Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go?





Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
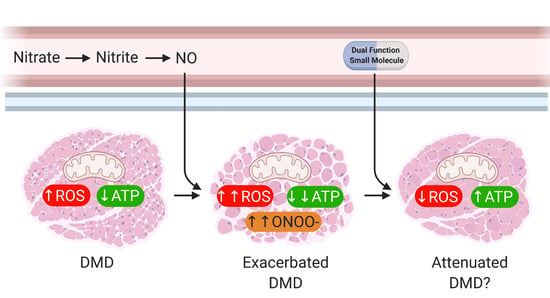
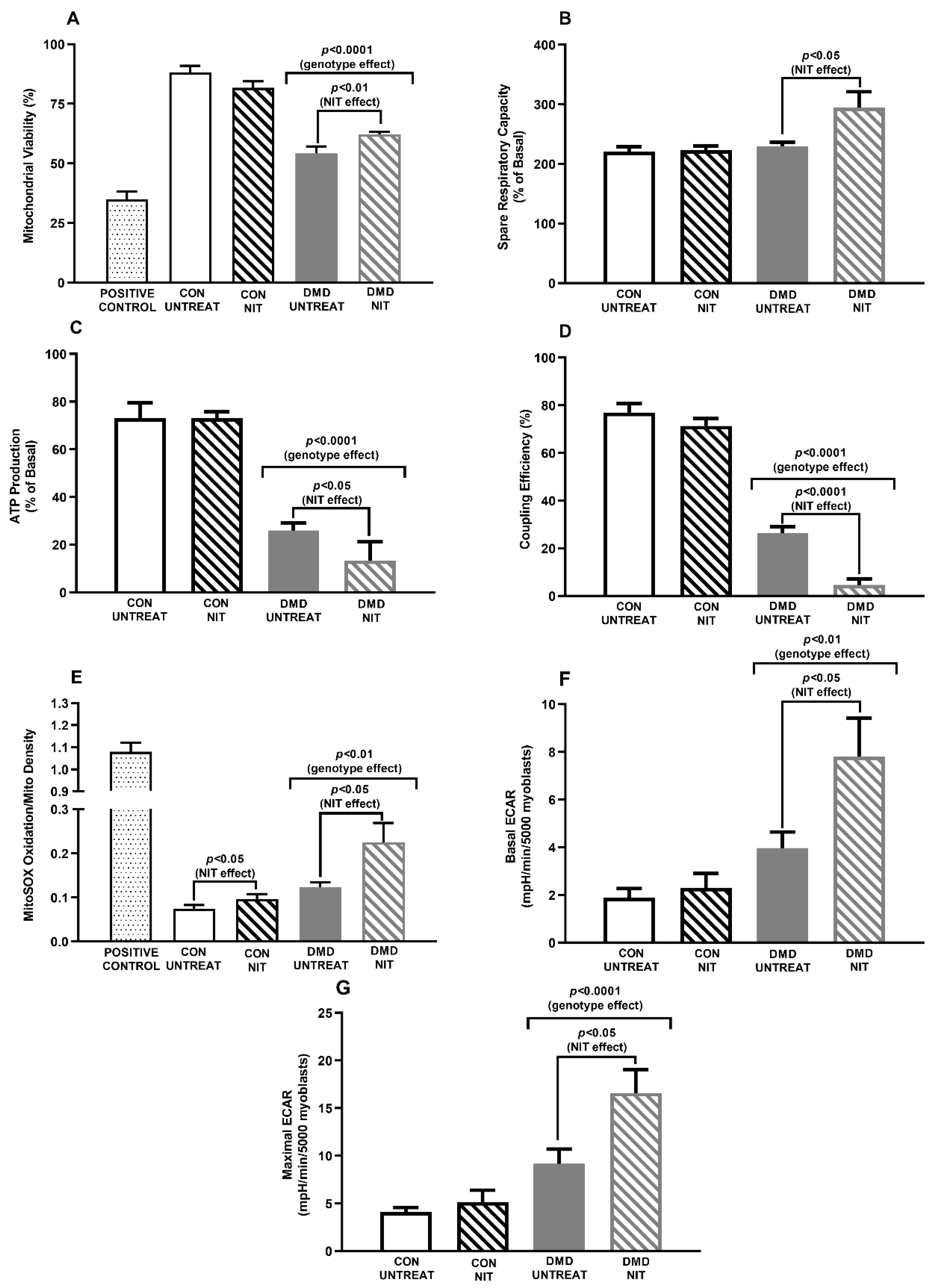
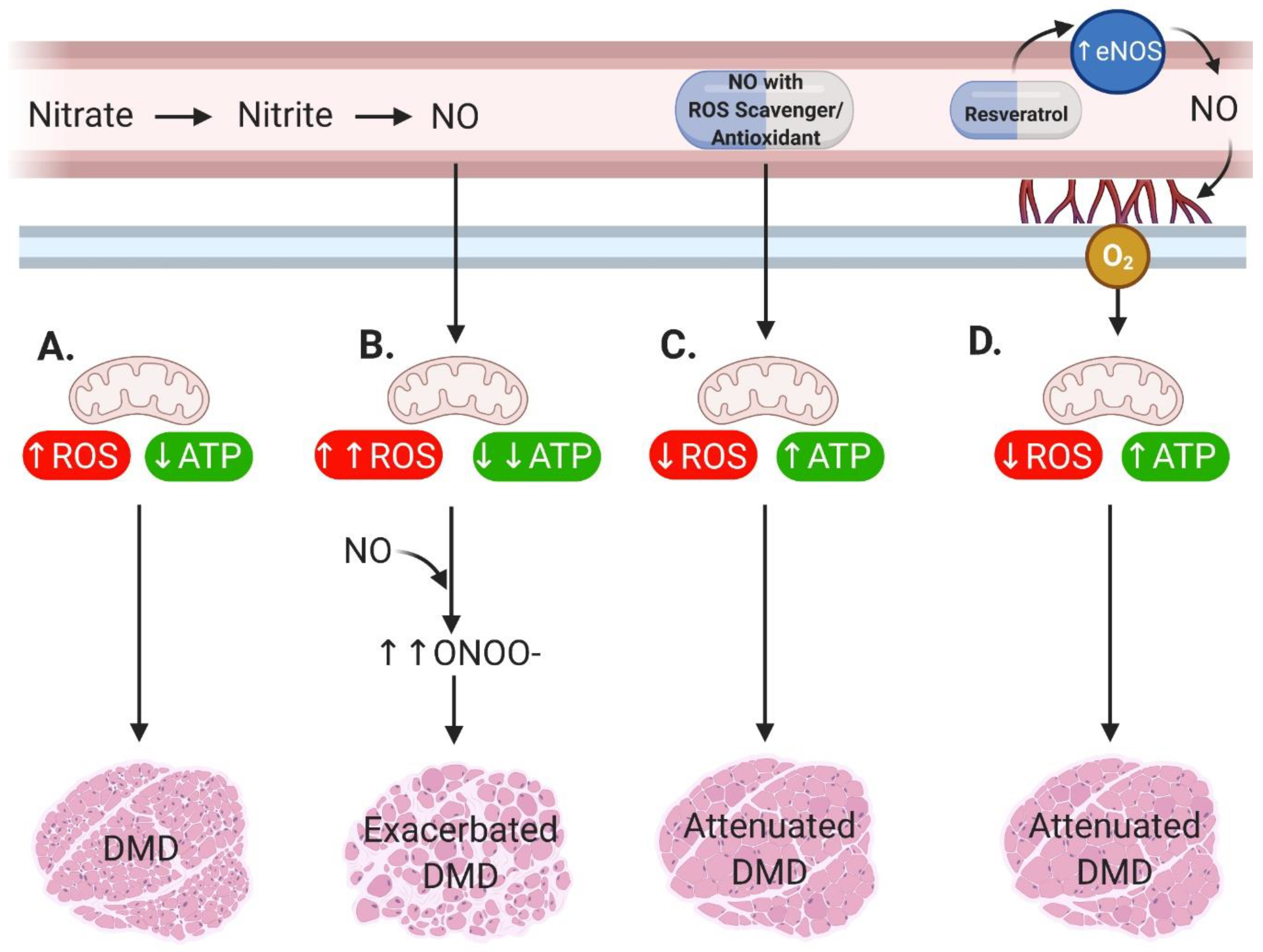
2. Main
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Nio, Y.; Tanaka, M.; Hirozane, Y.; Muraki, Y.; Okawara, M.; Hazama, M.; Matsuo, T. Phosphodiesterase 4 inhibitor and phosphodiesterase 5 inhibitor combination therapy has antifibrotic and anti-inflammatory effects in mdx mice with Duchenne muscular dystrophy. FASEB J. 2017, 31, 5307–5320. [Google Scholar] [CrossRef]
- De Arcangelis, V.; Strimpakos, G.; Gabanella, F.; Corbi, N.; Luvisetto, S.; Magrelli, A.; Onori, A.; Passananti, C.; Pisani, C.; Rome, S.; et al. Pathways Implicated in Tadalafil Amelioration of Duchenne Muscular Dystrophy. J. Cell Physiol. 2016, 231, 224–232. [Google Scholar] [CrossRef]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.M.; Dai, D.F.; Percival, J.M.; Minami, E.; Willis, M.S.; Patrucco, E.; Froehner, S.C.; Beavo, J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 19079–19083. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Vohra, R.S.; Chrzanowski, S.M.; Hammers, D.W.; Lott, D.J.; Vandenborne, K.; Walter, G.A.; Forbes, S.C. Effects of PDE5 inhibition on dystrophic muscle following an acute bout of downhill running and endurance training. J. Appl. Physiol. 2019, 126, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Therapeutic strategies to address neuronal nitric oxide synthase deficiency and the loss of nitric oxide bioavailability in Duchenne Muscular Dystrophy. Orphanet J. Rare Dis. 2017, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Victor, R.G.; Sweeney, H.L.; Finkel, R.; McDonald, C.M.; Byrne, B.; Eagle, M.; Goemans, N.; Vandenborne, K.; Dubrovsky, A.L.; Topaloglu, H. A phase 3 randomized placebo-controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology 2017, 89, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.G.; Herzka, D.A.; Thompson, W.R.; He, B.; Bibat, G.; Tennekoon, G.; Russell, S.D.; Schuleri, K.H.; Lardo, A.C.; Kass, D.A. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann. Neurol. 2014, 76, 541–549. [Google Scholar] [CrossRef]
