Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Viability and Cytoprotective Analysis
2.3. The Constitutive Expression of the ER Stress-Related Genes in Selected Cell Lines
2.4. The mRNA Expression of the Examined Responsive Genes to TU
2.5. The Protein Expression of the Examined Responsive Genes to TU
2.6. Flow Cytometry Analysis of Cell Apoptosis and Intracellular ROS Levels
2.7. Luciferase Reporter Assays of ARE-Driven Gene Trans-Activity
2.8. Statistical Analysis
3. Results
3.1. Both Nrf1α and Nrf2 Contribute to Differential Expression of Responsive Genes to the Basal ER Stress in Four Different Genotypic Cell Lines
3.2. Distinctive Effects of Nrf1α and Nrf2 on Basal Expression of Antioxidant and UPR-Related Proteins, and Their Responsive Expression at the Early Stages of TU-Stimulated ER Stress
3.3. Nrf1α and Nrf2 Mediate Distinct Transcriptional Responses of Antioxidant and most UPR-Target Genes to the Long-Term TU-Stimulated ER Stress
3.4. Distinct Contributions of Nrf1α and Nrf2 to the Protein Expression of Antioxidant Responsive Genes to TU that Serves as a Classic ER Stressor
3.5. Distinctive Requirements of Nrf1α and Nrf2 in Differential Expression of the ER Stress-Responsive Genes Induced by TU
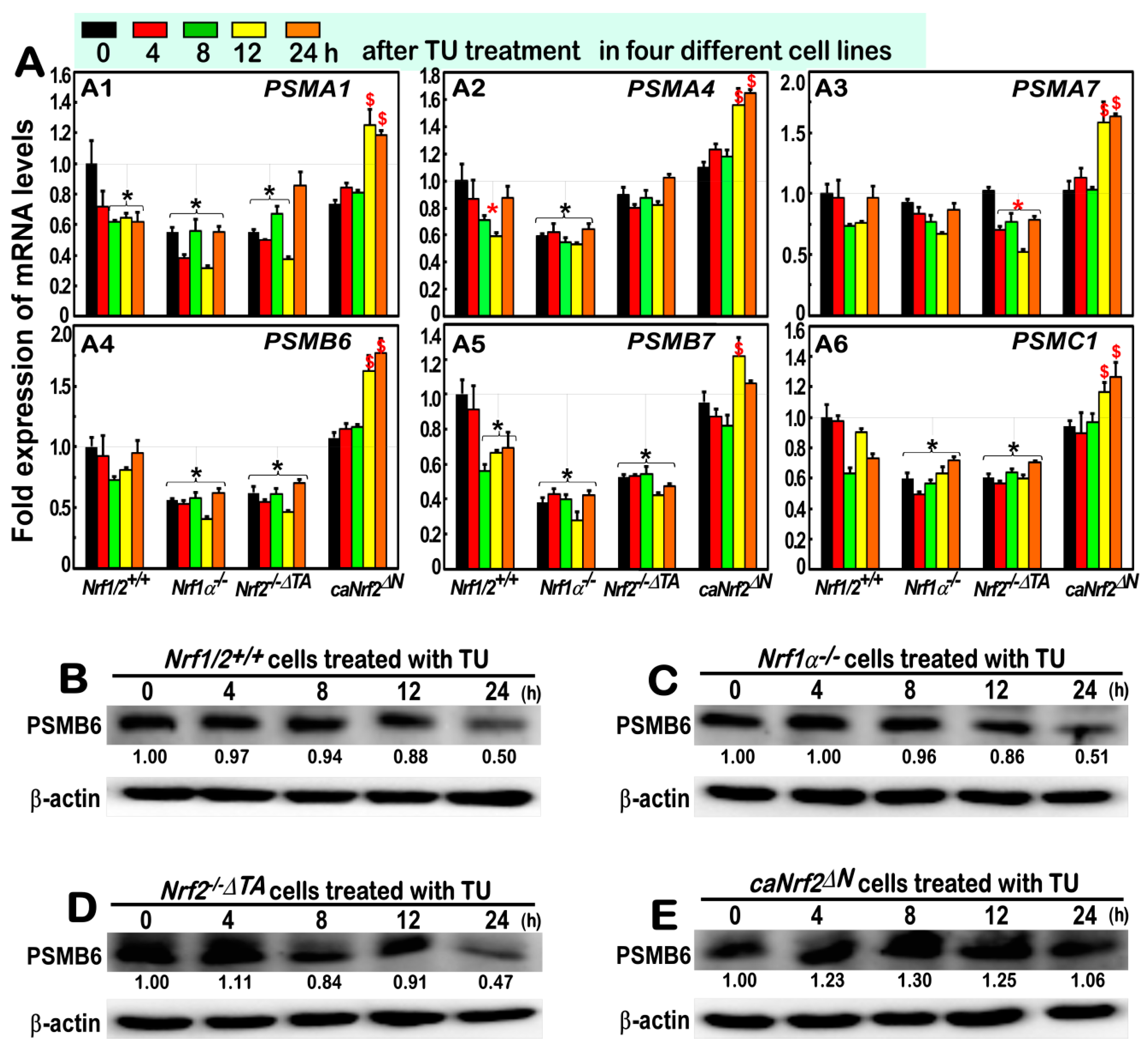
3.6. Almost No Induction of the Proteasomal (PSM) Subunit Genes Regulated by Nrf1 in the Response to TU
3.7. Nrf1α Is More Potent Than Nrf2 at Mediating Cytoprotective Response against the Cytototic Effects of TU
3.8. The Intracellular ROS Levels Are Increased in Nrf1α−/− cells, but Decreased in Nrf2−/−ΔTA Cells, with Different Responses to TU Alone or Plus tBHQ
3.9. Nrf1 and Nrf2 Bi-Directionally Mediate Transcriptional Expression of Distinct ARE-Driven UPR-Luc Reporter Genes
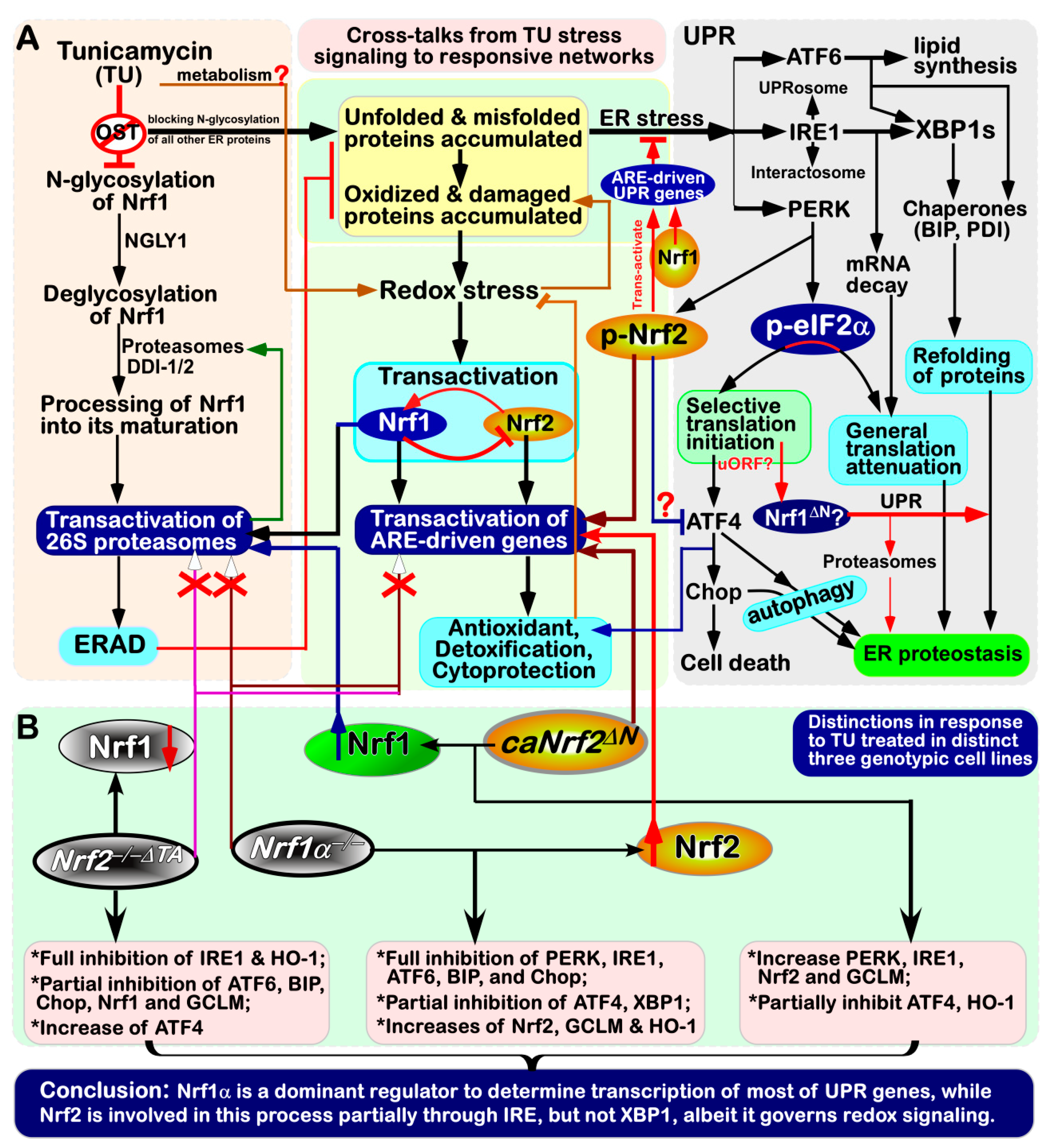
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hegde, R.S.; Ploegh, H.L. Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol. 2010, 22, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Ron, D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033894. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.; Chevalier, M.W.; Aragon, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Gogala, M.; Becker, T.; Beatrix, B.; Armache, J.P.; Barrio-Garcia, C.; Berninghausen, O.; Beckmann, R. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 2014, 506, 107–110. [Google Scholar] [CrossRef]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem. J. 2016, 473, 961–1000. [Google Scholar] [CrossRef]
