Lion’s Mane Mushroom, Hericium erinaceus (Bull.: Fr.) Pers. Suppresses H2O2-Induced Oxidative Damage and LPS-Induced Inflammation in HT22 Hippocampal Neurons and BV2 Microglia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Hot Water and Ethanolic Extracts
2.3. Estimation of Total Phenolic Content (TPC)
2.4. 2,2-Diphenyl-1-Picrylhydrazyl (DPPH) Free Radical Scavenging Assay
2.5. Cell Culture
2.6. MTS Cell Viability Assay
2.7. Nitric Oxide (NO) Level Assay
2.8. Reactive Oxygen Species (ROS) Assay
2.9. Tetramethylrhodamine Ethyl Ester (TMRE) Staining of Mitochondrial Membrane Potential (MMP)
2.10. Hoechst 33258 Nuclear Apoptotic Staining
2.11. Measurement of Catalase (CAT) and Glutathione (GSH) Level
2.12. Measurement of Mitochondrial Toxicity and Adenosine Triphosphate (ATP) Levels
2.13. Measurement of Caspase 3 Activity
2.14. Quantitative Polymerase Chain Gene (qPCR) Expression Analysis
| Nrf2: |
| Forward 5′-TCTCCTCGCTGGAAAAAGAA-3′ |
| Reverse 5′-AATGTGCTGGCTGTGCTTTA-3′ |
| HO-1: |
| Forward 5′-AGG TGT CCA GAG AAG GCT T-3′ |
| Reverse 5′-ATC TTG CAC CAG GCT AGC A-3′ |
| NQO1: |
| Forward 5′-ATC CTT CCG AGT CAT CTC TA-3′ |
| Reverse 5′-CAA CGA ATC TTG AAT GGA GG-3′ |
| Bcl-2: |
| Forward 5′-TCGCAGAGATGTCCAGTCAG-3′ |
| Reverse 5′-ATGCCGGTTCAGGTACTCAG-3′ |
| Bax: |
| Forward 5′-CACAGCGTGGTGGTACCTTA-3′ |
| Reverse 5′-TCTTCTGTACGGCGGTCTCT-3′ |
| GAPDH: |
| Forward 5′-TCACCACCATGGAGAAGGC-3′ |
| Reverse 5′-GCTAAGCAGTTGGTGGTGCA-3′ |
2.15. Statistical Analysis
3. Results
3.1. Total Phenolic Content (TPC) and 2,2-Diphenyl-1-Picrylhydrazyl (DPPH) Scavenging Activities of Hot Water (HE-HWA) and Ethanolic (HE-ETH) Extracts
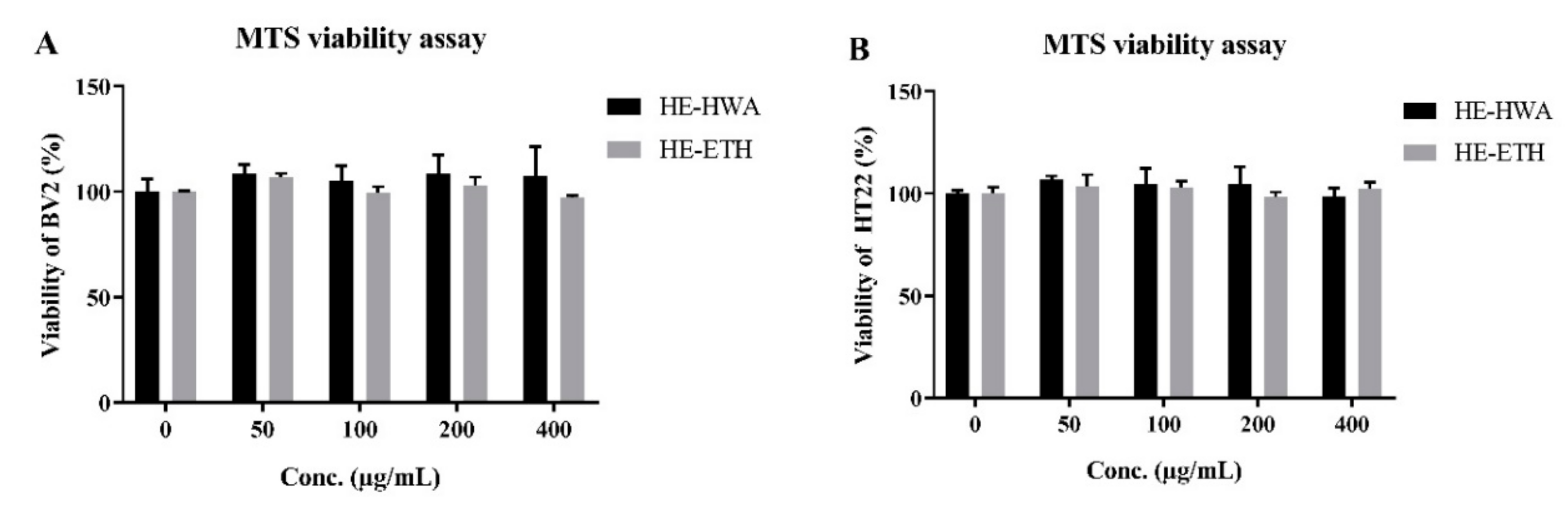
3.2. Effects of Hot Water (HE-HWA) and Ethanolic (HE-ETH) Extracts on the Viability of BV2 and HT22
3.3. Effects of Hot Water (HE-HWA) and Ethanolic (HE-ETH) Extracts on Lipopolysaccharide (LPS)-Induced NO Production in BV2
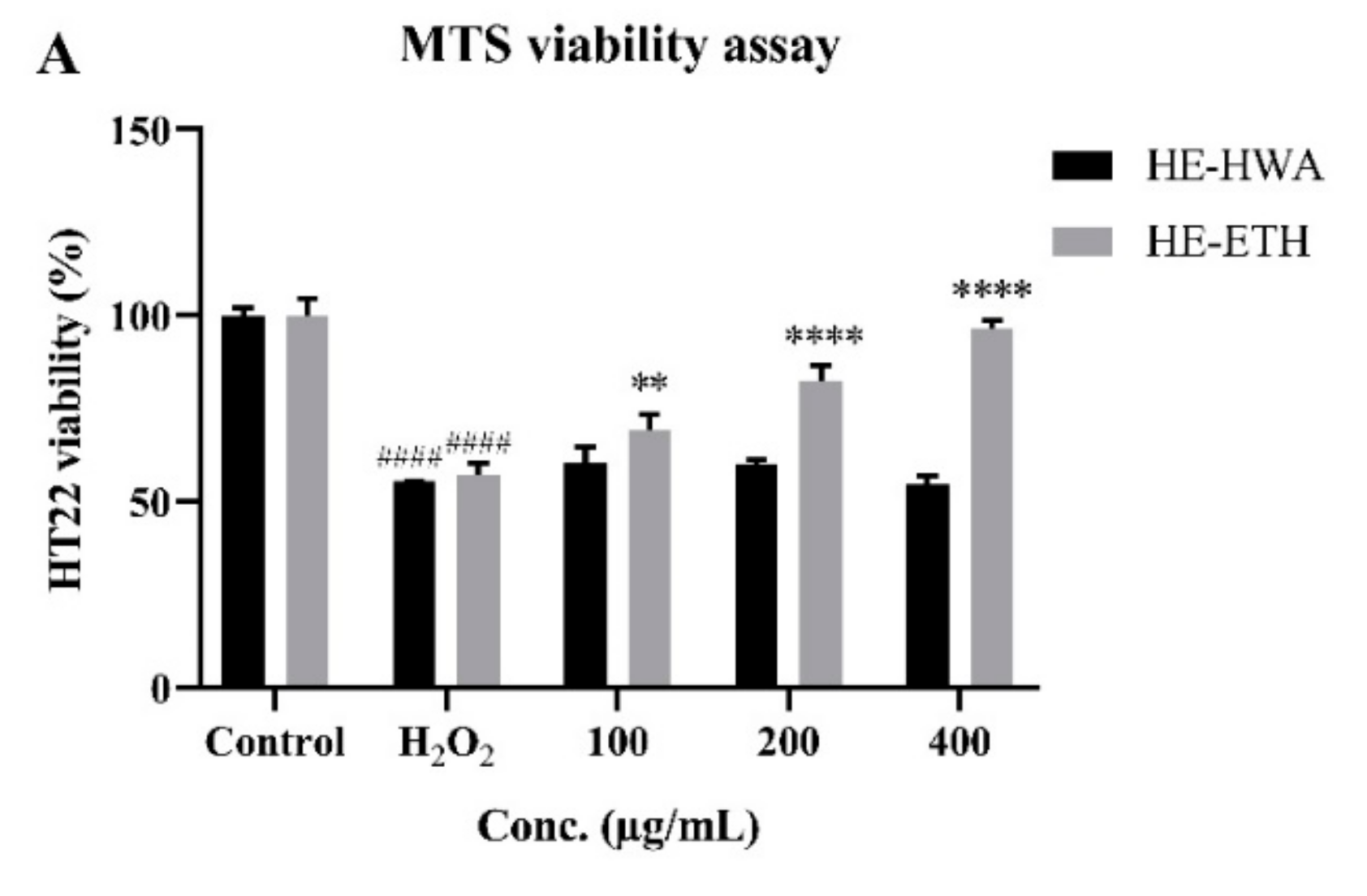
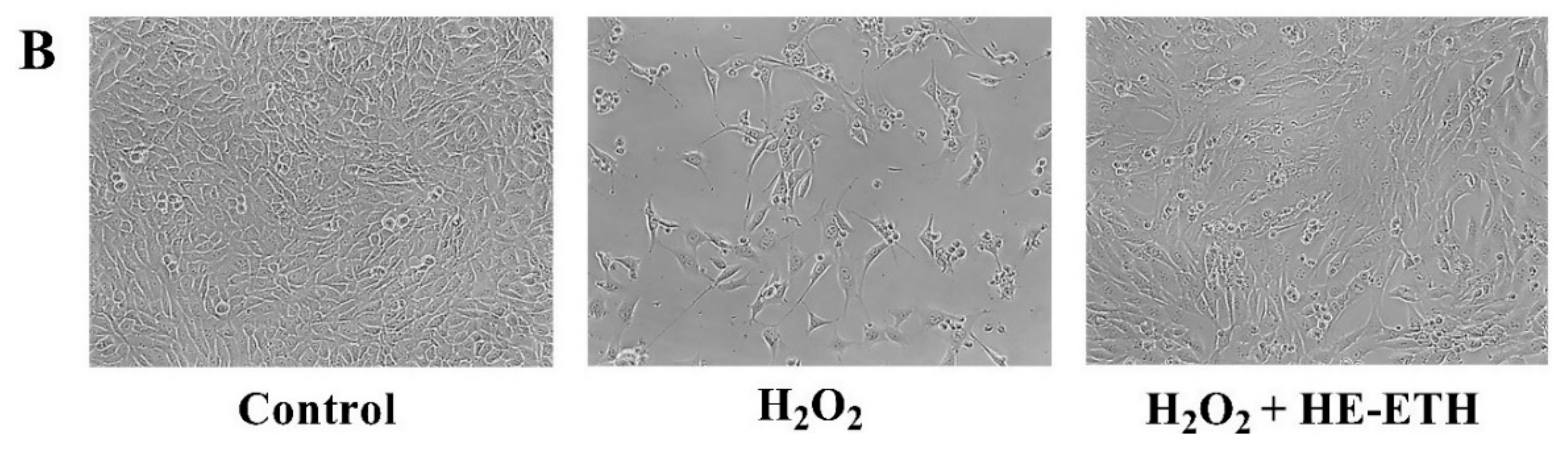
3.4. Effects of Hot Water (HE-HWA) and Ethanolic (HE-ETH) Extracts on the Viability of H2O2-Treated HT22
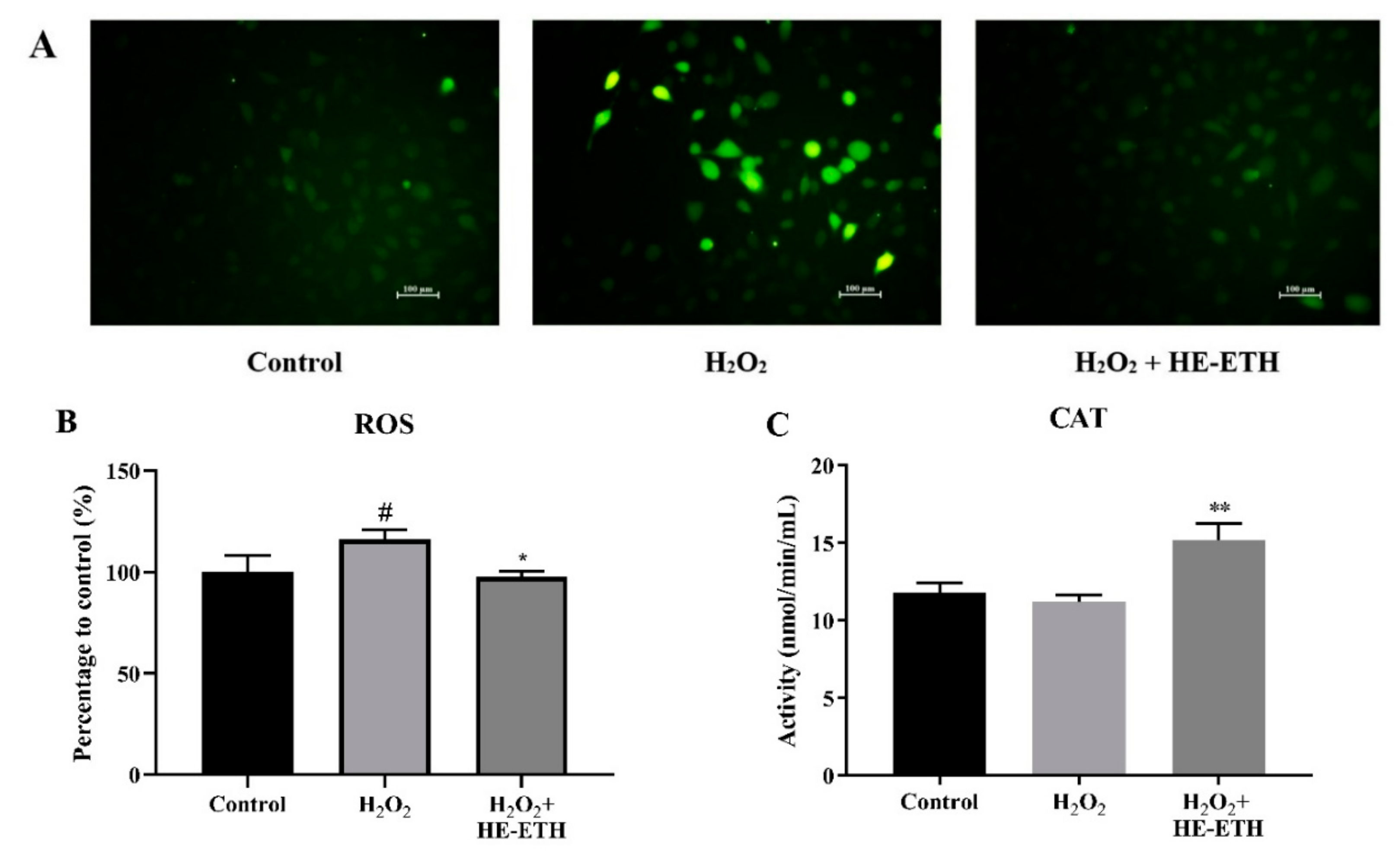
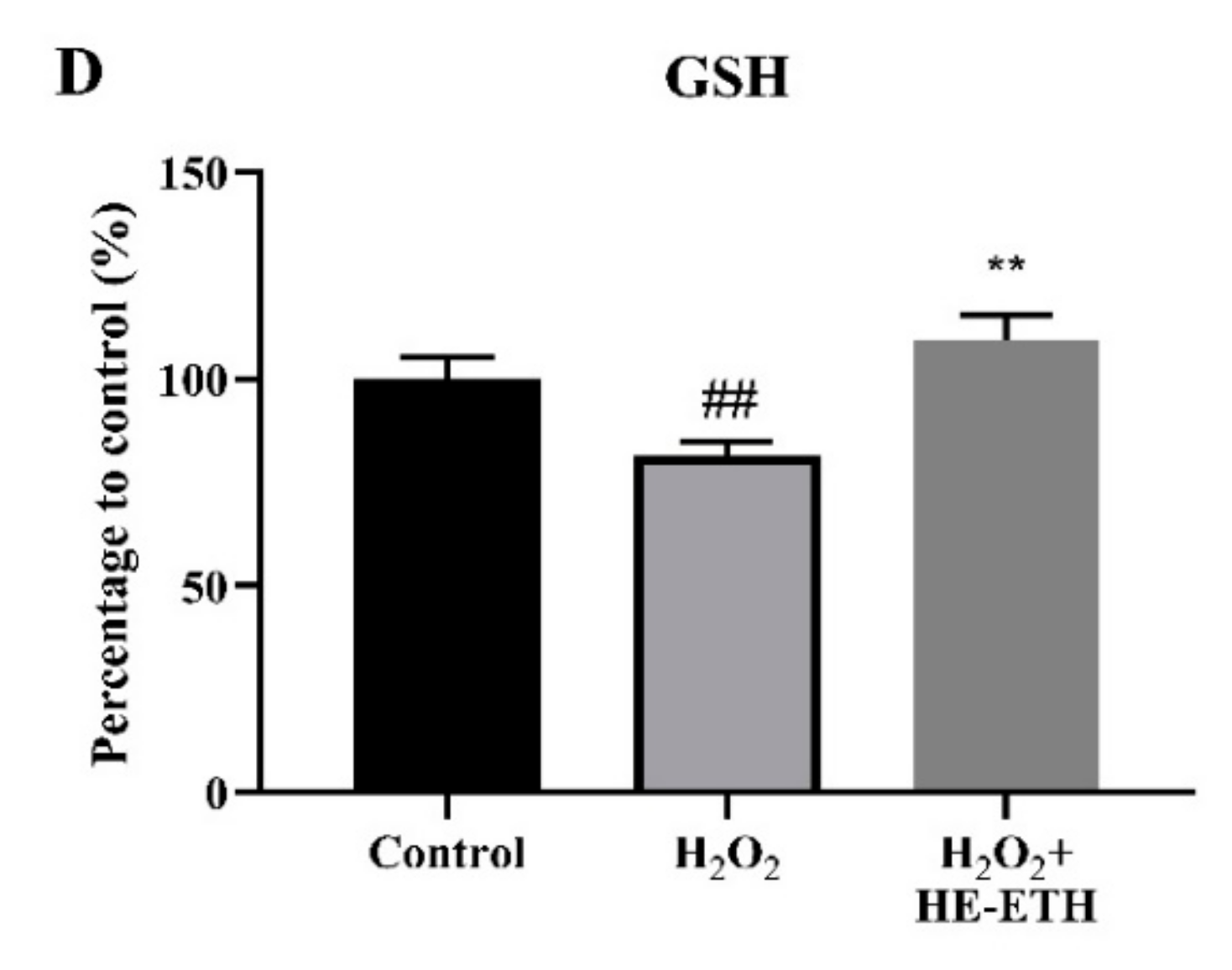
3.5. Effects of Ethanolic Extract (HE-ETH) on the Antioxidant Activities in H2O2-Treated HT22
3.6. Effects of Ethanolic Extract (HE-ETH) on the Mitochondrial Functions in H2O2-Treated HT22
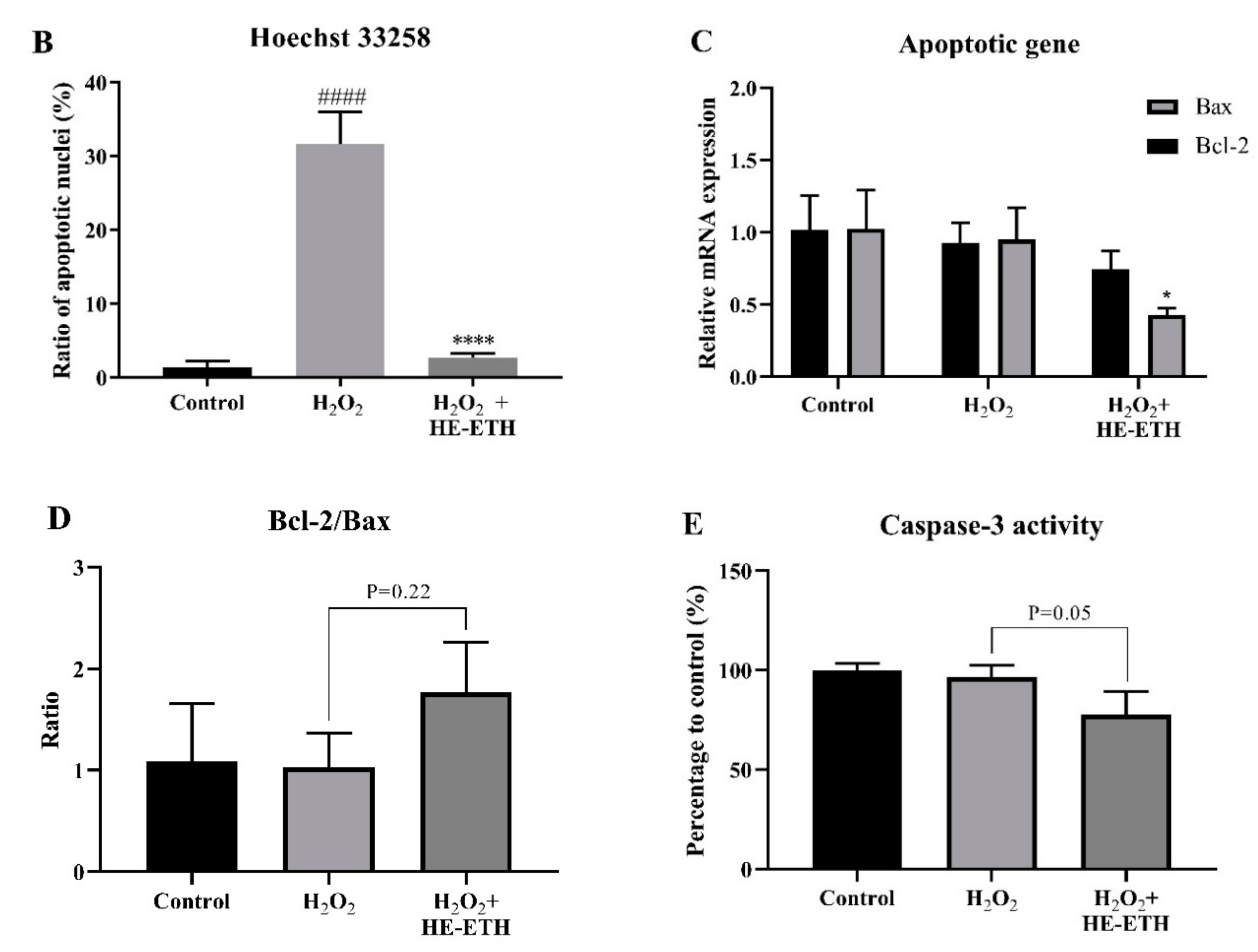
3.7. Effects of Ethanolic Extract (HE-ETH) on the Anti-Apoptosis in H2O2-Treated HT22
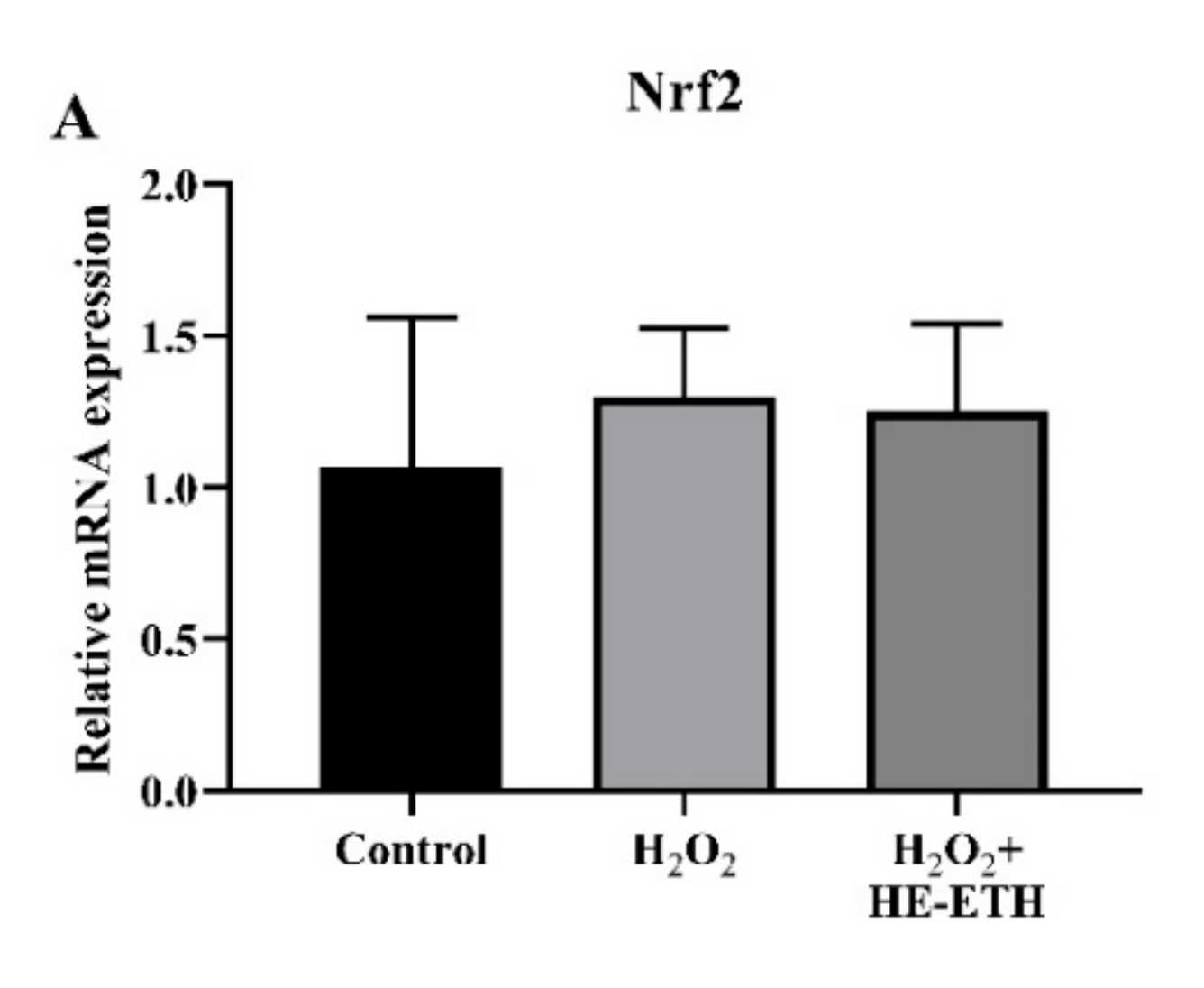
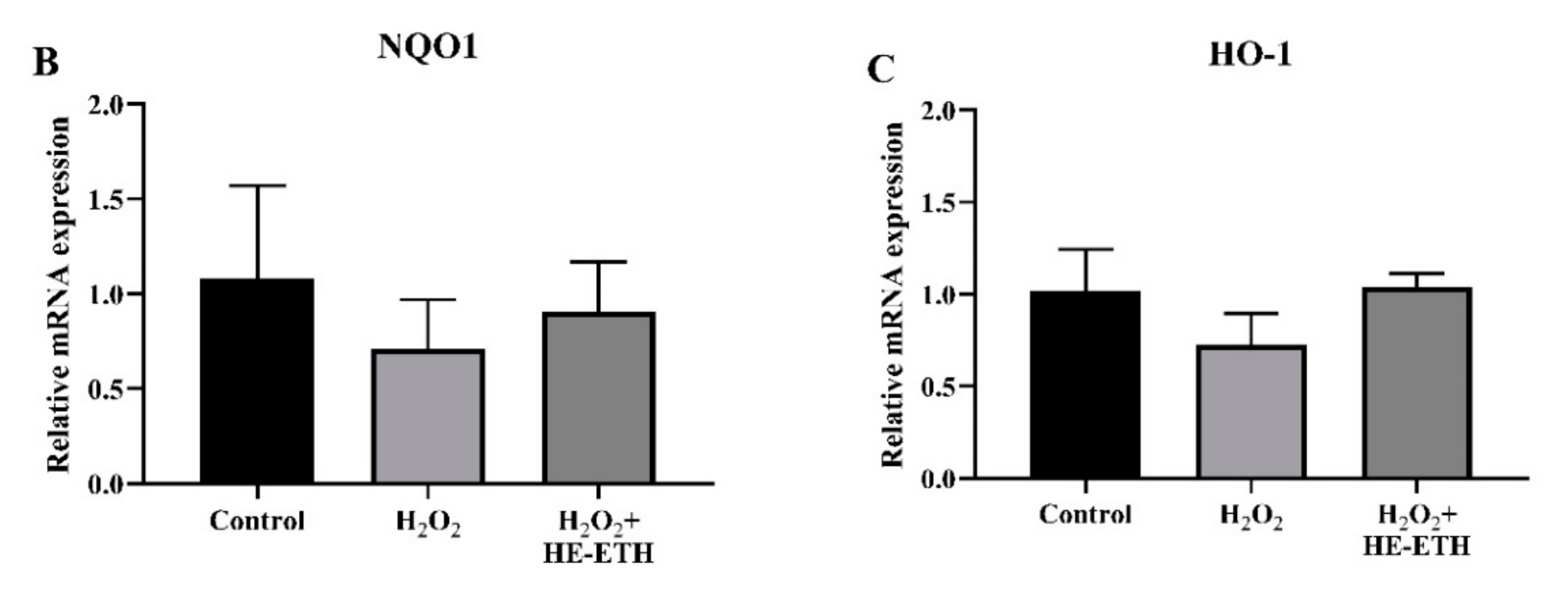
3.8. Effects of Ethanolic Extract (HE-ETH) on the Transcriptional Expression of Nrf2/NQO1/HO1 Antioxidant Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S265–S279. [Google Scholar] [CrossRef]
