Suppression of Light-Induced Retinal Degeneration by Quercetin via the AP-1 Pathway in Rats

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Quercetin Administration
2.3. Light Exposure Model
2.4. ERGs
2.5. Preparation of Retinal Paraffin Sections
2.6. Light-Induced Retinal Damage
2.7. Terminal Deoxynucleotidyl Transferase (TdT)-Mediated 2′-Deoxyuridine-5′-Triphosphate (dUTP) Nick End Labeling (TUNEL)
2.8. Immunohistochemistry for 8-OHdG
2.9. Electron Microscopy
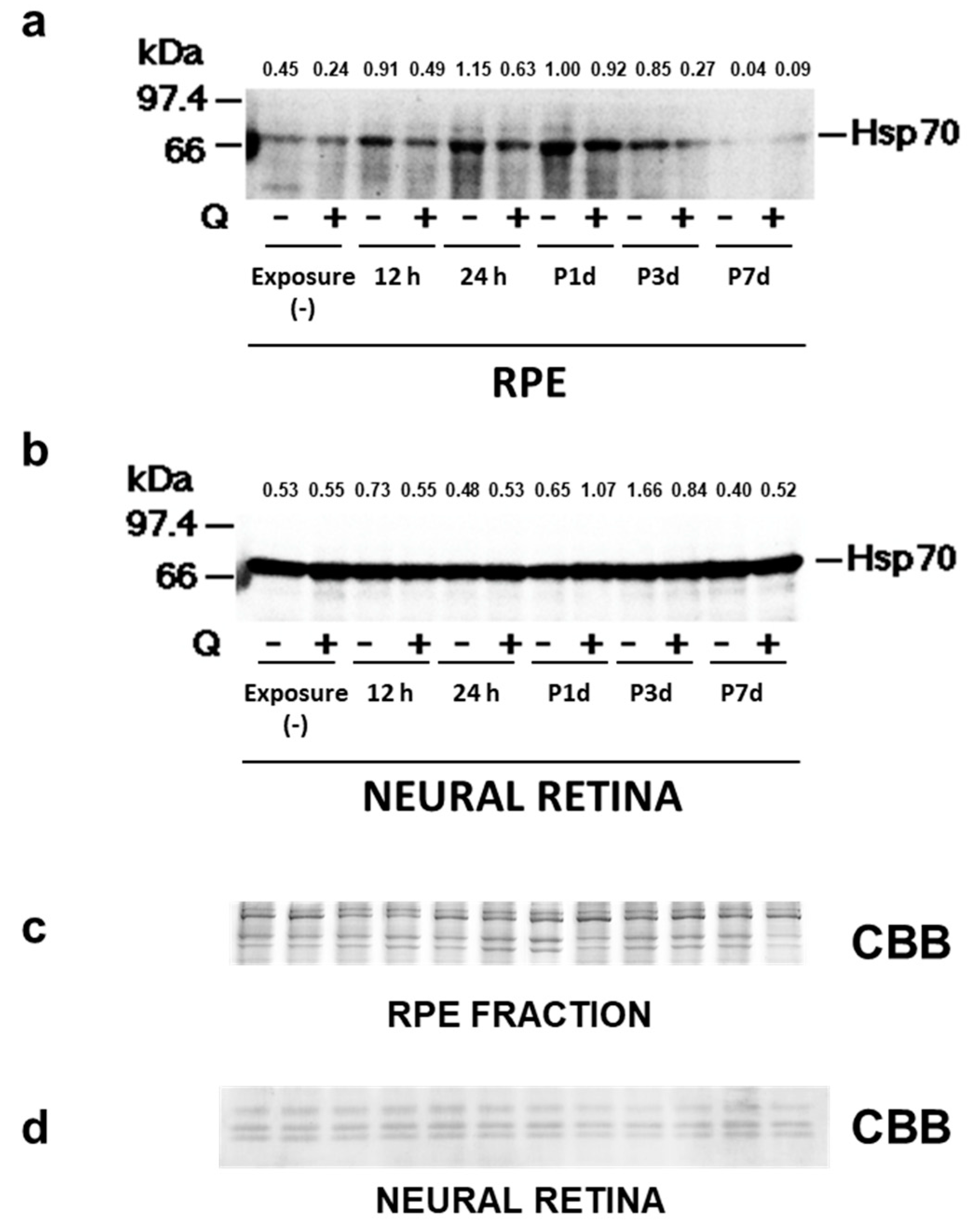
2.10. Western Blotting for Hsp70