- Ashabi, G.; Alamdary, S.Z.; Ramin, M.; Khodagholi, F. Reduction of hippocampal apoptosis by intracerebroventricular administration of extracellular signal-regulated protein kinase and/or p38 inhibitors in amyloid beta rat model of Alzheimer’s disease: Involvement of nuclear-related factor-2 and nuclear factor-kappaB. Basic Clin. Pharmacol. Toxicol. 2013, 112, 145–155. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Ron, D. Endoplasmic reticulum stress and the development of diabetes: A review. Diabetes 2002, 51, S455–S461. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef]
- Choe, K.P.; Leung, C.K. SKN-1/Nrf, a new unfolded protein response factor? PLoS Genet. 2013, 9, e1003827. [Google Scholar] [CrossRef]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013, 9, e1003701. [Google Scholar] [CrossRef]
- Zhang, Y.; Kobayashi, A.; Yamamoto, M.; Hayes, J.D. The Nrf3 transcription factor is a membrane-bound glycoprotein targeted to the endoplasmic reticulum through its N-terminal homology box 1 sequence. J. Biol. Chem. 2009, 284, 3195–3210. [Google Scholar] [CrossRef]
- Zhang, Y.; Lucocq, J.M.; Hayes, J.D. The Nrf1 CNC/bZIP protein is a nuclear envelope-bound transcription factor that is activated by t-butyl hydroquinone but not by endoplasmic reticulum stressors. Biochem. J. 2009, 418, 293–310. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Xue, P.; Woods, C.G.; Fu, J.; Feng, B.; Guan, D.; Sun, G.; Chan, J.Y.; Waalkes, M.P.; et al. Long isoforms of NRF1 contribute to arsenic-induced antioxidant response in human keratinocytes. Environ. Health Perspect. 2011, 119, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Goldberg, A.L. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Wang, M.; Hu, S.; Qiu, L.; Yang, F.; Zhang, Z.; Yu, S.; Pi, J.; Zhang, Y. Mechanisms controlling the multistage post-translational processing of endogenous Nrf1alpha/TCF11 proteins to yield distinct isoforms within the coupled positive and negative feedback circuits. Toxicol. Appl. Pharmacol. 2018, 360, 212–235. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef]
- Lee, C.S.; Ho, D.V.; Chan, J.Y. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 2013, 280, 3609–3620. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, S.; Zhang, Y. Nrf1 is paved as a new strategic avenue to prevent and treat cancer, neurodegenerative and other diseases. Toxicol. Appl. Pharmacol. 2018, 360, 273–283. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Herrmann, M.; et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef]
