Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Study
2.2. Animal Study
2.3. Serum and Tissue Lysate Biochemical Measurements
2.4. Immunohistochemical (IHC) Staining
2.5. Western Blot Analysis
2.6. Statistical Analysis
3. Results
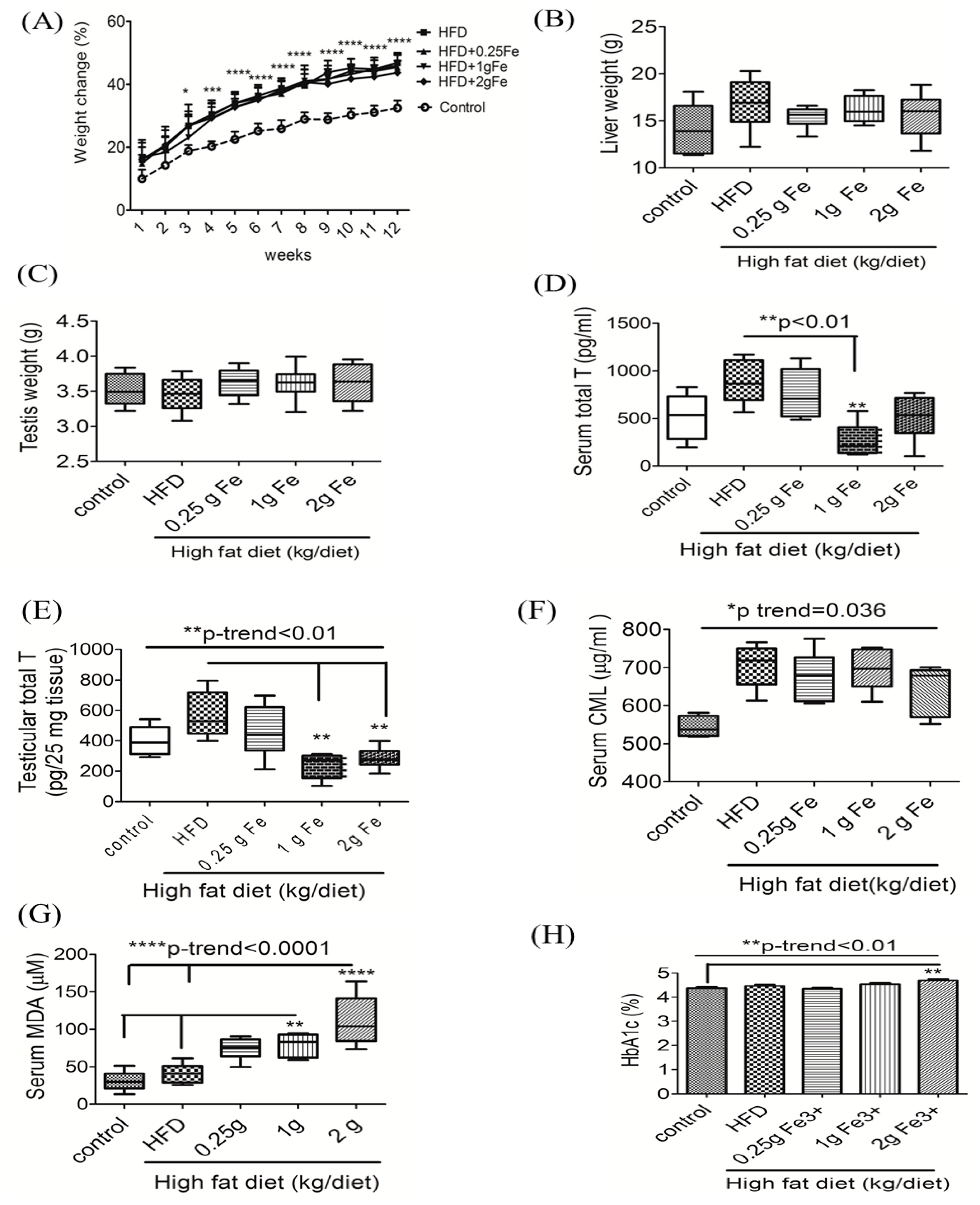
3.1. Ferric Citrate Supplementation Decreases Testicular Total T Levels and Increases Serum MDA in Obese Male Rats
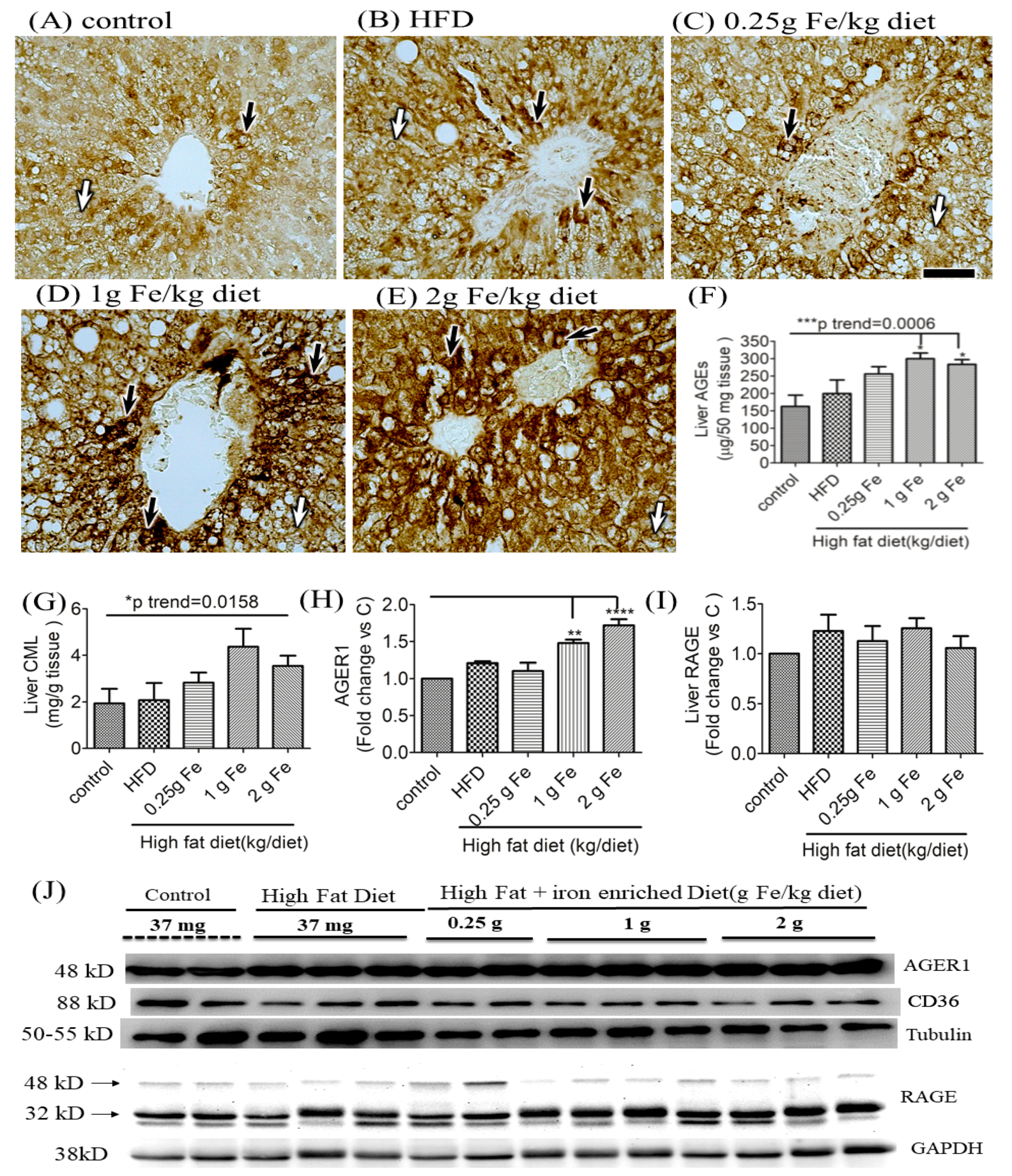
3.2. Impacts of Ferric Citrate Supplementation on the Hepatic AGE-RAGE Axis
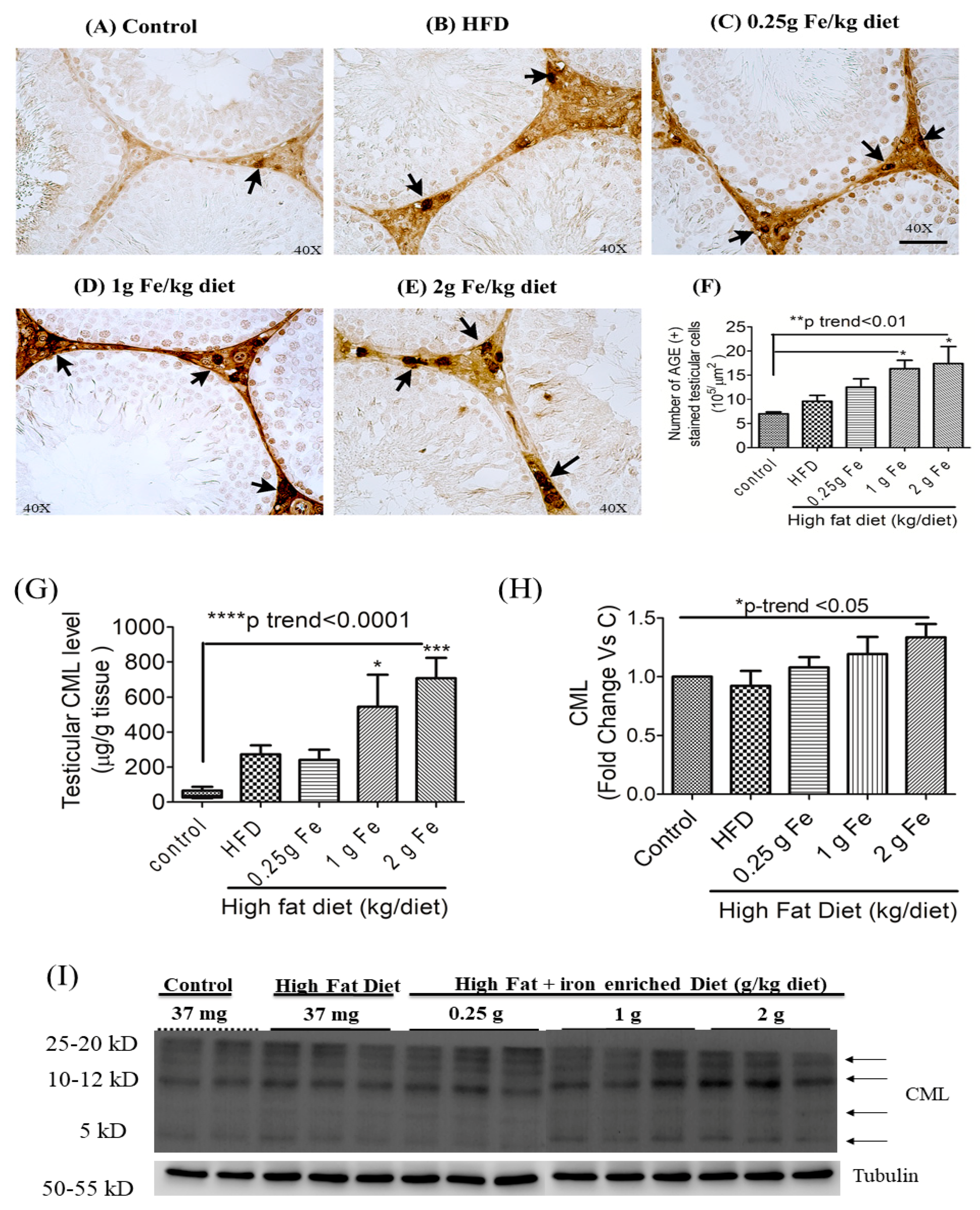
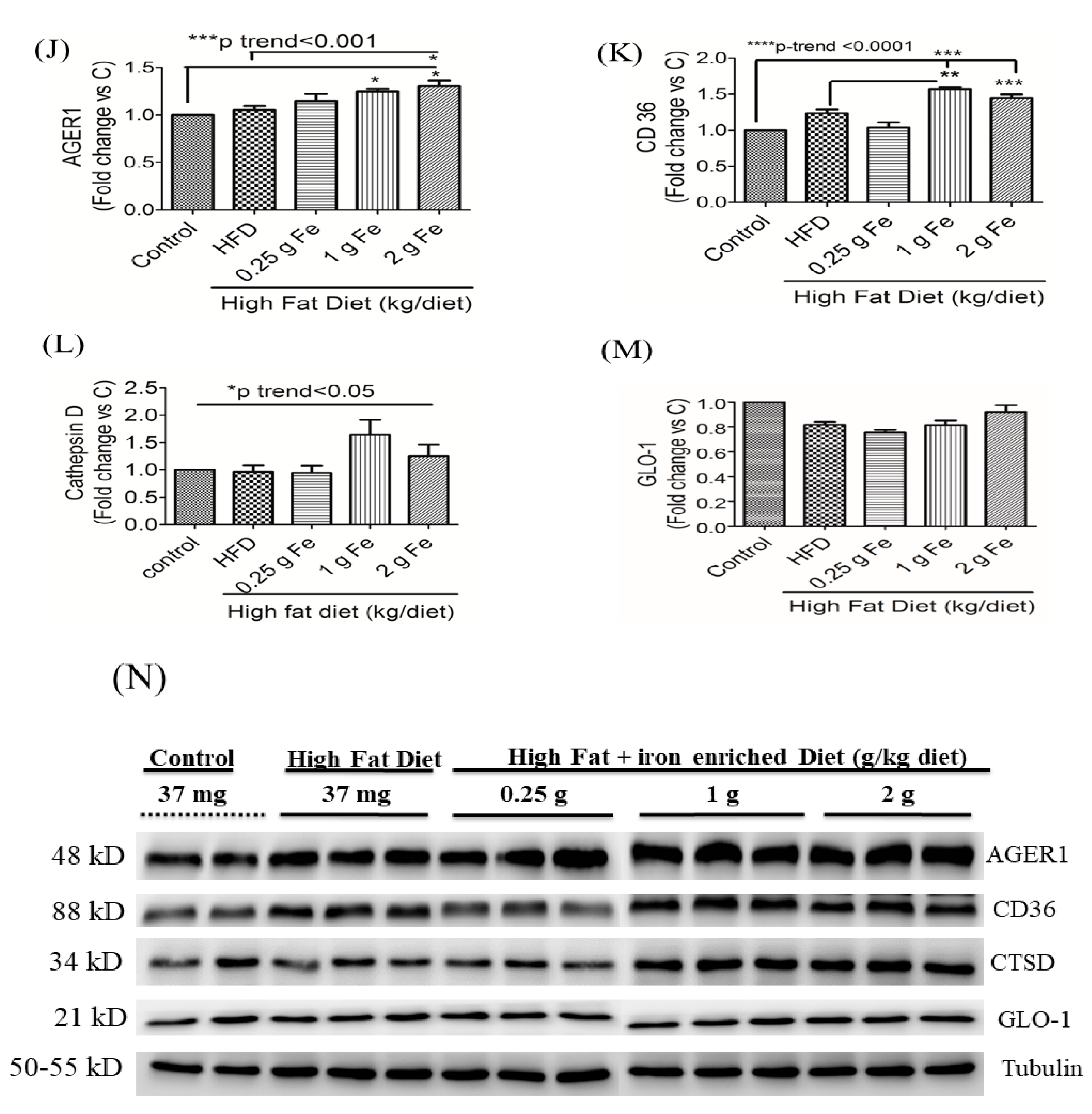
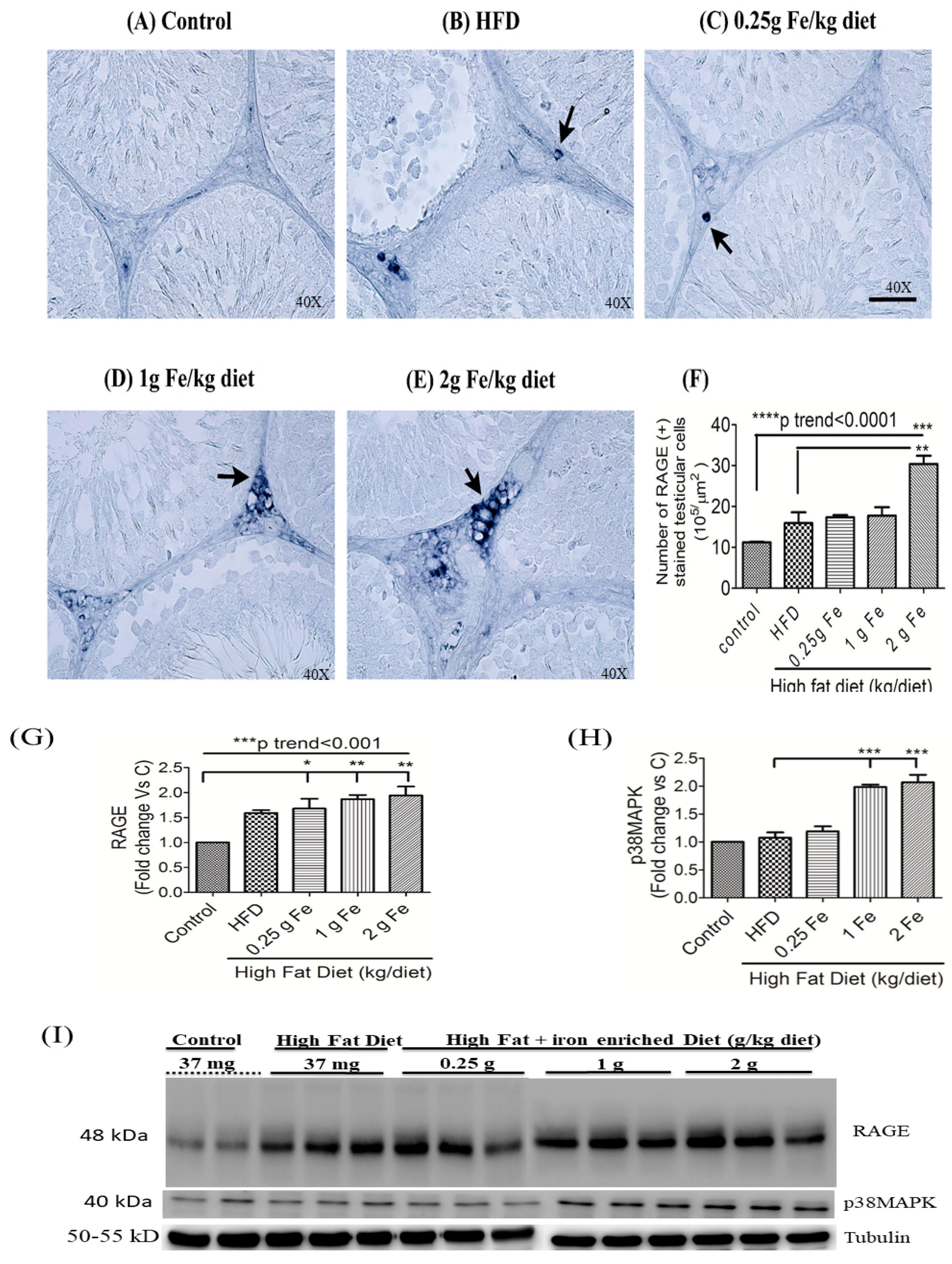
3.3. Impacts of Ferric Citrate Supplementation on the Testicular AGE-RAGE Axis
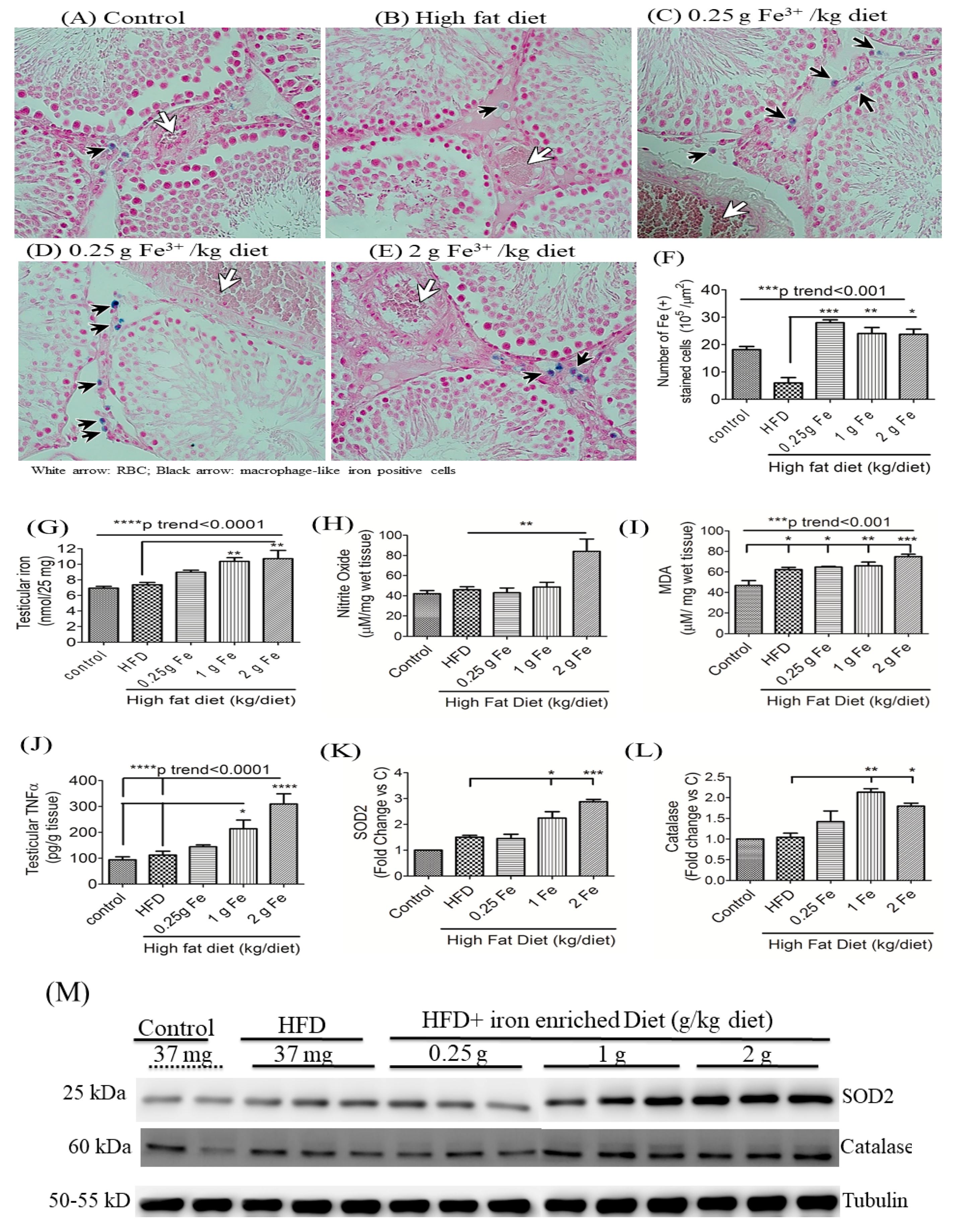
3.4. Impacts of Ferric Citrate Supplementation on Testicular Iron, TNF-α, NO, and MDA Levels
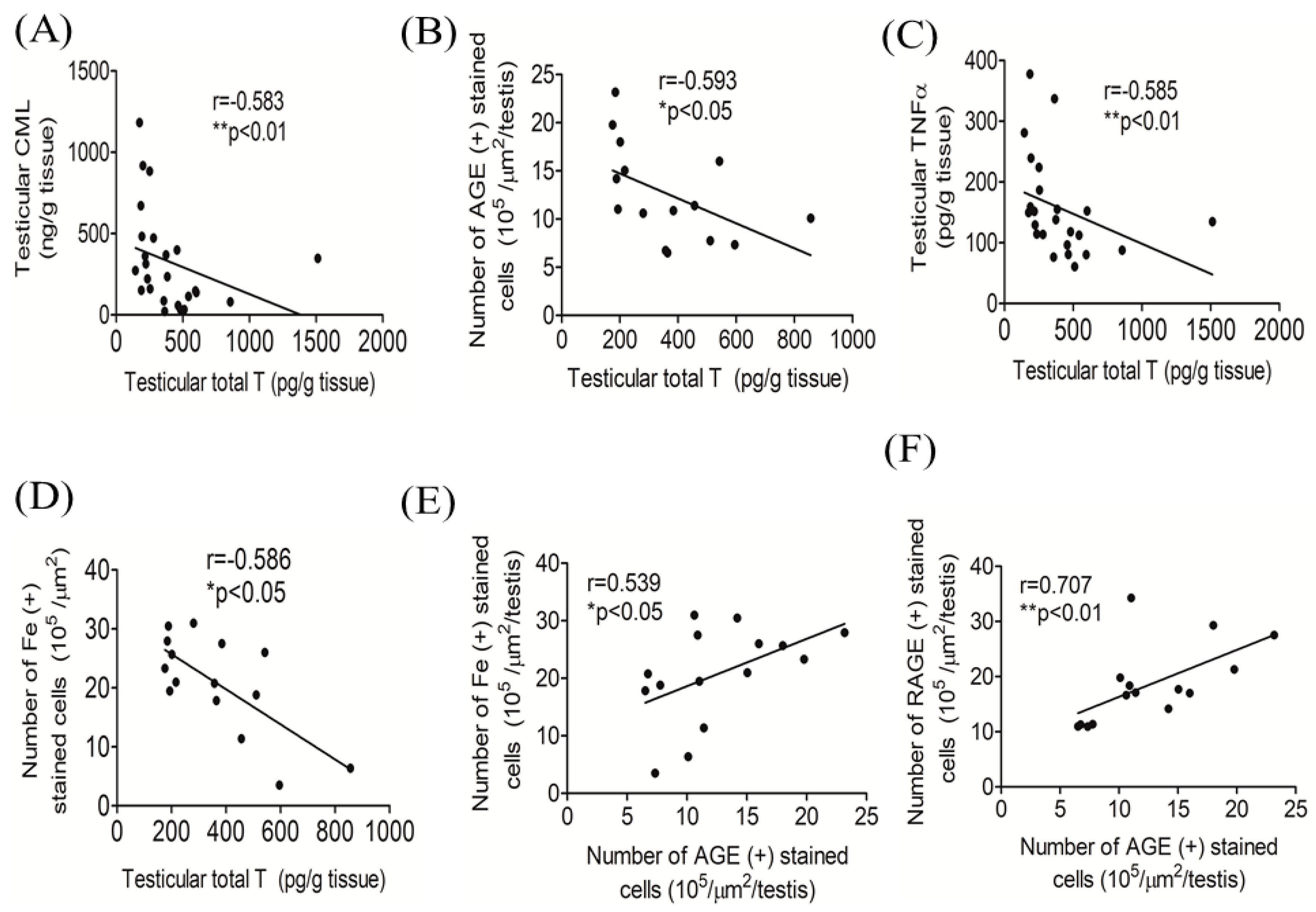
3.5. Correlations among Testicular Total T, AGEs, and Iron
3.6. Human Study: Correlations among Serum Total T, Iron and AGEs Biomarkers
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lopez, D.S.; Qiu, X.; Advani, S.; Tsilidis, K.K.; Khera, M.; Kim, J.; Morgentaler, A.; Wang, R.; Canfield, S. Double trouble: Co-occurrence of testosterone deficiency and body fatness associated with all-cause mortality in US men. Clin. Endocrinol. 2018, 88, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, M.; Zhu, C.; Zhang, Y.; Liu, Q.; Wang, X.; Li, L.; Zhou, D.; Qu, S. The Change in the Percent of Android and Gynoid Fat Mass Correlated with Increased Testosterone After Laparoscopic Sleeve Gastrectomy in Chinese Obese Men: A 6-Month Follow-Up. Obes. Surg. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.S.; Zhao, M.J.; An, Q.; Jia, Y.F.; Fu, L.L.; Xu, J.F.; Gu, Y.Q. Effects of testosterone supplementation therapy on lipid metabolism in hypogonadal men with T2DM: A meta-analysis of randomized controlled trials. Andrology 2018, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.J.; Chang, H.Y.; Pan, W.H. Time trend of obesity, the metabolic syndrome and related dietary pattern in Taiwan: From NAHSIT 1993-1996 to NAHSIT 2005-2008. Asia Pac. J. Clin. Nutr. 2011, 20, 292–300. [Google Scholar]
- Liu, C.C.; Wu, W.J.; Lee, Y.C.; Wang, C.J.; Ke, H.L.; Li, W.M.; Hsiao, H.L.; Yeh, H.C.; Li, C.C.; Chou, Y.H.; et al. The prevalence of and risk factors for androgen deficiency in aging Taiwanese men. J. Sex. Med. 2009, 6, 936–946. [Google Scholar] [CrossRef]