- Witting, N.; Kruuse, C.; Nyhuus, B.; Prahm, K.P.; Citirak, G.; Lundgaard, S.J.; von Huth, S.; Vejlstrup, N.; Lindberg, U.; Krag, T.O.; et al. Effect of sildenafil on skeletal and cardiac muscle in Becker muscular dystrophy. Ann. Neurol. 2014, 76, 550–557. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Gücüyener, K.; Ergenekon, E.; Erbas, D.; Pinarli, G.; Serdaroğlu, A. The serum nitric oxide levels in patients with Duchenne muscular dystrophy. Brain Dev. 2000, 22, 181–183. [Google Scholar] [CrossRef]
- Kasai, T.; Abeyama, K.; Hashiguchi, T.; Fukunaga, H.; Osame, M.; Maruyama, K. Decreased total nitric oxide production in patients with Duchenne muscular dystrophy. J. Biomed. Sci. 2004, 11, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Oltmann, M.; Rinaldi, C.; Myung, K.H.; Tidball, J.G. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 3439–3451. [Google Scholar] [CrossRef]
- Austin, L.; De Niese, M.; McGregor, A.; Arthur, H.; Gurusinghe, A.; Gould, M. Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscul. Disord. 1992, 2, 27–33. [Google Scholar] [CrossRef]
- Cole, M.; Rafael, J.; Taylor, D.; Lodi, R.; Davies, K.; Styles, P. A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypothesis 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Barton, E.R.; Morris, L.; Kawana, M.; Bish, L.T.; Toursel, T. Systemic administration of L-arginine benefits mdx skeletal muscle function. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2005, 32, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Voisin, V.; Sébrié, C.; Matecki, S.; Yu, H.; Gillet, B.; Ramonatxo, M.; Israël, M.; De la Porte, S. L-arginine improves dystrophic phenotype in mdx mice. Neurobiol. Dis. 2005, 20, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Kapil, V.; Khambata, R.S.; Jones, D.A.; Rathod, K.; Primus, C.; Massimo, G.; Fukuto, J.M.; Ahluwalia, A. The Noncanonical Pathway for In Vivo Nitric Oxide Generation: The Nitrate-Nitrite-Nitric Oxide Pathway. Pharmacol. Rev. 2020, 72, 692–766. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.J.; Schiffer, T.A.; Borniquel, S.; Sahlin, K.; Ekblom, B.; Lundberg, J.O.; Weitzberg, E. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2011, 13, 149–159. [Google Scholar] [CrossRef]
- Timpani, C.A.; Trewin, A.J.; Stojanovska, V.; Robinson, A.; Goodman, C.A.; Nurgali, K.; Betik, A.C.; Stepto, N.; Hayes, A.; McConell, G.K. Attempting to compensate for reduced neuronal nitric oxide synthase protein with nitrate supplementation cannot overcome metabolic dysfunction but rather has detrimental effects in dystrophin-deficient mdx muscle. Neurotherapeutics 2017, 14, 429–446. [Google Scholar] [CrossRef]
- Rando, T.A.; Disatnik, M.-H.; Yu, Y.; Franco, A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 1998, 8, 14–21. [Google Scholar] [CrossRef]
- Disatnik, M.-H.; Dhawan, J.; Yu, Y.; Beal, M.F.; Whirl, M.M.; Franco, A.A.; Rando, T.A. Evidence of oxidative stress in mdx mouse muscle: Studies of the pre-necrotic state. J. Neurol. Sci. 1998, 161, 77–84. [Google Scholar] [CrossRef]
- Timpani, C.A.; Goodman, C.A.; Stathis, C.G.; White, J.D.; Mamchaoui, K.; Butler-Browne, G.; Gueven, N.; Hayes, A.; Rybalka, E. Adenylosuccinic acid therapy ameliorates murine Duchenne Muscular Dystrophy. Sci. Rep. 2020, 10, 1125. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cheregi, B.D.; Sorensen, J.C.; Nurgali, K.; Hayes, A. Chemotherapeutic agents induce mitochondrial superoxide production and toxicity but do not alter respiration in skeletal muscle in vitro. Mitochondrion 2018, 42, 33–49. [Google Scholar] [CrossRef]