- Chen, L.; Kwong, M.; Lu, R.; Ginzinger, D.; Lee, C.; Leung, L.; Chan, J.Y. Nrf1 is critical for redox balance and survival of liver cells during development. Mol. Cell Biol. 2003, 23, 4673–4686. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wang, M.; Hu, S.; Ru, X.; Ren, Y.; Zhang, Z.; Yu, S.; Zhang, Y. Oncogenic activation of Nrf2, though as a master antioxidant transcription factor, liberated by specific knockout of the full-length Nrf1alpha that acts as a dominant tumor repressor. Cancers 2018, 10, 520. [Google Scholar] [CrossRef]
- Ren, Y.; Qiu, L.; Lu, F.; Ru, X.; Li, S.; Xiang, Y.; Yu, S.; Zhang, Y. TALENs-directed knockout of the full-length transcription factor Nrf1alpha that represses malignant behaviour of human hepatocellular carcinoma (HepG2) cells. Sci. Rep. 2016, 6, e23775. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qiu, L.; Ru, X.; Song, Y.; Zhang, Y. Distinct isoforms of Nrf1 diversely regulate different subsets of its cognate target genes. Sci. Rep. 2019, 9, e2960. [Google Scholar] [CrossRef]
- Xiang, Y.; Halin, J.; Fan, Z.; Hu, S.; Wang, M.; Qiu, L.; Zhang, Z.; Mattjus, P.; Zhang, Y. Topovectorial mechanisms control the juxtamembrane proteolytic processing of Nrf1 to remove its N-terminal polypeptides during maturation of the CNC-bZIP factor. Toxicol. Appl. Pharmacol. 2018, 360, 160–184. [Google Scholar] [CrossRef]
- Zhang, Y.; Hayes, J.D. The membrane-topogenic vectorial behaviour of Nrf1 controls its post-translational modification and transactivation activity. Sci. Rep. 2013, 3, e2006. [Google Scholar] [CrossRef]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef]
- Xiao, H.F.L.; Zhang, Y. Mechanisms underlying chemopreventive effects of flavonoids via multiple signaling nodes within Nrf2-ARE and AhR-XRE gene regulatory networks. Curr. Chem. Biol. 2013, 7, 37. [Google Scholar] [CrossRef]