- Guest, J.; Grant, R.; Mori, T.A.; Croft, K.D. Changes in oxidative damage, inflammation and [NAD(H)] with age in cerebrospinal fluid. PLoS ONE 2014, 9, e85335. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.L.; Wilhelmus, M.M.M.; de Vries, H.E.; Drukarch, B.; Hoozemans, J.J.M.; van Horssen, J. Antioxidative defense mechanisms controlled by Nrf2: State-of-the-art and clinical perspectives in neurodegenerative diseases. Arch. Toxicol. 2014, 88, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Huang, S.; Mao, J.; Ding, K.; Zhou, Y.; Zeng, X.; Yang, W.; Wang, P.; Zhao, C.; Yao, J.; Xia, P.; et al. Polysaccharides from Ganoderma lucidum Promote Cognitive Function and Neural Progenitor Proliferation in Mouse Model of Alzheimer’s Disease. Stem Cell Rep. 2017, 8, 84–94. [Google Scholar] [CrossRef]
- Bai, R.; Zhang, C.-C.; Yin, X.; Wei, J.; Gao, J.-M. Striatoids A–F, Cyathane Diterpenoids with Neurotrophic Activity from Cultures of the Fungus Cyathus striatus. J. Nat. Prod. 2015, 78, 783–788. [Google Scholar] [CrossRef]
- Yang, W.; Yu, J.; Zhao, L.; Ma, N.; Fang, Y.; Pei, F.; Mariga, A.M.; Hu, Q. Polysaccharides from Flammulina velutipes improve scopolamine-induced impairment of learning and memory of rats. J. Funct. Foods 2015, 18, 411–422. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Jin, G.; Yang, X.; Zhang, Y. Polysaccharides from Pleurotus ostreatus alleviate cognitive impairment in a rat model of Alzheimer’s disease. Int. J. Biol. Macromol. 2016, 92, 935–941. [Google Scholar] [CrossRef]
- Kawagishi, H.; Ando, M.; Sakamoto, H.; Yoshida, S.; Ojima, F.; Ishiguro, Y.; Ukai, N.; Furukawa, S. Hericenones C, D and E, stimulators of nerve growth factor (NGF)-synthesis, from the mushroom Hericium erinaceum. Tetrahedron Lett. 1991, 32, 4561–4564. [Google Scholar] [CrossRef]
- Hao-Yu, T.; Xia, Y.; Cheng-Chen, Z.; Qian, J.; Jin-Ming, G. Structure Diversity, Synthesis, and Biological Activity of Cyathane Diterpenoids in Higher Fungi. Curr. Med. Chem. 2015, 22, 2375–2391. [Google Scholar] [CrossRef]
- Abdullah, N.; Ismail, S.M.; Aminudin, N.; Shuib, A.S.; Lau, B.F. Evaluation of Selected Culinary-Medicinal Mushrooms for Antioxidant and ACE Inhibitory Activities. Evid. Based Complement. Altern. Med. 2012, 2012, 464238. [Google Scholar] [CrossRef]
- Han, Z.-H.; Ye, J.-M.; Wang, G.-F. Evaluation of in vivo antioxidant activity of Hericium erinaceus polysaccharides. Int. J. Biol. Macromol. 2013, 52, 66–71. [Google Scholar] [CrossRef]
- Wong, J.-Y.; Abdulla, M.A.; Raman, J.; Phan, C.-W.; Kuppusamy, U.R.; Golbabapour, S.; Sabaratnam, V. Gastroprotective Effects of Lion’s Mane Mushroom Hericium erinaceus (Bull.:Fr.) Pers. (Aphyllophoromycetideae) Extract against Ethanol-Induced Ulcer in Rats. Evid. Based Complement. Altern. Med. 2013, 2013, 492976. [Google Scholar] [CrossRef]
- Zhang, Z.; Lv, G.; Pan, H.; Pandey, A.; He, W.; Fan, L. Antioxidant and hepatoprotective potential of endo-polysaccharides from Hericium erinaceus grown on tofu whey. Int. J. Biol. Macromol. 2012, 51, 1140–1146. [Google Scholar] [CrossRef]
