin B2 as a reference [
28]. Briefly, the procedure by Schofield et al. includes the serial dilution of samples (or standard), followed by the addition of an acid butanol reagent (
n-butanol/concentrated HCl, 95:5 by volume) and iron reagent (2% ferric ammonium sulphate dodecahydrate salt, FeNH
4(SO
4)
2·12H
2O in HCl 2 M). After 50 min at 95 °C, the absorbance was registered at 550 nm on a Cary 60 UV-VIS spectrophotometer (Agilent Technologies, Santa Clara, CA, USA).
2.11. HPLC-DAD Analysis
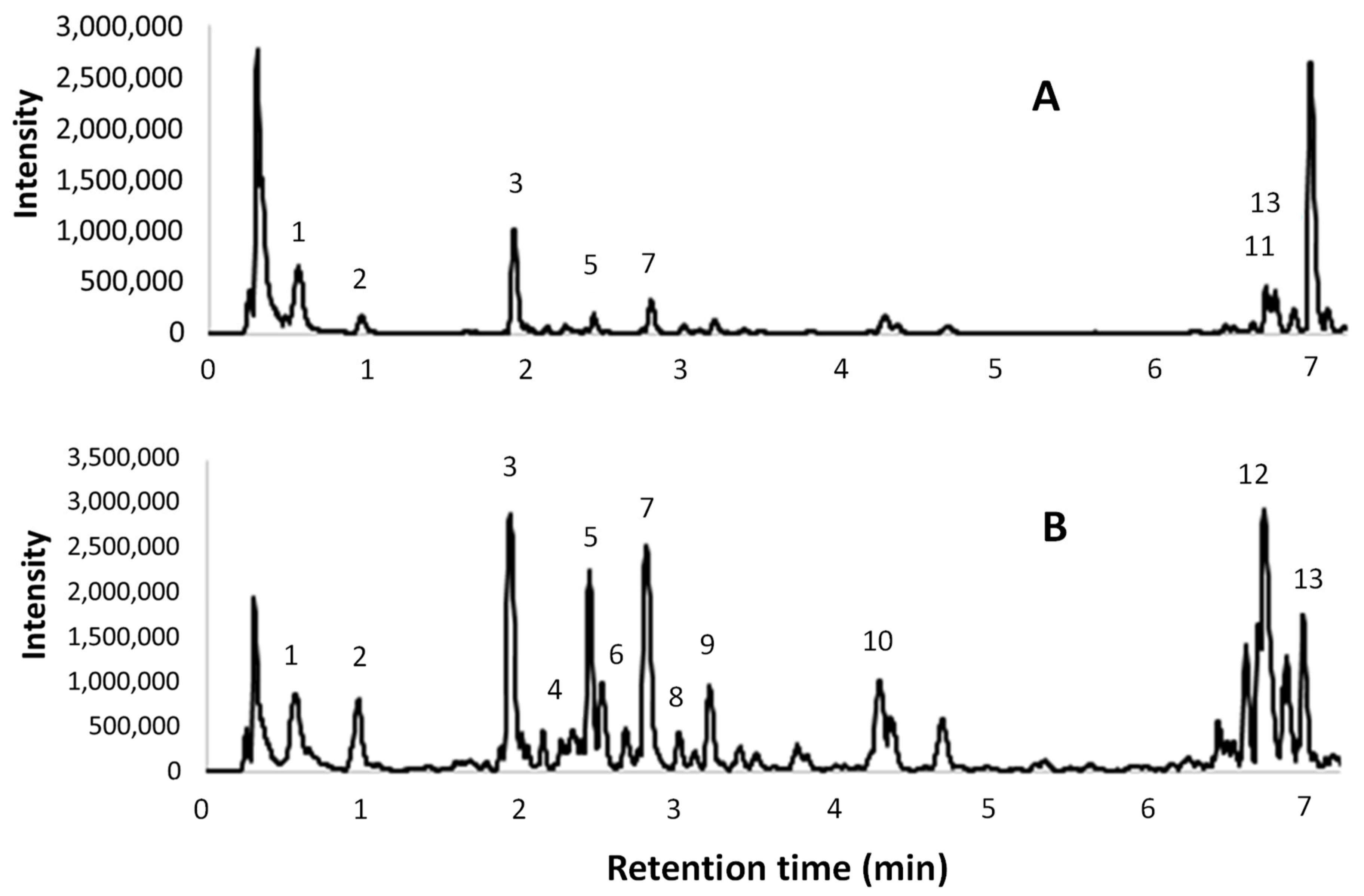
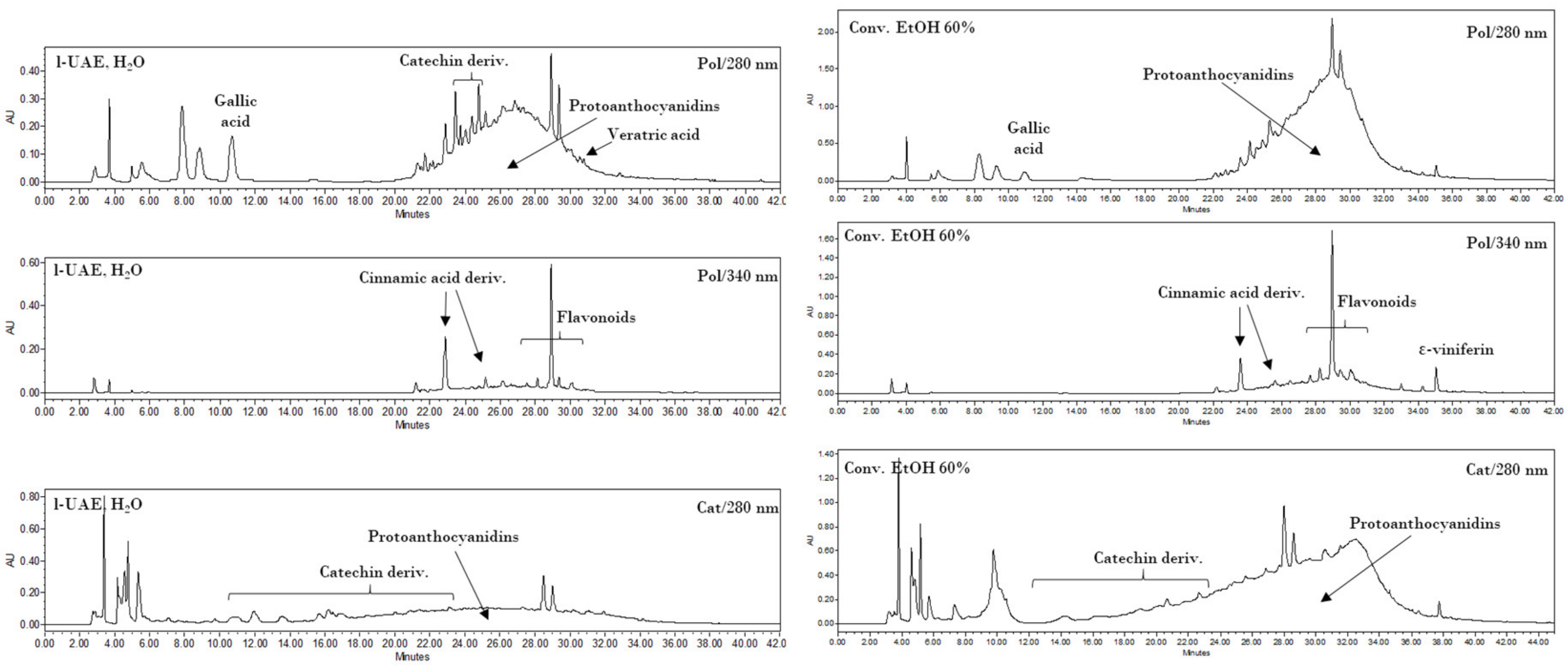
The HPLC analyses were performed on a Waters 1525 pump linked to a 2998 PDA (Waters Corp., Milford, CT, USA), using a Synergi Hydro RP C18 column (250 mm, 4.6 mm, 4 μm; Phenomenex, Torrance, CA, USA), with 2% AcOH (A) and MeCN (B) as the mobile phases. The monitored wavelengths were 280 (benzoic acids, catechins, and proanthocyanidins detection) and 340 nm (cinnamic derivatives and flavonoids detection), while three-dimensional data were acquired in the 200–600 nm range. Two different methods were used. The first gradient program (Pol) started from 0% B, which was maintained for 6.5 min, up to 50% B over the 6.5–30 min period, from 50% to 100% B for 30–36 min, followed by a 100% B step at 36–42 min. The second method (Cat) started from 8% B, which was maintained for 3 min, up to 50% B over the 3–21 min period, from 50% to 100% B for 21–33 min, followed by a 100% B step at 33–45 min.