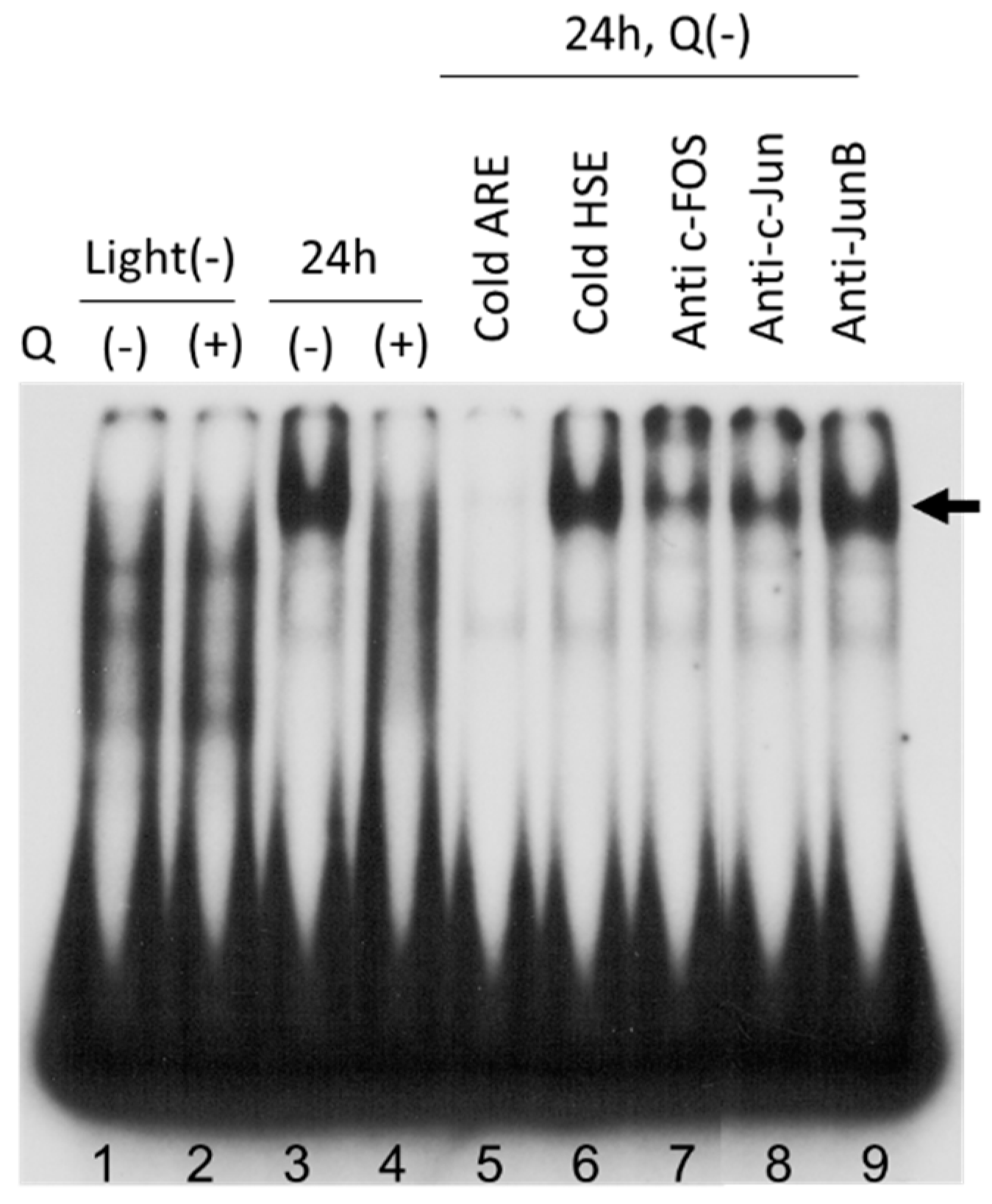
2.11. EMSA
2.12. Statistical Analysis
3. Results
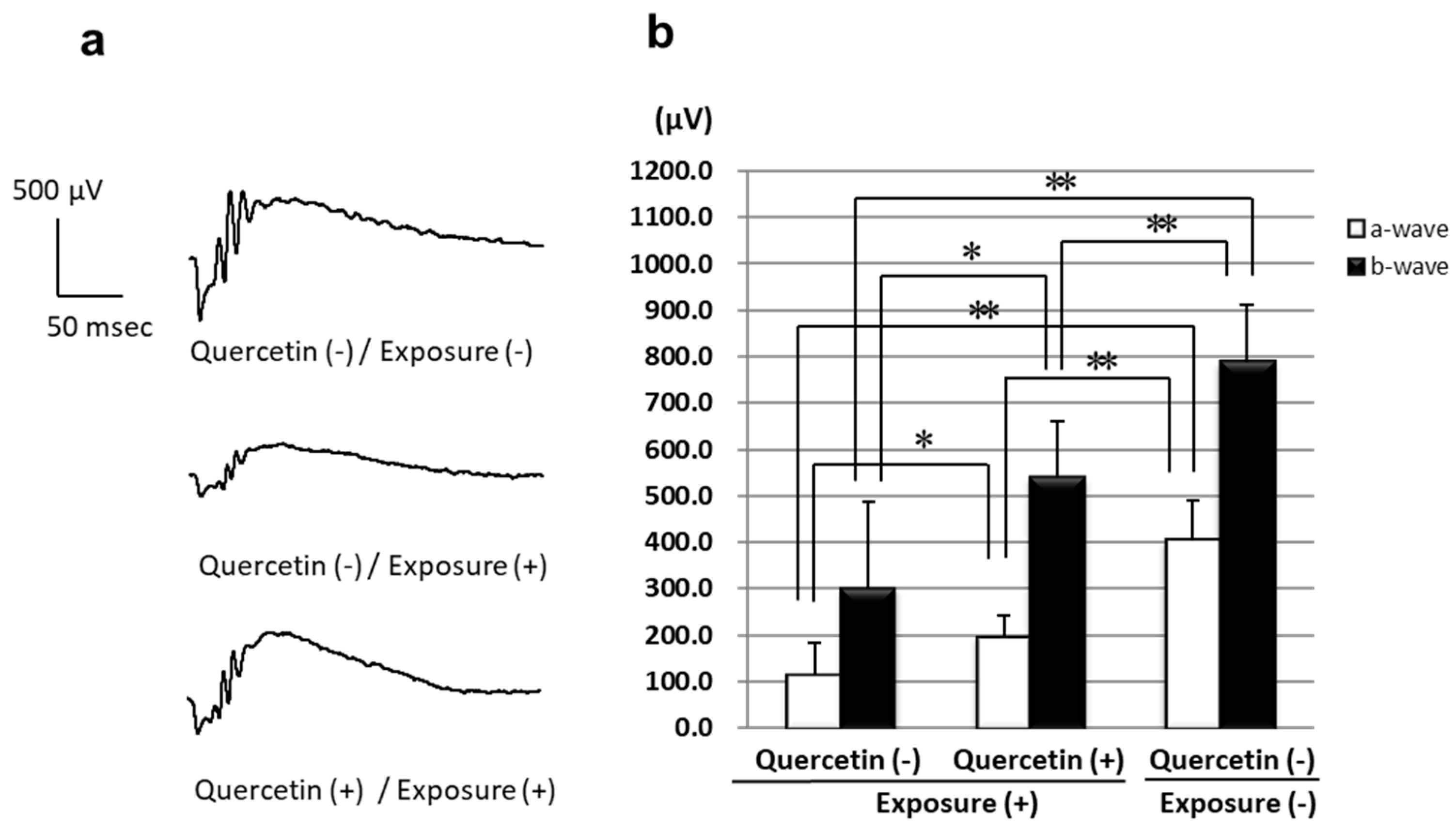