- Cheng, J.-H.; Tsai, C.-L.; Lien, Y.-Y.; Lee, M.-S.; Sheu, S.-C. High molecular weight of polysaccharides from Hericium erinaceus against amyloid beta-induced neurotoxicity. BMC Complement. Altern. Med. 2016, 16, 170. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Q.; Cui, J.; Wang, L.; Xiong, L.; Wang, W.; Li, D.; Liu, N.; Wu, Y.; Mao, C. Systemic Screening of Strains of the Lion’s Mane Medicinal Mushroom Hericium erinaceus (Higher Basidiomycetes) and Its Protective Effects on Aβ-Triggered Neurotoxicity in PC12 Cells. Int. J. Med. Mushrooms 2015, 17, 219–229. [Google Scholar] [CrossRef]
- Kuo, H.-C.; Lu, C.-C.; Shen, C.-H.; Tung, S.-Y.; Hsieh, M.C.; Lee, K.-C.; Lee, L.-Y.; Chen, C.-C.; Teng, C.-C.; Huang, W.-S.; et al. Hericium erinaceus mycelium and its isolated erinacine A protection from MPTP-induced neurotoxicity through the ER stress, triggering an apoptosis cascade. J. Transl. Med. 2016, 14, 78. [Google Scholar] [CrossRef]
- Chang, C.-H.; Chen, Y.; Yew, X.-X.; Chen, H.-X.; Kim, J.-X.; Chang, C.-C.; Peng, C.-C.; Peng, R.Y. Improvement of erinacine A productivity in Hericium erinaceus mycelia and its neuroprotective bioactivity against the glutamate-insulted apoptosis. LWT-Food Sci. Technol. 2016, 65, 1100–1108. [Google Scholar] [CrossRef]
- Zhang, J.; An, S.; Hu, W.; Teng, M.; Wang, X.; Qu, Y.; Liu, Y.; Yuan, Y.; Wang, D. The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2016, 17, 1810. [Google Scholar] [CrossRef]
- Kim, Y.O.; Lee, S.W.; Oh, C.H.; Rhee, Y.H. Hericium erinaceus suppresses LPS-induced pro-inflammation gene activation in RAW264.7 macrophages. Immunopharmacol. Immunotoxicol. 2012, 34, 504–512. [Google Scholar] [CrossRef]
- Li, Q.Z.; Wu, D.; Chen, X.; Zhou, S.; Liu, Y.; Yang, Y.; Cui, F. Chemical Compositions and Macrophage Activation of Polysaccharides from Leon’s Mane Culinary-Medicinal Mushroom Hericium erinaceus (Higher Basidiomycetes) in Different Maturation Stages. Int. J. Med. Mushrooms 2015, 17, 443–452. [Google Scholar] [CrossRef]
- Noh, H.J.; Yoon, J.Y.; Kim, G.S.; Lee, S.E.; Lee, D.Y.; Choi, J.H.; Kim, S.Y.; Kang, K.S.; Cho, J.Y.; Kim, K.H. Benzyl alcohol derivatives from the mushroom Hericium erinaceum attenuate LPS-stimulated inflammatory response through the regulation of NF-κB and AP-1 activity. Immunopharmacol. Immunotoxicol. 2014, 36, 349–354. [Google Scholar] [CrossRef]
- Mori, K.; Ouchi, K.; Hirasawa, N. The Anti-Inflammatory Effects of Lion’s Mane Culinary-Medicinal Mushroom, Hericium erinaceus (Higher Basidiomycetes) in a Coculture System of 3T3-L1 Adipocytes and RAW264 Macrophages. Int. J. Med. Mushrooms 2015, 17, 609–618. [Google Scholar] [CrossRef]
- Li, Q.-Z.; Wu, D.; Zhou, S.; Liu, Y.-F.; Li, Z.-P.; Feng, J.; Yang, Y. Structure elucidation of a bioactive polysaccharide from fruiting bodies of Hericium erinaceus in different maturation stages. Carbohydr. Polym. 2016, 144, 196–204. [Google Scholar] [CrossRef]
- Lai, P.L.; Naidu, M.; Sabaratnam, V.; Wong, K.H.; David, R.P.; Kuppusamy, U.R.; Abdullah, N.; Malek, S.N. Neurotrophic properties of the Lion’s mane medicinal mushroom, Hericium erinaceus (Higher Basidiomycetes) from Malaysia. Int. J. Med. Mushrooms 2013, 15, 539–554. [Google Scholar] [CrossRef]