- Pearse, B.R.; Hebert, D.N. Cotranslocational degradation: Utilitarianism in the ER stress response. Mol. Cell 2006, 23, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Ann. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Brown, N.J.; Pockley, A.G.; Wyld, L. The unfolded protein response and cancer: A brighter future unfolding? J. Mol. Med. 2007, 85, 331–341. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Choi, H.; Jackson, N.L.; Shaw, D.R.; Emanuel, P.D.; Liu, Y.L.; Tousson, A.; Meng, Z.; Blume, S.W. Mrtl-A translation/localization regulatory protein encoded within the human c-myc locus and distributed throughout the endoplasmic and nucleoplasmic reticular network. J. Cell Biochem. 2008, 105, 1092–1108. [Google Scholar] [CrossRef]
- Wethmar, K.; Smink, J.J.; Leutz, A. Upstream open reading frames: Molecular switches in (patho)physiology. Bioessays 2010, 32, 885–893. [Google Scholar] [CrossRef]
- Mielnicki, L.M.; Hughes, R.G.; Chevray, P.M.; Pruitt, S.C. Mutated Atf4 suppresses c-Ha-ras oncogene transcript levels and cellular transformation in NIH3T3 fibroblasts. Biochem. Biophys. Res. Commun. 1996, 228, 586–595. [Google Scholar] [CrossRef]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordonez-Rueda, D.; et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef]
- Van Keulen, B.J.; Rotteveel, J.; Finken, M.J.J. Unexplained death in patients with NGLY1 mutations may be explained by adrenal insufficiency. Physiol. Rep. 2019, 7, e13979. [Google Scholar] [CrossRef] [PubMed]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Owings, K.G.; Lowry, J.B.; Bi, Y.; Might, M.; Chow, C.Y. Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 2018, 27, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Endoplasmic reticulum-associated SKN-1A/Nrf1 mediates a cytoplasmic unfolded protein response and promotes longevity. eLife 2019, 8, e44425. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Breen, P.C.; Ruvkun, G. Protein sequence editing of SKN-1A/Nrf1 by peptide: N-Glycanase controls proteasome gene expression. Cell 2019, 177, 737–750. [Google Scholar] [CrossRef]
- Zhang, Y.; Lucocq, J.M.; Yamamoto, M.; Hayes, J.D. The NHB1 (N-terminal homology box 1) sequence in transcription factor Nrf1 is required to anchor it to the endoplasmic reticulum and also to enable its asparagine-glycosylation. Biochem. J. 2007, 408, 161–172. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, Y.; Li, S.; Hayes, J.D. Transcription factor Nrf1 is topologically repartitioned across membranes to enable target gene transactivation through its acidic glucose-responsive domains. PLoS ONE 2014, 9, e93458. [Google Scholar] [CrossRef]