- Chang, J.S.; Lin, S.M.; Chao, J.C.; Chen, Y.C.; Wang, C.M.; Chou, N.H.; Pan, W.H.; Bai, C.H. Serum ferritin contributes to racial or geographic disparities in metabolic syndrome in Taiwan. Public Health Nutr. 2014, 17, 1498–1506. [Google Scholar] [CrossRef]
- Chang, J.S.; Lin, S.M.; Huang, T.C.; Chao, J.C.; Chen, Y.C.; Pan, W.H.; Bai, C.H. Serum ferritin and risk of the metabolic syndrome: A population-based study. Asia Pac. J. Clin. Nutr. 2013, 22, 400–407. [Google Scholar]
- Hawkins, W.W.; Speck, E.; Leonard, V.G. Variation of the hemoglobin level with age and sex. Blood 1954, 9, 999–1007. [Google Scholar] [CrossRef]
- Tajar, A.; Huhtaniemi, I.T.; O‘Neill, T.W.; Finn, J.D.; Pye, S.R.; Lee, D.M.; Bartfai, G.; Boonen, S.; Casanueva, F.F.; Forti, G.; et al. Characteristics of androgen deficiency in late-onset hypogonadism: Results from the European Male Aging Study (EMAS). J. Clin. Endocrinol. Metab. 2012, 97, 1508–1516. [Google Scholar] [CrossRef]
- Bhatia, V.; Chaudhuri, A.; Tomar, R.; Dhindsa, S.; Ghanim, H.; Dandona, P. Low testosterone and high C-reactive protein concentrations predict low hematocrit in type 2 diabetes. Diabetes Care 2006, 29, 2289–2294. [Google Scholar] [CrossRef]
- Bachman, E.; Feng, R.; Travison, T.; Li, M.; Olbina, G.; Ostland, V.; Ulloor, J.; Zhang, A.; Basaria, S.; Ganz, T.; et al. Testosterone suppresses hepcidin in men: A potential mechanism for testosterone-induced erythrocytosis. J. Clin. Endocrinol. Metab. 2010, 95, 4743–4747. [Google Scholar] [CrossRef] [PubMed]
- Beggs, L.A.; Yarrow, J.F.; Conover, C.F.; Meuleman, J.R.; Beck, D.T.; Morrow, M.; Zou, B.; Shuster, J.J.; Borst, S.E. Testosterone alters iron metabolism and stimulates red blood cell production independently of dihydrotestosterone. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E456–E461. [Google Scholar] [CrossRef] [PubMed]
- Bachman, E.; Travison, T.G.; Basaria, S.; Davda, M.N.; Guo, W.; Li, M.; Connor Westfall, J.; Bae, H.; Gordeuk, V.; Bhasin, S. Testosterone induces erythrocytosis via increased erythropoietin and suppressed hepcidin: Evidence for a new erythropoietin/hemoglobin set point. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Pak, M.; Lopez, M.A.; Gabayan, V.; Ganz, T.; Rivera, S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006, 108, 3730–3735. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Yassin, M.A.; Di Maio, S.; Daar, S.; Elsedfy, H.; Soliman, N.; Kattamis, C. Hypogonadism in male thalassemia major patients: Pathophysiology, diagnosis and treatment. Acta Bio-Med. Atenei Parm. 2018, 89, 6–15. [Google Scholar]