- Phan, C.-W.; Lee, G.-S.; Hong, S.-L.; Wong, Y.-T.; Brkljača, R.; Urban, S.; Abd Malek, S.N.; Sabaratnam, V. Hericium erinaceus (Bull.: Fr) Pers. Cultivated under tropical conditions: Isolation of hericenones and demonstration of NGF-mediated neurite outgrowth in PC12 cells via MEK/ERK and PI3K-Akt signaling pathways. Food Funct. 2014, 5, 3160–3169. [Google Scholar] [CrossRef]
- Kah Hui, W.; Sabaratnam, V.; Abdullah, N.; Naidu, M.; Keynes, R. Activity of Aqueous Extracts of Lion’s Mane Mushroom Hericium erinaceus (Bull.: Fr.) Pers. (Aphyllophoromycetideae) on the Neural Cell Line NG108-15. Int. J. Med. Mushrooms 2007, 9, 57–65. [Google Scholar]
- Singleton, V.L.; Rossi, J.A. Colorimetry of Total Phenolics with Phosphomolybdic-Phosphotungstic Acid Reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- Blois, M.S. Antioxidant Determinations by the Use of a Stable Free Radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Pepper, C.; Hoy, T.; Bentley, D.P. Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. Br. J. Cancer 1997, 76, 935–938. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2665–2672. [Google Scholar] [CrossRef]
- Chaiyasut, C.; Sivamaruthi, B.S. Anti-hyperglycemic property of Hericium erinaceus—A mini review. Asian Pac. J. Trop. Biomed. 2017, 7, 1036–1040. [Google Scholar] [CrossRef]
- Kawagishi, H.; Ando, M.; Shinba, K.; Sakamoto, H.; Yoshida, S.; Ojima, F.; Ishiguro, Y.; Ukai, N.; Furukawa, S. Chromans, hericenones F, G and H from the mushroom Hericium erinaceum. Phytochemistry 1992, 32, 175–178. [Google Scholar] [CrossRef]
- Kawagishi, H.; Ishiyama, D.; Mori, H.; Sakamoto, H.; Ishiguro, Y.; Furukawa, S.; Jingxuan, L. Dictyophorines A and B, two stimulators of NGF-synthesis from the mushroom Dictyophora indusiata. Phytochemistry 1997, 45, 1203–1205. [Google Scholar] [CrossRef]
- Kawagishi, H.; Shimada, A.; Hosokawa, S.; Mori, H.; Sakamoto, H.; Ishiguro, Y.; Sakemi, S.; Bordner, J.; Kojima, N.; Furukawa, S. Erinacines E, F, and G, stimulators of nerve growth factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1996, 37, 7399–7402. [Google Scholar] [CrossRef]
- Kawagishi, H.; Shimada, A.; Shirai, R.; Okamoto, K.; Ojima, F.; Sakamoto, H.; Ishiguro, Y.; Furukawa, S. Erinacines A, B and C, strong stimulators of nerve growth factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1994, 35, 1569–1572. [Google Scholar] [CrossRef]
- Kawagishi, H.; Simada, A.; Shizuki, K.; Mori, H.; Okamoto, K.; Sakamoto, H.; Furukawa, S. Erinacine D, a stimulator of NGF-synthesis, from the mycelia of Hericium erinaceum. Heterocycl. Commun. 1996, 2, 51–54. [Google Scholar] [CrossRef]
- Lee, E.W.; Shizuki, K.; Hosokawa, S.; Suzuki, M.; Suganuma, H.; Inakuma, T.; Li, J.; Ohnishi-Kameyama, M.; Nagata, T.; Furukawa, S.; et al. Two novel diterpenoids, erinacines H and I from the Mycelia of Hericium erinaceum. Biosci. Biotechnol. Biochem. 2000, 64, 2402–2405. [Google Scholar] [CrossRef]
- Phan, C.-W.; David, P.; Naidu, M.; Wong, K.-H.; Sabaratnam, V. Neurite outgrowth stimulatory effects of culinary-medicinal mushrooms and their toxicity assessment using differentiating Neuro-2a and embryonic fibroblast BALB/3T3. BMC Complement. Altern. Med. 2013, 13, 261. [Google Scholar] [CrossRef]