- Zhang, Y.; Hayes, J.D. Identification of topological determinants in the N-terminal domain of transcription factor Nrf1 that control its orientation in the endoplasmic reticulum membrane. Biochem. J. 2010, 430, 497–510. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 is an ER membrane sensor that is central to cholesterol homeostasis. Cell 2017, 171, 1094–1109. [Google Scholar] [CrossRef]
- Wang, X.; Sato, R.; Brown, M.S.; Hua, X.; Goldstein, J.L. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 1994, 77, 53–62. [Google Scholar] [CrossRef]
- Sakai, J.; Duncan, E.A.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Bogdanov, M.; Vitrac, H.; Dowhan, W. Flip-Flopping Membrane Proteins: How the Charge Balance Rule Governs Dynamic Membrane Protein Topology. In Biogenesis of Fatty Acids, Lipids and Membranes; Springer: Cham, Switzerland, 2018; pp. 1–28. [Google Scholar]
- Baird, L.; Tsujita, T.; Kobayashi, E.H.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yamamoto, M. A Homeostatic Shift Facilitates Endoplasmic Reticulum Proteostasis through Transcriptional Integration of Proteostatic Stress Response Pathways. Mol. Cell Biol. 2017, 37, e00439-16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | Forward Primers (from 5′ to 3′-ends) | Reverse Primers (from 5′ to 3′-ends) |
|---|---|---|---|
| 60 | β-actin | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
| 468 | ATF4 | CCCTTCACCTTCTTACAACCTC | TGCCCAGCTCTAAACTAAAGGA |
| 22926 | ATF6 | AGCAGCACCCAAGACTCAAAC | GCATAAGCGTTGGTACTGTCTGA |
| 3309 | BIP | GAACGTCTGATTGGCGATGC | ACCACCTTGAACGGCAAGAA |
| 1649 | Chop | GGAAACAGAGTGGTCATTCCC | CTGCTTGAGCCGTTCATTCTC |
| 2730 | GCLM | GTGTGATGCCACCAGATTTGAC | CACAATGACCGAATACCGCAGT |
| 3162 | HO-1 | CAGAGCCTGGAAGACACCCTAA | AAACCACCCCAACCCTGCTAT |
| 2081 | IRE1 | GAGACCCTGCGCTATCTGAC | CTTGGCCTCTGTCTCCTTGG |
| 4779 | Nrf1 | GCTGGACACCATCCTGAATC | CCTTCTGCTTCATCTGTCGC |
| 4780 | Nrf2 | TCAGCGACGGAAAGAGTATGA | CCACTGGTTTCTGACTGGATGT |
| 9451 | PERK | CTTCCAGTGGGACCAAGACC | CGAGGTCCGACAGCTCTAAC |
| 5682 | PSMA1 | ATTCATCAAATTGAATATGCAAT | CTCTGATTGCGCCCTTTTCAA |
| 5685 | PSMA4 | TTGCTGTACATTGGCTGGGA | ACACAGCTGCAGCGCTATTA |
| 5688 | PSMA7 | TACATCACCCGCTACATCGC | AGAGCCTAGGAGTGCCATCA |
| 5694 | PSMB6 | TCAAGAAGGAGGGCAGGTGT | GTAAAGTGGCAACGGCGAA |
| 5695 | PSMB7 | CTGTGTCGGTGTATGCTCCA | TGCCAGTTTTCCGGACCTTT |
| 5700 | PSMC1 | ACAAGGTGCATGCCGTGATA | CTGTGCCAGGTGGACCATAG |
| 7494 | XBP1 | CCCTCCAGAACATCTCCCCAT | ACATGACTGGGTCCAAGTTGT |