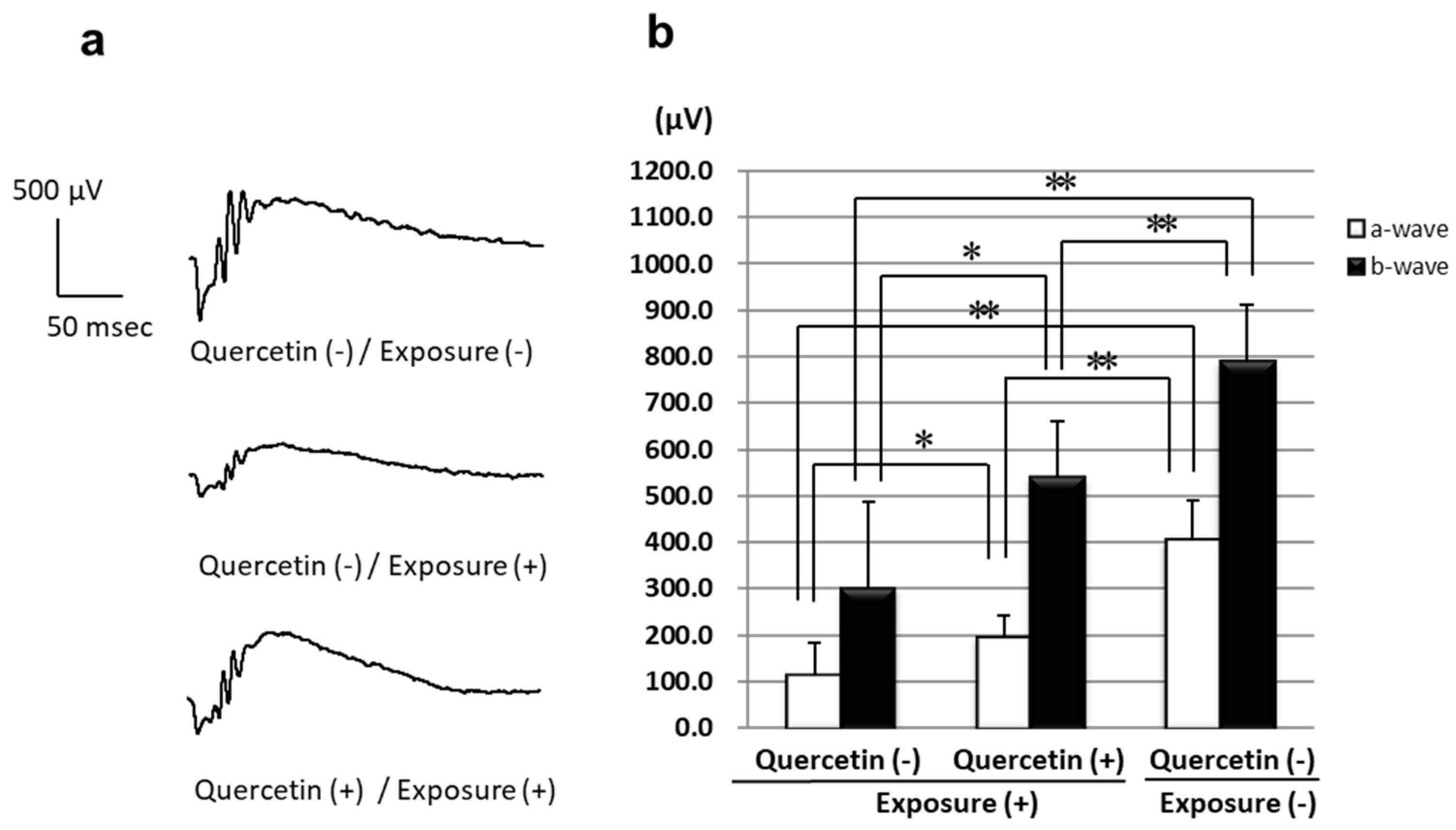
3.1. ERGs
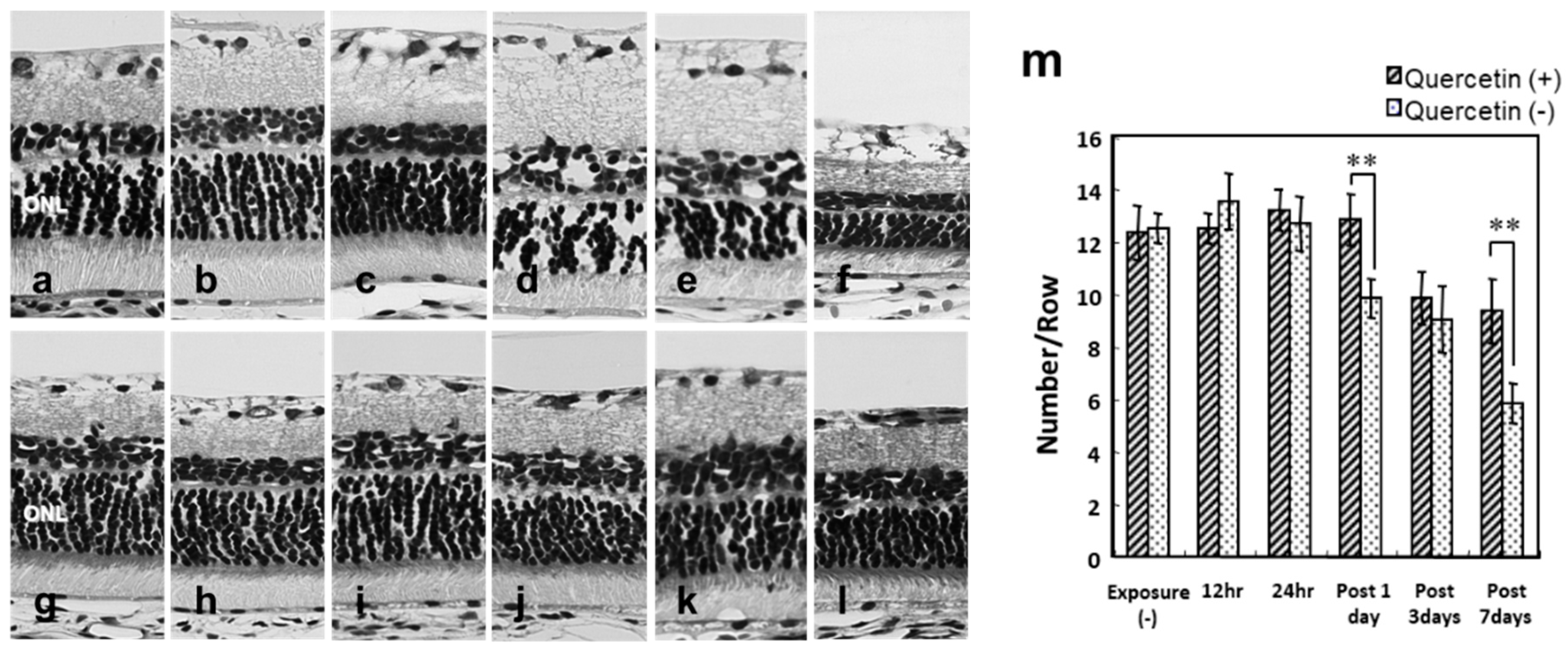
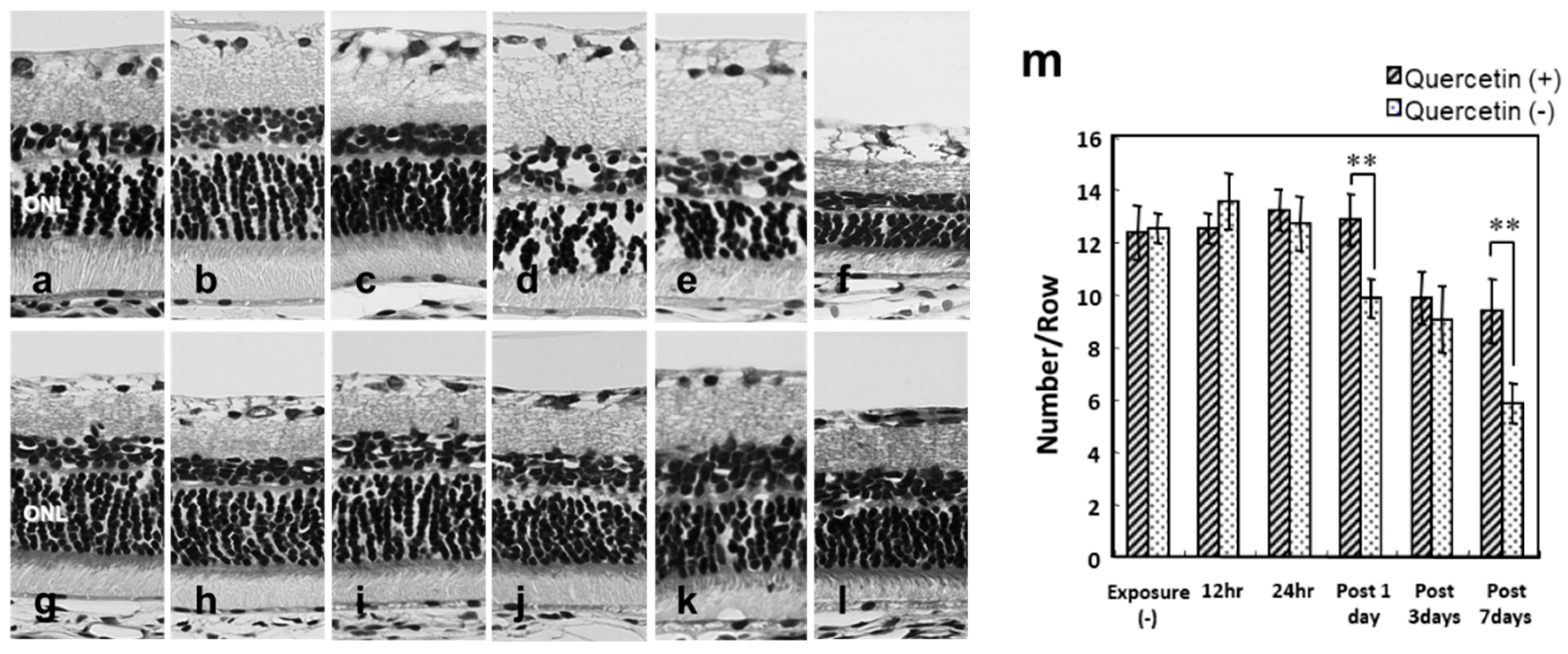