- Samberkar, S.; Gandhi, S.; Naidu, M.; Wong, K.H.; Raman, J.; Sabaratnam, V. Lion’s mane, hericium erinaceus and tiger milk, lignosus rhinocerotis (Higher basidiomycetes) medicinal mushrooms stimulate neurite outgrowth in dissociated cells of brain, spinal cord, and retina: An in vitro study. Int. J. Med. Mushrooms 2015, 17, 1047–1054. [Google Scholar] [CrossRef]
- Wang, M.; Kanako, N.; Zhang, Y.; Xiao, X.; Gao, Q.; Tetsuya, K. A unique polysaccharide purified from Hericium erinaceus mycelium prevents oxidative stress induced by H2O2 in human gastric mucosa epithelium cell. PLoS ONE 2017, 12, e0181546. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Yang, S.; Zhao, D.; Wang, M. A polysaccharide from cultured mycelium of Hericium erinaceus relieves ulcerative colitis by counteracting oxidative stress and improving mitochondrial function. Int. J. Biol. Macromol. 2019, 125, 572–579. [Google Scholar] [CrossRef]
- Bhebhe, M.; Füller, T.; Chipurura, B.; Muchuweti, M. Effect of Solvent Type on Total Phenolic Content and Free Radical Scavenging Activity of Black Tea and Herbal Infusions. Food Anal. Methods 2015, 9, 1060–1067. [Google Scholar] [CrossRef]
- Pasrija, D.; Anandharamakrishnan, C. Techniques for extraction of green tea polyphenols: A review. Food Bioprocess Technol. 2015, 8, 935–950. [Google Scholar] [CrossRef]
- Lin, C.-W.; Yu, C.-W.; Wu, S.-C.; Yih, K.-H. DPPH Free-Radical Scavenging Activity, Total Phenolic Contents and Chemical Composition Analysis of Forty-Two Kinds of Essential Oils. J. Food Drug Anal. 2009, 17, 386–395. [Google Scholar]
- Dai, J.; Mumper, R.J. Plant phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Aschbacher, K.; O’Donovan, A.; Wolkowitz, O.M.; Dhabhar, F.S.; Su, Y.; Epel, E. Good stress, bad stress and oxidative stress: Insights from anticipatory cortisol reactivity. Psychoneuroendocrinology 2013, 38, 1698–1708. [Google Scholar] [CrossRef]
- Bergamini, C.M.; Gambetti, S.; Dondi, A.; Cervellati, C. Oxygen, reactive oxygen species and tissue damage. Curr. Pharm. Des. 2004, 10, 1611–1626. [Google Scholar] [CrossRef]
- Singhal, G.; Baune, B.T. Microglia: An Interface between the Loss of Neuroplasticity and Depression. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef]
- Kim, H.D.; Li, H.; Han, E.Y.; Jeong, H.J.; Lee, J.H.; Ryu, J.-H. Modulation of Inducible Nitric Oxide Synthase Expression in LPS-Stimulated BV-2 Microglia by Prenylated Chalcones from Cullen corylifolium (L.) Medik. Through Inhibition of I-κBα Degradation. Molecules 2018, 23, 109. [Google Scholar] [CrossRef]
- Hwang, G.H.; Jeon, Y.J.; Han, H.J.; Park, S.H.; Baek, K.M.; Chang, W.; Kim, J.S.; Kim, L.K.; Lee, Y.-M.; Lee, S.; et al. Protective effect of butylated hydroxylanisole against hydrogen peroxide-induced apoptosis in primary cultured mouse hepatocytes. J. Vet. Sci. 2015, 16, 17–23. [Google Scholar] [CrossRef]