- Liu, Z.; Ye, F.; Zhang, H.; Gao, Y.; Tan, A.; Zhang, S.; Xiao, Q.; Zhang, B.; Huang, L.; Ye, B.; et al. The association between the levels of serum ferritin and sex hormones in a large scale of Chinese male population. PLoS ONE 2013, 8, e75908. [Google Scholar] [CrossRef] [PubMed]
- Chao, K.C.; Chang, C.C.; Chiou, H.Y.; Chang, J.S. Serum Ferritin Is Inversely Correlated with Testosterone in Boys and Young Male Adolescents: A Cross-Sectional Study in Taiwan. PLoS ONE 2015, 10, e0144238. [Google Scholar] [CrossRef]
- Noetzli, L.J.; Panigrahy, A.; Mittelman, S.D.; Hyderi, A.; Dongelyan, A.; Coates, T.D.; Wood, J.C. Pituitary iron and volume predict hypogonadism in transfusional iron overload. Am. J. Hematol. 2012, 87, 167–171. [Google Scholar] [CrossRef]
- Gautier, A.; Laine, F.; Massart, C.; Sandret, L.; Piguel, X.; Brissot, P.; Balkau, B.; Deugnier, Y.; Bonnet, F. Liver iron overload is associated with elevated SHBG concentration and moderate hypogonadotrophic hypogonadism in dysmetabolic men without genetic haemochromatosis. Eur. J. Endocrinol. 2011, 165, 339–343. [Google Scholar] [CrossRef]
- Soliman, A.; Yassin, M.; De Sanctis, V. Intravenous iron replacement therapy in eugonadal males with iron-deficiency anemia: Effects on pituitary gonadal axis and sperm parameters; A pilot study. Indian J. Endocrinol. Metab. 2014, 18, 310–316. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Qi, Y.W.; Hu, C.Y.; Chen, S.H.; Liu, Y. Advanced glycation end products inhibit testosterone secretion by rat Leydig cells by inducing oxidative stress and endoplasmic reticulum stress. Int. J. Mol. Med. 2016, 38, 659–665. [Google Scholar] [CrossRef]
- Mallidis, C.; Agbaje, I.; Rogers, D.; Glenn, J.; McCullough, S.; Atkinson, A.B.; Steger, K.; Stitt, A.; McClure, N. Distribution of the receptor for advanced glycation end products in the human male reproductive tract: Prevalence in men with diabetes mellitus. Hum. Reprod. 2007, 22, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Vistoli, G.; Stefek, M.; Chondrogianni, N.; Grune, T.; Sereikaite, J.; Sadowska-Bartosz, I.; Bartosz, G. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic. Res. 2013, 47, 93–137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Snooks, H.; Sang, S. Complexity of Advanced Glycation End Products in Foods: Where Are We Now? J. Agric. Food Chem. 2018, 66, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Goldberg, T.; Cai, W.; Rayfield, E.; Vlassara, H. Glycotoxins: A missing link in the relationship of dietary fat and meat intake in relation to risk of type 2 diabetes in men. Diabetes Care 2002, 25, 1898–1899. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Chao, K.C.; Chen, S.H.; Chang, C.C.; Lee, Y.C.; Wang, C.M.; Chang, J.S. Effects of ferric citrate supplementation on advanced glycation end products in a rat model of streptozotocin/nicotinamide-induced diabetes. Mol. Nutr. Food Res. 2017, 61, 1600753. [Google Scholar] [CrossRef]
- Chen, Y.; Filipov, N.M.; Guo, T.L. Dietary Glycation Products Regulate Immune Homeostasis: Early Glycation Products Promote Prostate Cancer Cell Proliferation through Modulating Macrophages. Mol. Nutr. Food Res. 2018, 62, 1700641. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Lu, C.; Vlassara, H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 13801–13806. [Google Scholar] [CrossRef]
- Xu, S.; Li, L.; Yan, J.; Ye, F.; Shao, C.; Sun, Z.; Bao, Z.; Dai, Z.; Zhu, J.; Jing, L.; et al. CML/CD36 accelerates atherosclerotic progression via inhibiting foam cell migration. Biomed Pharm. 2018, 97, 1020–1031. [Google Scholar] [CrossRef]
- Lin, J.; Tang, Y.; Kang, Q.; Chen, A. Curcumin eliminates the inhibitory effect of advanced glycation end-products (AGEs) on gene expression of AGE receptor-1 in hepatic stellate cells in vitro. Lab. Investig. 2012, 92, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.M.; Seiquer, I.; Delgado-Andrade, C.; Galdo, G.; Navarro, M.P. Intake of Maillard reaction products reduces iron bioavailability in male adolescents. Mol. Nutr. Food Res. 2009, 53, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Roca, B.; Navarro, M.P.; Seiquer, I. Antioxidant properties and metal chelating activity of glucose-lysine heated mixtures: Relationships with mineral absorption across Caco-2 cell monolayers. J. Agric. Food Chem. 2008, 56, 9056–9063. [Google Scholar] [CrossRef] [PubMed]