- Chi, M.M.Y.; Hintz, C.S.; McKee, D.; Felder, S.; Grant, N.; Kaiser, K.K.; Lowry, O.H. Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 1987, 36, 761–767. [Google Scholar] [CrossRef]
- De Young, L.; Yu, D.; Freeman, D.; Brock, G.B. Effect of PDE5 inhibition combined with free oxygen radical scavenger therapy on erectile function in a diabetic animal model. Int. J. Impot. Res. 2003, 15, 347–354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wallerath, T.; Deckert, G.; Ternes, T.; Anderson, H.; Li, H.; Witte, K.; Förstermann, U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation 2002, 106, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, L.; Formoso, L.; Torelli, L.; Crea, E.; Bergamo, A.; Sava, G.; Giacomello, E. Long-term resveratrol treatment improves the capillarization in the skeletal muscles of ageing C57BL/6J mice. Int. J. Food Sci. Nutr. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sitzia, C.; Farini, A.; Colleoni, F.; Fortunato, F.; Razini, P.; Erratico, S.; Tavelli, A.; Fabrizi, F.; Belicchi, M.; Meregalli, M.; et al. Improvement of endurance of DMD animal model using natural polyphenols. BioMed Res. Int. 2015, 2015, 680615. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timpani, C.A.; Mamchaoui, K.; Butler-Browne, G.; Rybalka, E. Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go? Antioxidants 2020, 9, 1268. https://doi.org/10.3390/antiox9121268
Timpani CA, Mamchaoui K, Butler-Browne G, Rybalka E. Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go? Antioxidants. 2020; 9(12):1268. https://doi.org/10.3390/antiox9121268
Chicago/Turabian StyleTimpani, Cara A., Kamel Mamchaoui, Gillian Butler-Browne, and Emma Rybalka. 2020. "Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go?" Antioxidants 9, no. 12: 1268. https://doi.org/10.3390/antiox9121268
APA StyleTimpani, C. A., Mamchaoui, K., Butler-Browne, G., & Rybalka, E. (2020). Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go? Antioxidants, 9(12), 1268. https://doi.org/10.3390/antiox9121268