Several compounds were screened, as described in
Section 2.1. The quantification of polyphenols was performed using calibration curves obtained with external standards. The analytical standard solutions were analysed using HPLC (20 μL injection) to give linear regressions with
R2 > 0.999. The equation curves, the wavelength used for the quantification, R
2, linearity range, limit of detection (LOD), and limit of quantification (LOQ) for each standard analysed are indicated in
Appendix A. The extract samples (l-UAE, C-horn, and conventional hydroalcoholic extraction) were dissolved in H
2O/MeOH 8:2 before injection (20 μL), giving concentrations of 5–10 mg/mL.
2.12. UPLC-ESI-MS/MS Analysis of Polyphenols
An Acquity UPLC system (Waters Corp., Singapore), coupled with a quadrupole-time of flight (Q-TOF) MS instrument (UPLC/Synapt Q-TOF MS, Waters, Milford, MA, USA) and an electrospray ionization (ESI) source, was used. The UPLC column (2.1 mm × 50 mm i.d., 1.7 µm, BEHC18, Waters Acquity) was used at a flow rate of 0.35 mL∙min
−1. The mobile phases were water with 0.1% formic acid (A) and acetonitrile (B). The gradient program was 0–1 min, 5–15% (B); 1–5 min, 20–25% (B); 5–6 min, 25–75% (B); and 6–7 min, 75–80%. The injection volume was 1 μL. The major operating parameters for Q-TOF MS were set as follows: capillary voltage, 2.5 kV; cone voltage, 60 V; cone gas flow, 100 L/h; source temperature, 120 °C; desolvation temperature, 400 °C; collision gas, argon; desolvation gas, nitrogen; flow rate, 600 L/h; data-acquisition range,
m/z 50–1200 Da; and ionization mode, negative. The detected compounds were identified by means of MS fragmentations, according to the data reported from a previous work [
29].
2.13. Statistical Analysis
The statistical analyses were performed using R software (R Foundation for Statistical Computing, Vienna, Austria), version 3.6.3 (2020-02-29). The analyses were all performed in triplicate. Residuals were checked for normality using the Shapiro–Wilk and Kolmogorov–Smirnov tests, and variances were checked for homogeneity using Levene’s test.
The data in
Section 3.2.3 and
Section 3.3.1 were processed by ANOVA statistical techniques. In details. Single way, two-way (two factors, 3 × 2 levels), and three-way (three factors, 3 × 2 × 2 levels) were investigated to find significant interactions (
p < 0.01). Tukey’s HSD post-hoc multiple comparison test was used with the Bonferroni adjustment, a confidence level of 0.95, and a significance level α = 0.05 in order to determine the differences between the different sample means. Multiple comparisons were made one factor at a time. The confidence level and
p value adjustments with the Bonferroni method were based on the number of tests and estimates performed (two or three, based on the factor considered).
A linear model (two-way ANOVA, two factors, 3 × 2 levels) was applied in order to determine the contribution of the time and granulometry, and their interaction (p < 0.001). Different linear regressions for the extracted TPC were calculated according to the different granulometries in the time. The SE are specified in the tables at a confidence level of 0.95.
5. Conclusions
This piece of work investigated the UAE in water of polyphenols from grape-stalk residues. Lab-scale screenings were performed with both I-horn and C-horn at RT and 45 °C on two different matrix granulometries (<1000 µm and 212 µm). Physical models of the extraction trends could be extrapolated from the kinetic studies on the base of TPC yields. C-horn was the best performing lab-scale device. The effect of the starting material size, the comminution effect of US, and the extraction temperature have been discussed [
32]. TPC yields showed how the extraction temperature and acoustic cavitation can mainly override the granulometry influence.