- Roncero-Ramos, I.; Pastoriza, S.; Navarro, M.P.; Delgado-Andrade, C. Assessing the effects of model Maillard compound intake on iron, copper and zinc retention and tissue delivery in adult rats. Food Funct. 2016, 7, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Patel, K.V.; Sun, K.; Guralnik, J.M.; Ershler, W.B.; Longo, D.L.; Ferrucci, L. Association between serum carboxymethyl-lysine, a dominant advanced glycation end product, and anemia in adults: The Baltimore longitudinal study of aging. J. Am. Geriatr. Soc. 2008, 56, 2145–2147. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.N.; Semba, R.D.; Sun, K.; Bandinelli, S.; Varadhan, R.; Patel, K.V.; Guralnik, J.M.; Ferrucci, L. Circulating selenium and carboxymethyl-lysine, an advanced glycation endproduct, are independent predictors of anemia in older community-dwelling adults. Nutrition 2012, 28, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Mirlohi, M.S.; Yaghooti, H.; Shirali, S.; Aminasnafi, A.; Olapour, S. Increased levels of advanced glycation end products positively correlate with iron overload and oxidative stress markers in patients with beta-thalassemia major. Ann. Hematol. 2018, 97, 679–684. [Google Scholar] [CrossRef]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef]
- Smith, P.R.; Thornalley, P.J. Mechanism of the degradation of non-enzymatically glycated proteins under physiological conditions. Studies with the model fructosamine, N epsilon-(1-deoxy-D-fructos-1-yl)hippuryl-lysine. Eur. J. Biochem. 1992, 210, 729–739. [Google Scholar] [CrossRef]
- Paduch, D.A.; Brannigan, R.E.; Fuchs, E.F.; Kim, E.D.; Marmar, J.L.; Sandlow, J.I. The laboratory diagnosis of testosterone deficiency. Urology 2014, 83, 980–988. [Google Scholar] [CrossRef]
- Awasthi, S.; Gayathiri, S.K.; Ramya, R.; Duraichelvan, R.; Dhason, A.; Saraswathi, N.T. Advanced Glycation-Modified Human Serum Albumin Evokes Alterations in Membrane and Eryptosis in Erythrocytes. Appl. Biochem. Biotechnol. 2015, 177, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ernst, L.; Grotzinger, N.; Hohn, A.; Breusing, N.; Reinheckel, T.; Grune, T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic. Res. 2010, 44, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Liu, K.L.; Chang, C.S.; Su, C.T.; Chen, S.H.; Lee, Y.C.; Chang, J.S. Ferric Citrate Supplementation Reduces Red-Blood-Cell Aggregation and Improves CD163+ Macrophage-Mediated Hemoglobin Metabolism in a Rat Model of High-Fat-Diet-Induced Obesity. Mol. Nutr. Food Res. 2018, 62, 1700442. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.J.; Matsumoto, A.M.; Araujo, A.B.; McKinlay, J.B. The effect of diurnal variation on clinical measurement of serum testosterone and other sex hormone levels in men. J. Clin. Endocrinol. Metab. 2009, 94, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.Y.; Chen, Y.C.; Lin, P.; Shih, C.K.; Bai, C.H.; Yuan, K.C.; Lee, S.Y.; Chang, J.S. Testosterone-Associated Dietary Pattern Predicts Low Testosterone Levels and Hypogonadism. Nutrients 2018, 10, 1786. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, A.L.; Lee, Y.C.; Shih, C.K.; Hsieh, R.H.; Chen, S.H.; Chang, J.S. Alteration in iron efflux affects male sex hormone testosterone biosynthesis in a diet-induced obese rat model. Food Funct. 2019, 10, 4113–4123. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.J.; O‘Donnell, A.B.; Matsumoto, A.M.; McKinlay, J.B. Intraindividual variation in levels of serum testosterone and other reproductive and adrenal hormones in men. Clin. Endocrinol. 2007, 67, 853–862. [Google Scholar] [CrossRef]
- Gaens, K.H.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Blaak, E.E.; et al. Nepsilon-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arter. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef]
- Gaens, K.H.; Ferreira, I.; van de Waarenburg, M.P.; van Greevenbroek, M.M.; van der Kallen, C.J.; Dekker, J.M.; Nijpels, G.; Rensen, S.S.; Stehouwer, C.D.; Schalkwijk, C.G. Protein-Bound Plasma Nepsilon-(Carboxymethyl)lysine Is Inversely Associated With Central Obesity and Inflammation and Significantly Explain a Part of the Central Obesity-Related Increase in Inflammation: The Hoorn and CODAM Studies. Arter. Thromb. Vasc. Biol. 2015, 35, 2707–2713. [Google Scholar] [CrossRef]
- Foroumandi, E.; Alizadeh, M.; Kheirouri, S.; Asghari Jafarabadi, M. Exploring the role of body mass index in relationship of serum nitric oxide and advanced glycation end products in apparently healthy subjects. PLoS ONE 2019, 14, e0213307. [Google Scholar] [CrossRef]