3.2. Photoreceptor Degeneration with Light Exposure
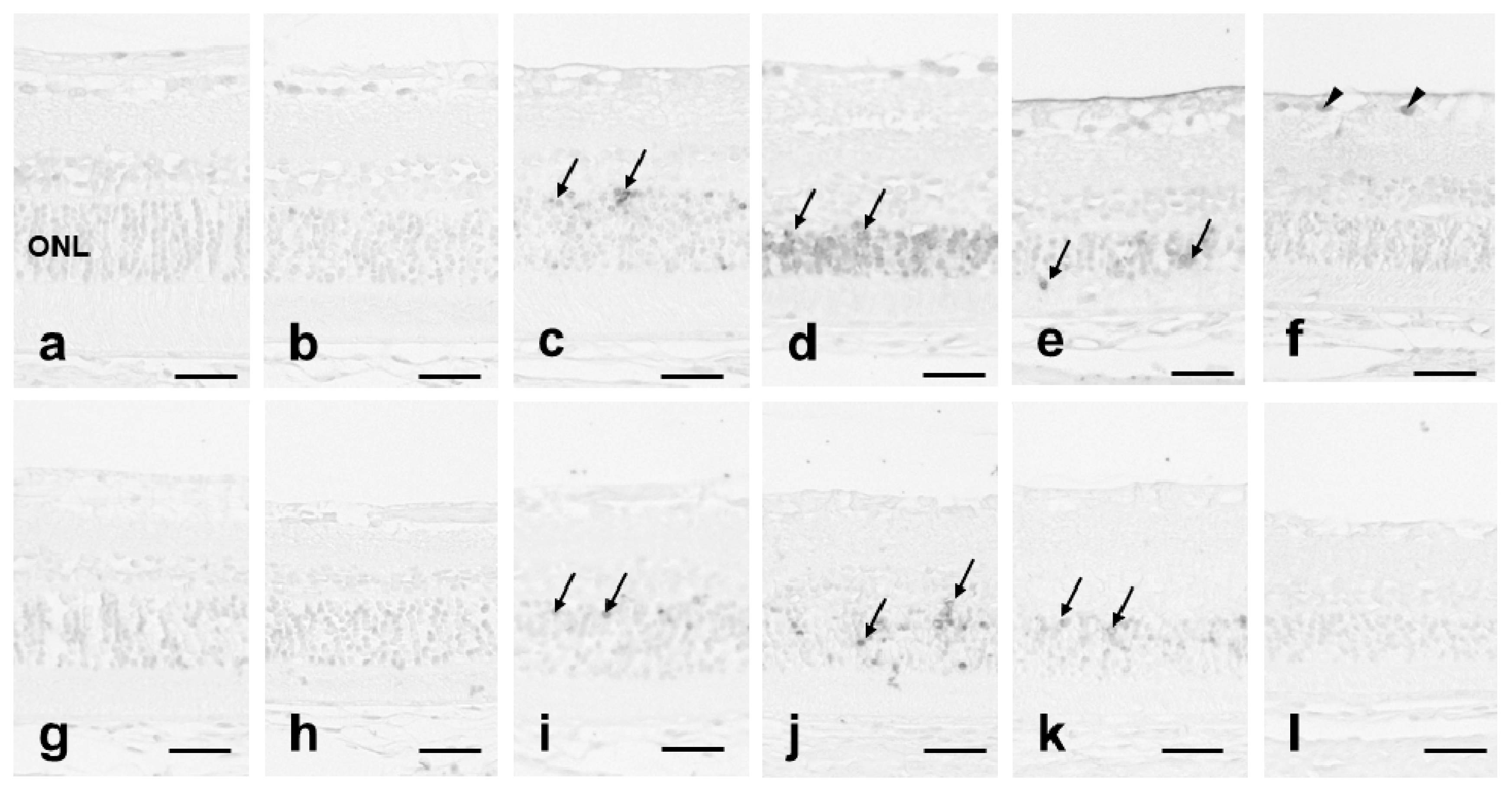
3.3. 8-OHdG Expression