The extract obtained with the best performing protocol has been characterized by means of HPLC-DAD, UPLC-ESI-MS/MS, and OPC quantification. Two different analytical methods for HPLC revealed interesting contents in the hydroxycinnamic and hydroxybenzoic acids (i.e., gallic and veratric acids), with a large variety of catechin derivatives, assigned to OPC and OPC fragmentation, being observed. This last consideration is supported by qualitative UPLC and colorimetric OPC quantification.
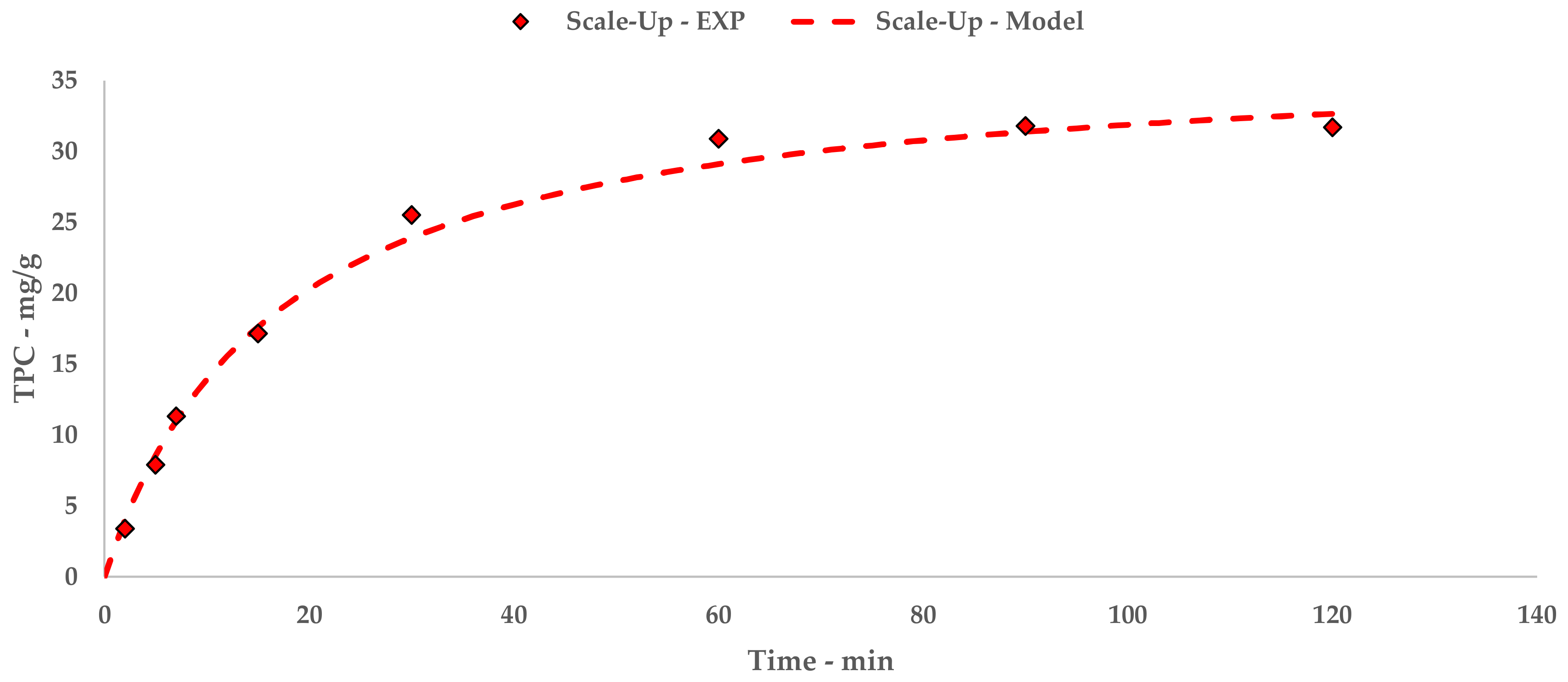
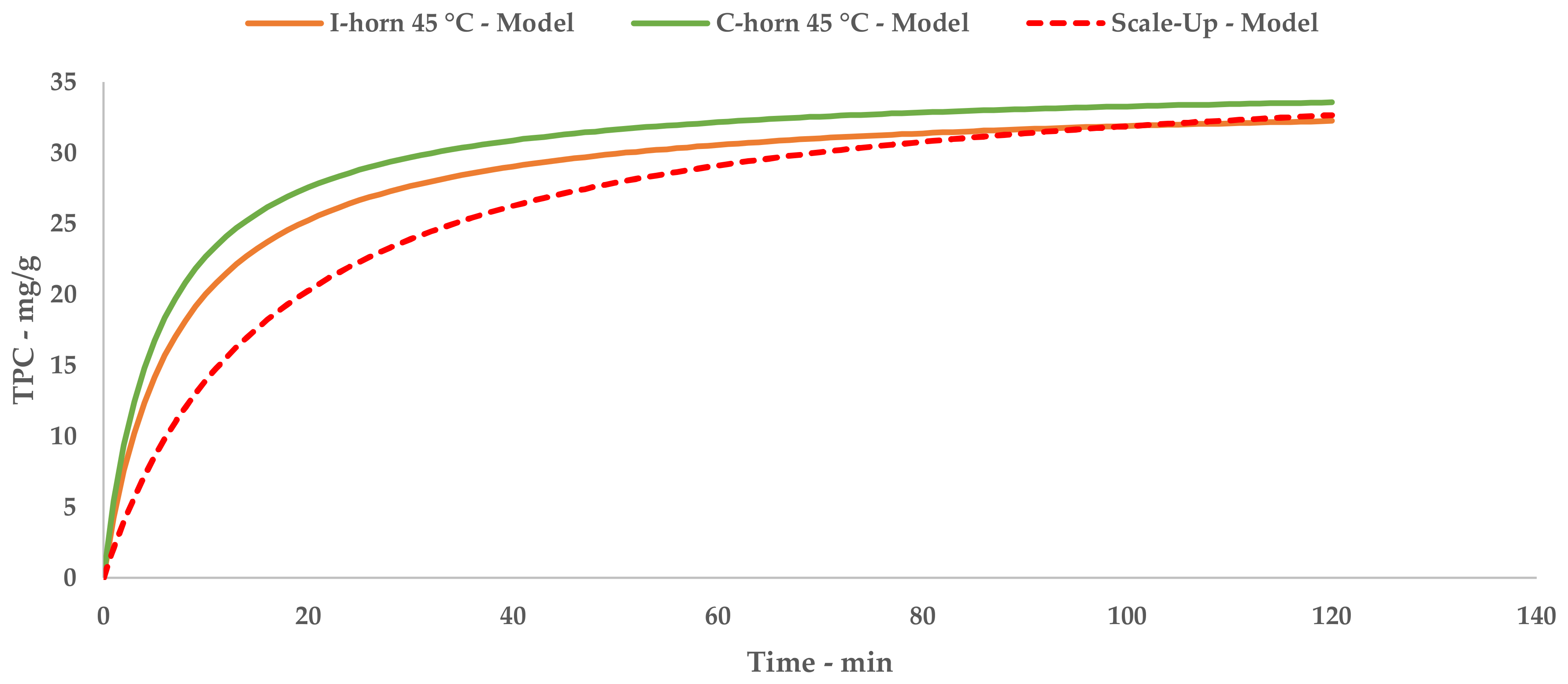
The results achieved on the lab scale made it possible to evaluate scale-up in a US recirculating flow system. We have proposed a kilo-scale extraction strategy, in which dedicated work-up procedures entail pre-industrial facilities, such as decanter and bag-filter units. Moreover, the increase in the quantity of starting material also requires a significant amount of water. We have therefore proposed using an NF protocol to deal with the large volumes in a sustainable way. The pilot NF skid concentrated the crude liquid extract by up to 355.91%, saving 80% of the water for possible reuse in the extraction process. TPC and antioxidant power evaluations confirmed that UAE can be successfully transposed from the gram- to kilo-scales, with limited losses in terms of yield and activity.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