- Kwon, S.-H.; Hong, S.-I.; Ma, S.-X.; Lee, S.-Y.; Jang, C.-G. 3′, 4′, 7-Trihydroxyflavone prevents apoptotic cell death in neuronal cells from hydrogen peroxide-induced oxidative stress. Food Chem. Toxicol. 2015, 80, 41–51. [Google Scholar] [CrossRef]
- Dai, S.-H.; Chen, T.; Wang, Y.-H.; Zhu, J.; Luo, P.; Rao, W.; Yang, Y.-F.; Fei, Z.; Jiang, X.-F. Sirt3 attenuates hydrogen peroxide-induced oxidative stress through the preservation of mitochondrial function in HT22 cells. Int. J. Mol. Med. 2014, 34, 1159–1168. [Google Scholar] [CrossRef]
- Kang, S.-M.; Cha, S.-H.; Ko, J.-Y.; Kang, M.-C.; Kim, D.; Heo, S.-J.; Kim, J.-S.; Heu, M.S.; Kim, Y.-T.; Jung, W.-K.; et al. Neuroprotective effects of phlorotannins isolated from a brown alga, Ecklonia cava, against H2O2-induced oxidative stress in murine hippocampal HT22 cells. Environ. Toxicol. Pharmacol. 2012, 34, 96–105. [Google Scholar] [CrossRef]
- Xu, H.; Luo, P.; Zhao, Y.; Zhao, M.; Yang, Y.; Chen, T.; Huo, K.; Han, H.; Fei, Z. Iduna protects HT22 cells from hydrogen peroxide-induced oxidative stress through interfering poly(ADP-ribose) polymerase-1-induced cell death (parthanatos). Cell. Signal. 2013, 25, 1018–1026. [Google Scholar] [CrossRef]
- Li, L.; Ng, T.; Song, M.; Yuan, F.; Liu, Z.; Wang, C.; Jiang, Y.; Fu, M.; Liu, F. A polysaccharide–peptide complex from abalone mushroom (Pleurotus abalonus) fruiting bodies increases activities and gene expression of antioxidant enzymes and reduces lipid peroxidation in senescence-accelerated mice. Appl. Microbiol. Biotechnol. 2007, 75, 863–869. [Google Scholar] [CrossRef]
- Govindan, S.; Johnson, E.E.R.; Christopher, J.; Shanmugam, J.; Thirumalairaj, V.; Gopalan, J. Antioxidant and anti-aging activities of polysaccharides from Calocybe indica var. APK2. Exp. Toxicol. Pathol. 2016, 68, 329–334. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, X.; Hu, Y.-S.; Wu, Y.; Wang, Q.-Z.; Li, N.-N.; Guo, Q.-C.; Dong, X.-C. Evaluation of in vivo antioxidant activities of Ganoderma lucidum polysaccharides in STZ-diabetic rats. Food Chem. 2009, 115, 32–36. [Google Scholar] [CrossRef]
- Veena, R.K.; Ajith, T.A.; Janardhanan, K.K. Lingzhi or reishi medicinal mushroom, Ganoderma lucidum (Agaricomycetes), prevents doxorubicin-induced cardiotoxicity in rats. Int. J. Med. Mushrooms 2018, 20, 761–774. [Google Scholar] [CrossRef]
- Xu, N.; Gao, Z.; Zhang, J.; Jing, H.; Li, S.; Ren, Z.; Wang, S.; Jia, L. Hepatoprotection of enzymatic-extractable mycelia zinc polysaccharides by Pleurotus eryngii var. tuoliensis. Carbohydr. Polym. 2017, 157, 196–206. [Google Scholar] [CrossRef]
- Krishnamoorthy, D.; Sankaran, M. Modulatory effect of Pleurotus ostreatus on oxidant/antioxidant status in 7, 12-dimethylbenz (a) anthracene induced mammary carcinoma in experimental rats-A dose-response study. J. Cancer Res. Ther. 2016, 12, 386. [Google Scholar]
- Anandhi, R.; Annadurai, T.; Anitha, T.S.; Muralidharan, A.R.; Najmunnisha, K.; Nachiappan, V.; Thomas, P.A.; Geraldine, P. Antihypercholesterolemic and antioxidative effects of an extract of the oyster mushroom, Pleurotus ostreatus, and its major constituent, chrysin, in Triton WR-1339-induced hypercholesterolemic rats. J. Physiol. Biochem. 2013, 69, 313–323. [Google Scholar] [CrossRef]