- Wang, C.; Jackson, G.; Jones, T.H.; Matsumoto, A.M.; Nehra, A.; Perelman, M.A.; Swerdloff, R.S.; Traish, A.; Zitzmann, M.; Cunningham, G. Low testosterone associated with obesity and the metabolic syndrome contributes to sexual dysfunction and cardiovascular disease risk in men with type 2 diabetes. Diabetes Care 2011, 34, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Yan, W.J.; Yin, T.L.; Zhang, Y.; Li, J.; Yang, J. Diet-induced obesity impairs spermatogenesis: A potential role for autophagy. Sci. Rep. 2017, 7, 43475. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.J.; Cheng, R.W.; Simmonds, M.; Richmond, W.; James, V.H. Dietary lipids: An additional regulator of plasma levels of sex hormone binding globulin. J. Clin. Endocrinol. Metab. 1987, 64, 1083–1085. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Gomez, A.L.; Love, D.M.; Avery, N.G.; Sharman, M.J.; Kraemer, W.J. Effects of a high-fat diet on postabsorptive and postprandial testosterone responses to a fat-rich meal. Metab. Clin. Exp. 2001, 50, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Liu, Y.; Sigmon, V.K.; McCort, A.; Ren, J. High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetesobesity Metab. 2005, 7, 448–454. [Google Scholar] [CrossRef]
- Davis, K.E.; Prasad, C.; Vijayagopal, P.; Juma, S.; Adams-Huet, B.; Imrhan, V. Contribution of dietary advanced glycation end products (AGE) to circulating AGE: Role of dietary fat. Br. J. Nutr. 2015, 114, 1797–1806. [Google Scholar] [CrossRef]
- Sowndhar Rajan, B.; Manivasagam, S.; Dhanusu, S.; Chandrasekar, N.; Krishna, K.; Kalaiarasu, L.P.; Babu, A.A.; Vellaichamy, E. Diet with high content of advanced glycation end products induces systemic inflammation and weight gain in experimental mice: Protective role of curcumin and gallic acid. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 114, 237–245. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Body Weight Status | p-Value | |
|---|---|---|---|
| Normal Weight (n = 44) | OW/Obese (n = 67) | ||
| Basic characteristic | |||
| Age (years) | 39.40 ± 12.35 | 42.20 ± 10.84 | 0.164 |
| BMI (kg/m2) | 21.86 ± 1.48 | 28.26 ± 3.65 | <0.001 |
| Hypogonadism (n,%) | 5 (11.3) | 13 (19.0) | <0.001 |
| Iron overload (n,%) | 7(15.9) | 23 (29.8) | <0.001 |
| AGE biomakers | |||
| CML (μg/mL) | 270.98 ± 109.28 | 363.18 ± 129.87 | 0.032 |
| sRAGE (pg/mL) | 361.58 ± 255.41 | 372.10 ± 190.03 | 0.293 |
| CTSD (ng/mL) | 2147.21 ± 1547.46 | 1720.48 ± 1485.99 | 0.200 |
| MDA (μM) | 43.11 ± 20.96 | 46.68 ± 27.79 | 0.626 |
| Iron biomarkers | |||