| ID | Name | ARE/EpRE (5′-TGAC/GnnnGC-3′) | TRE/AP1 Site (5′-TGAC/GTCA-3′) |
|---|---|---|---|
| 3309 | BIP/GRP78 | TGGCGCAATCTCAGCTC (–4344 to –4328) ATTTTGACCAGGCTGGT (–3811 to –3795) TGGTGCGATCTCAGCTC (–2848 to –2832) TAAGTGACTGTGCTTTG (–2480 to –2464) GAGCTGAGATTGCACTA (–1339 to –1323) | CTCTTGAGTCACCAG (–2104 to –2090) GTACTGAGTCACAGG (–2048 to –2034) |
| 9451 | PERK | GGTTTGAGTTCGCTCAT (–2728 to –2712) TCTAGCAAACTCATATA (–1844 to –1828) GCGTGCCAGGTCAGAGT (–719 to –703) CCAATGAGAGAGCAAAC (+60 to +76) | |
| 22926 | ATF6 | TCTTGCTCTGTCACCCA (–3459 to –3443) GAGCTGAGATGGCTCCA (–2054 to –2038) GTTCTGAGATAGCCACG (–343 to –327) | |
| 1649 | Chop | CACAGCTTGGTCATGTC (–4521 to –4505) AAGGGCTACCTCAGTCA (–4384 to –4368) AGGCGCCCTGTCACCCA (–2780 to –2764) TCTCGCTCTGTCACCCA (–935 to –919) AAGCTGAGTTGGCCAGG (+2219 to +2235) | CGCATGACTCACCCA (–242 to –228) |
| 2081 | IRE1 | ACCCGCCACCTCAGCCT (–4079 to –4063) TGAGTGACTTGGCCGTG (–692 to –676) AGTCTGACGCGGCAGGT (–370 to –354) TGAGGCTCGGTCACCGC (+26 to +42) | |
| 7494 | XBP1 | TCCCTGACCGAGCTGGT (–4419 to –4403) CACTGCAGCCTCAATCT (–4205 to –4189) CTCAGCCTCCTCAGTAG (–3987 to –3971) ATGTTGACCAGGCTGGT (–3901 to –3885) CTGTTGACCAGGCTGGA (–2943 to –2927) CTGGTGACAGAGCCTGA (–869 to –853) AAATGCACGCTCATAGT (–701 to –685) | GGCATGAGTCACCGT (–4306 to –4292) |
| 468 | ATF4 | CTGCTGAGATTGCAGTA (–4933 to –4917) ATCTTGAGAGAGCTCAT (–4449 to –4433) ACCATGACTGGGCAAGC (–3612 to –3596) TTGCTGACTGTGCTCCC (–3105 to –3089) GGACTGACTTGGCTGAG (–2940 to –2924) ATTTGCACAGTCATCTG (–2230 to –2214) CCTCTGAGGCAGCAGGA (–1788 to –1773) CCATGCAGACTCAGCCG (–893 to –877) | GGCGTGAGTCAAGGG (+513 to +527) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.-p.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin. Antioxidants 2020, 9, 4. https://doi.org/10.3390/antiox9010004
Zhu Y-p, Zheng Z, Hu S, Ru X, Fan Z, Qiu L, Zhang Y. Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin. Antioxidants. 2020; 9(1):4. https://doi.org/10.3390/antiox9010004
Chicago/Turabian StyleZhu, Yu-ping, Ze Zheng, Shaofan Hu, Xufang Ru, Zhuo Fan, Lu Qiu, and Yiguo Zhang. 2020. "Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin" Antioxidants 9, no. 1: 4. https://doi.org/10.3390/antiox9010004
APA StyleZhu, Y.-p., Zheng, Z., Hu, S., Ru, X., Fan, Z., Qiu, L., & Zhang, Y. (2020). Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin. Antioxidants, 9(1), 4. https://doi.org/10.3390/antiox9010004