- Giannenas, I.; Pappas, I.; Mavridis, S.; Kontopidis, G.; Skoufos, J.; Kyriazakis, I. Performance and antioxidant status of broiler chickens supplemented with dried mushrooms (Agaricus bisporus) in their diet. Poult. Sci. 2010, 89, 303–311. [Google Scholar] [CrossRef]
- Jayakumar, T.; Thomas, P.A.; Ramesh, E.; Geraldine, P. An extract of the pleurotus ostreatus mushroom bolsters the glutathione redox system in various organs of aged rats. J. Med. Food 2010, 13, 771–778. [Google Scholar] [CrossRef]
- Wang, Y.; Li, B.; Zhang, X. Scutellaria barbata D. Don (SBD) protects oxygen glucose deprivation/reperfusion-induced injuries of PC12 cells by up-regulating Nrf2. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1797–1807. [Google Scholar] [CrossRef]
- Ren, J.; Yuan, L.; Wang, W.; Zhang, M.; Wang, Q.; Li, S.; Zhang, L.; Hu, K. Tricetin protects against 6-OHDA-induced neurotoxicity in Parkinson’s disease model by activating Nrf2/HO-1 signaling pathway and preventing mitochondria-dependent apoptosis pathway. Toxicol. Appl. Pharmacol. 2019, 378. [Google Scholar] [CrossRef]
- Wang, A.; Si, Z.; Xue, P.; Li, X.; Liu, J. Tacrolimus protects hippocampal neurons of rats with status epilepticus through suppressing oxidative stress and inhibiting mitochondrial pathway of apoptosis. Brain Res. 2019, 1715, 176–181. [Google Scholar] [CrossRef]
- Naoi, M.; Wu, Y.; Shamoto-Nagai, M.; Maruyama, W. Mitochondria in Neuroprotection by Phytochemicals: Bioactive Polyphenols Modulate Mitochondrial Apoptosis System, Function and Structure. Int. J. Mol. Sci. 2019, 20, 2451. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Yi, H.; Inaba-Hasegawa, K.; Akao, Y.; Shamoto-Nagai, M. Mitochondria in neurodegenerative disorders: Regulation of the redox state and death signaling leading to neuronal death and survival. J. Neural Transm. 2009, 116, 1371–1381. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef]
- Jeong, G.-S.; Li, B.; Lee, D.-S.; Kim, K.H.; Lee, I.K.; Lee, K.R.; Kim, Y.-C. Cytoprotective and anti-inflammatory effects of spinasterol via the induction of heme oxygenase-1 in murine hippocampal and microglial cell lines. Int. Immunopharmacol. 2010, 10, 1587–1594. [Google Scholar] [CrossRef]
- Bijjem, K.R.V.; Padi, S.S.; lal Sharma, P. Pharmacological activation of heme oxygenase (HO)-1/carbon monoxide pathway prevents the development of peripheral neuropathic pain in Wistar rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 79–90. [Google Scholar] [CrossRef]
- Tanigawa, S.; Fujii, M.; Hou, D.-X. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic. Biol. Med. 2007, 42, 1690–1703. [Google Scholar] [CrossRef]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H: Quinone oxidoreductase 1 (NQO1): Chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Koistinaho, J.; Miettinen, S.; Keinänen, R.; Vartiainen, N.; Roivainen, R.; Laitinen, J.T. Long-term induction of haem oxygenase-1 (HSP-32) in astrocytes and microglia following transient focal brain ischaemia in the rat. Eur. J. Neurosci. 1996, 8, 2265–2272. [Google Scholar] [CrossRef]