| Free Hb (μg/mL) | 139.08 ± 42.42 | 165.70 ± 53.71 | 0.003 |
| Ferritin (ng/mL) | 205.53 ± 151.63 | 242.66 ± 156.18 | 0.080 |
| Transferrin saturation (%) | 34.34 ± 11.19 | 32.87 ± 12.60 | 0.300 |
| Serum iron (μg/mL) | 107.5 ±32.5 | 108.3 ± 37.1 | 0.954 |
| RBC Aggregation CSS (mPa) | 262.78 ± 63.50 | 286.60 ± 62.85 | 0.044 |
| Sex hormone biomarkers | |||
| Total T (ng/mL) | 4.77 ± 1.30 | 3.62 ± 1.09 | <0.001 |
| Free T (pg/mL) | 90.58 ± 26.86 | 79.51 ± 24.15 | 0.042 |
| SHBG (nmol/L) | 36.21 ± 13.55 | 29.79 ± 14.69 | 0.003 |
| Variables | Total T (ng/mL) | Serum CML (μg/mL) | sRAGE (pg/mL) | |||
|---|---|---|---|---|---|---|
| r | p-Value | r | p-Value | r | p-Value | |
| Iron biomarkers | ||||||
| Transferrin saturation (%) | 0.242 | 0.007 | −0.132 | 0.207 | 0.0214 | 0.832 |
| RBC Aggregation CSS (mPa) | −0.419 | <0.0001 | 0.0957 | 0.359 | 0.0026 | 0.98 |
| Serum iron(μg/mL) | 0.039 | 0.665 | −0.0423 | 0.697 | 0.0114 | 0.912 |
| Serum ferritin (ng/mL) | −0.117 | 0.194 | 0.1343 | 0.204 | 0.0559 | 0.578 |
| Free Hb (μg/mL) | −0.1308 | 0.192 | −0.3653 | 0.0004 | −0.2809 | 0.004 |
| AGE biomakers | ||||||
| CML(μg/mL) | −0.209 | 0.030 | ||||
| Cathepsin D (ng/mL) | 0.3302 | 0.001 | −0.071 | 0.526 | 0.0583 | 0.581 |
| Malondialdehyde (μM) | 0.026 | 0.818 | 0.1537 | 0.168 | 0.0185 | 0.863 |
| sRAGE (pg/mL) | −0.1309 | 0.192 | 0.2132 | 0.042 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-H.; Yuan, K.-C.; Lee, Y.-C.; Shih, C.-K.; Tseng, S.-H.; Tinkov, A.A.; Skalny, A.V.; Chang, J.-S. Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity. Antioxidants 2020, 9, 261. https://doi.org/10.3390/antiox9030261
Chen S-H, Yuan K-C, Lee Y-C, Shih C-K, Tseng S-H, Tinkov AA, Skalny AV, Chang J-S. Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity. Antioxidants. 2020; 9(3):261. https://doi.org/10.3390/antiox9030261
Chicago/Turabian StyleChen, Seu-Hwa, Kuo-Ching Yuan, Yu-Chieh Lee, Chun-Kuang Shih, Sung-Hui Tseng, Alexey A. Tinkov, Anatoly V. Skalny, and Jung-Su Chang. 2020. "Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity" Antioxidants 9, no. 3: 261. https://doi.org/10.3390/antiox9030261
APA StyleChen, S.-H., Yuan, K.-C., Lee, Y.-C., Shih, C.-K., Tseng, S.-H., Tinkov, A. A., Skalny, A. V., & Chang, J.-S. (2020). Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity. Antioxidants, 9(3), 261. https://doi.org/10.3390/antiox9030261