- Beschorner, R.; Adjodah, D.; Schwab, J.M.; Mittelbronn, M.; Pedal, I.; Mattern, R.; Schluesener, H.J.; Meyermann, R. Long-term expression of heme oxygenase-1 (HO-1, HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol. 2000, 100, 377–384. [Google Scholar] [CrossRef]
- Schipper, H.; Liberman, A.; Stopa, E. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Premkumar, D.R.; Smith, M.A.; Richey, P.L.; Petersen, R.B.; Castellani, R.; Kutty, R.K.; Wiggert, B.; Perry, G.; Kalaria, R.N. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer’s disease. J. Neurochem. 1995, 65, 1399–1402. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Nguyen, T.L.X.; Kim, C.K.; Cho, J.-H.; Lee, K.-H.; Ahn, J.-Y. Neuroprotection signaling pathway of nerve growth factor and brain-derived neurotrophic factor against staurosporine induced apoptosis in hippocampal H19-7 cells. Exp. Amp Mol. Med. 2010, 42, 583–595. [Google Scholar] [CrossRef]
- Karmarkar, S.W.; Bottum, K.M.; Krager, S.L.; Tischkau, S.A. ERK/MAPK is essential for endogenous neuroprotection in SCN2.2 cells. PLoS ONE 2011, 6, e23493. [Google Scholar] [CrossRef]
- Lahiani, A.; Brand-Yavin, A.; Yavin, E.; Lazarovici, P. Neuroprotective Effects of Bioactive Compounds and MAPK Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells. Brain Sci. 2018, 8, 32. [Google Scholar] [CrossRef]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT Signalling Cascades inParkinson’s Disease. Int. J. Mol. Cell. Med. 2015, 4, 67–86. [Google Scholar]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kushairi, N.; Phan, C.W.; Sabaratnam, V.; David, P.; Naidu, M. Lion’s Mane Mushroom, Hericium erinaceus (Bull.: Fr.) Pers. Suppresses H2O2-Induced Oxidative Damage and LPS-Induced Inflammation in HT22 Hippocampal Neurons and BV2 Microglia. Antioxidants 2019, 8, 261. https://doi.org/10.3390/antiox8080261
Kushairi N, Phan CW, Sabaratnam V, David P, Naidu M. Lion’s Mane Mushroom, Hericium erinaceus (Bull.: Fr.) Pers. Suppresses H2O2-Induced Oxidative Damage and LPS-Induced Inflammation in HT22 Hippocampal Neurons and BV2 Microglia. Antioxidants. 2019; 8(8):261. https://doi.org/10.3390/antiox8080261
Chicago/Turabian StyleKushairi, Naufal, Chia Wei Phan, Vikineswary Sabaratnam, Pamela David, and Murali Naidu. 2019. "Lion’s Mane Mushroom, Hericium erinaceus (Bull.: Fr.) Pers. Suppresses H2O2-Induced Oxidative Damage and LPS-Induced Inflammation in HT22 Hippocampal Neurons and BV2 Microglia" Antioxidants 8, no. 8: 261. https://doi.org/10.3390/antiox8080261
APA StyleKushairi, N., Phan, C. W., Sabaratnam, V., David, P., & Naidu, M. (2019). Lion’s Mane Mushroom, Hericium erinaceus (Bull.: Fr.) Pers. Suppresses H2O2-Induced Oxidative Damage and LPS-Induced Inflammation in HT22 Hippocampal Neurons and BV2 Microglia. Antioxidants, 8(8), 261. https://doi.org/10.3390/antiox8080261