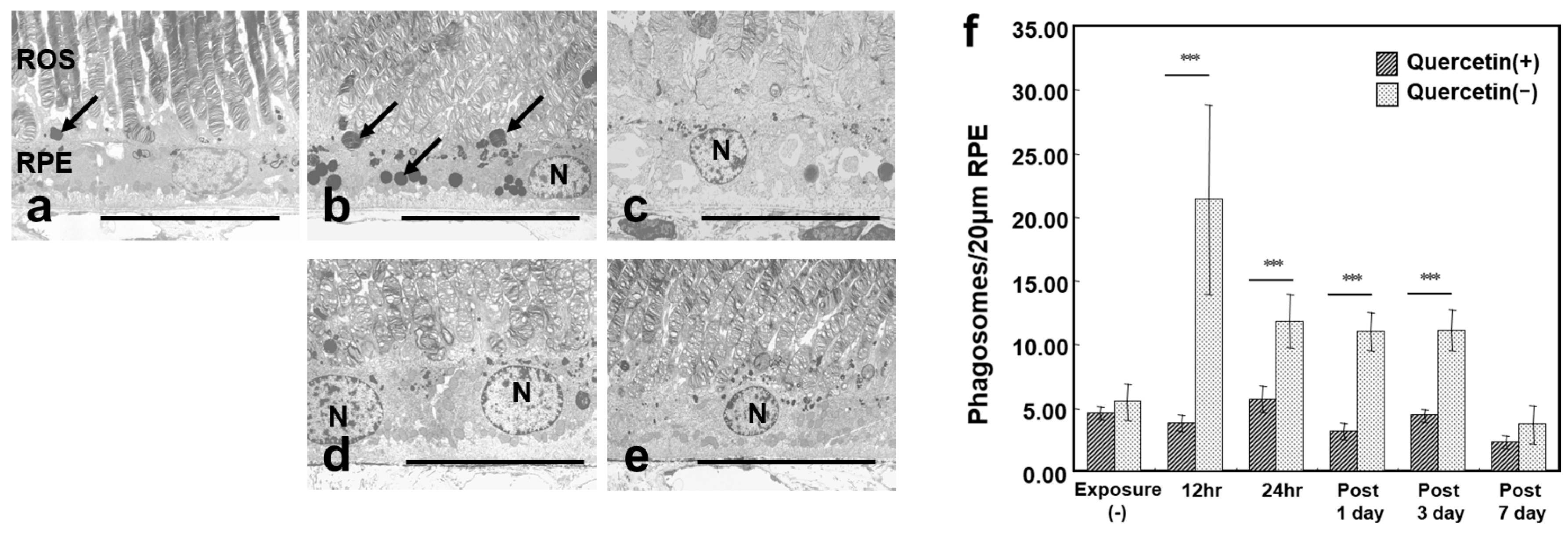
3.4. ROS Microstructure and Phagosomes in RPE
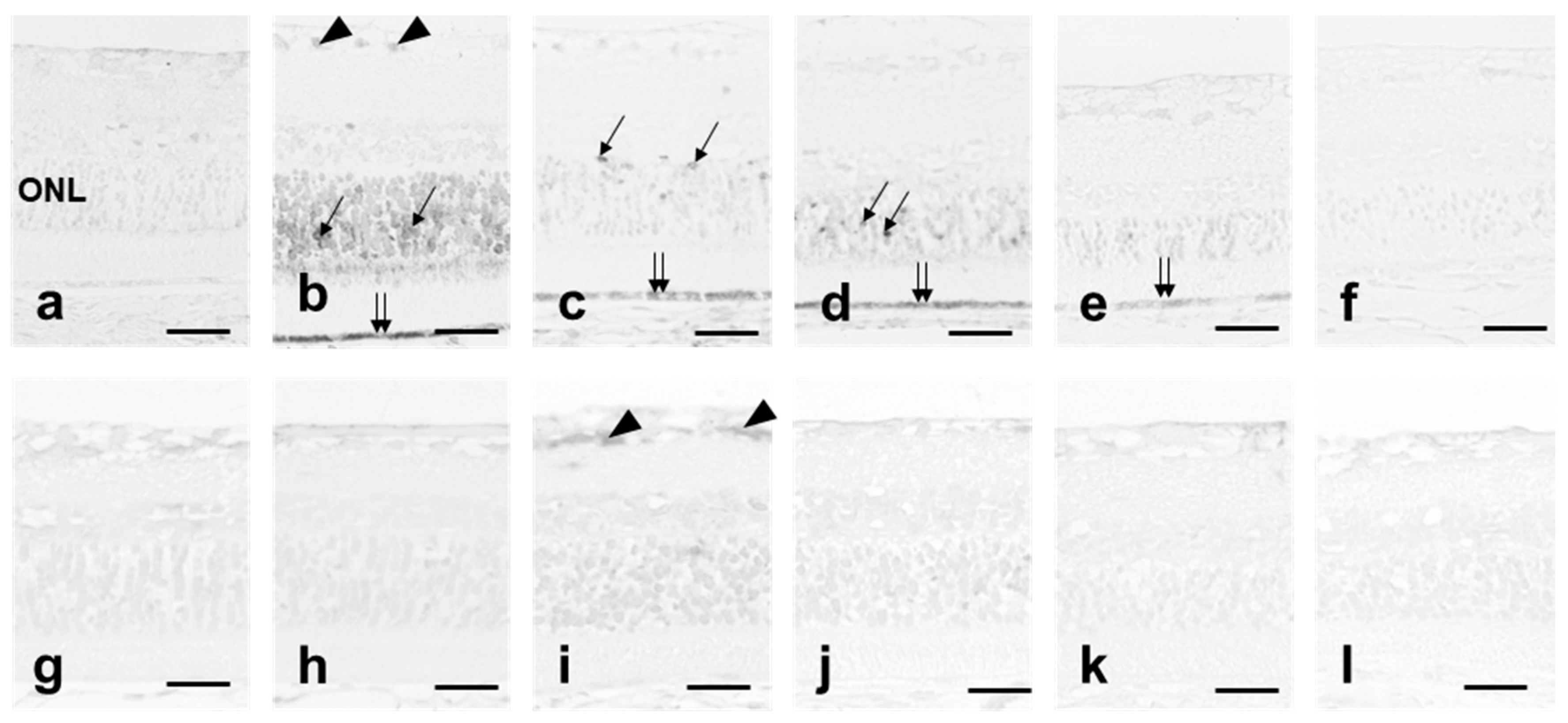
3.5. Hsp70 Expression
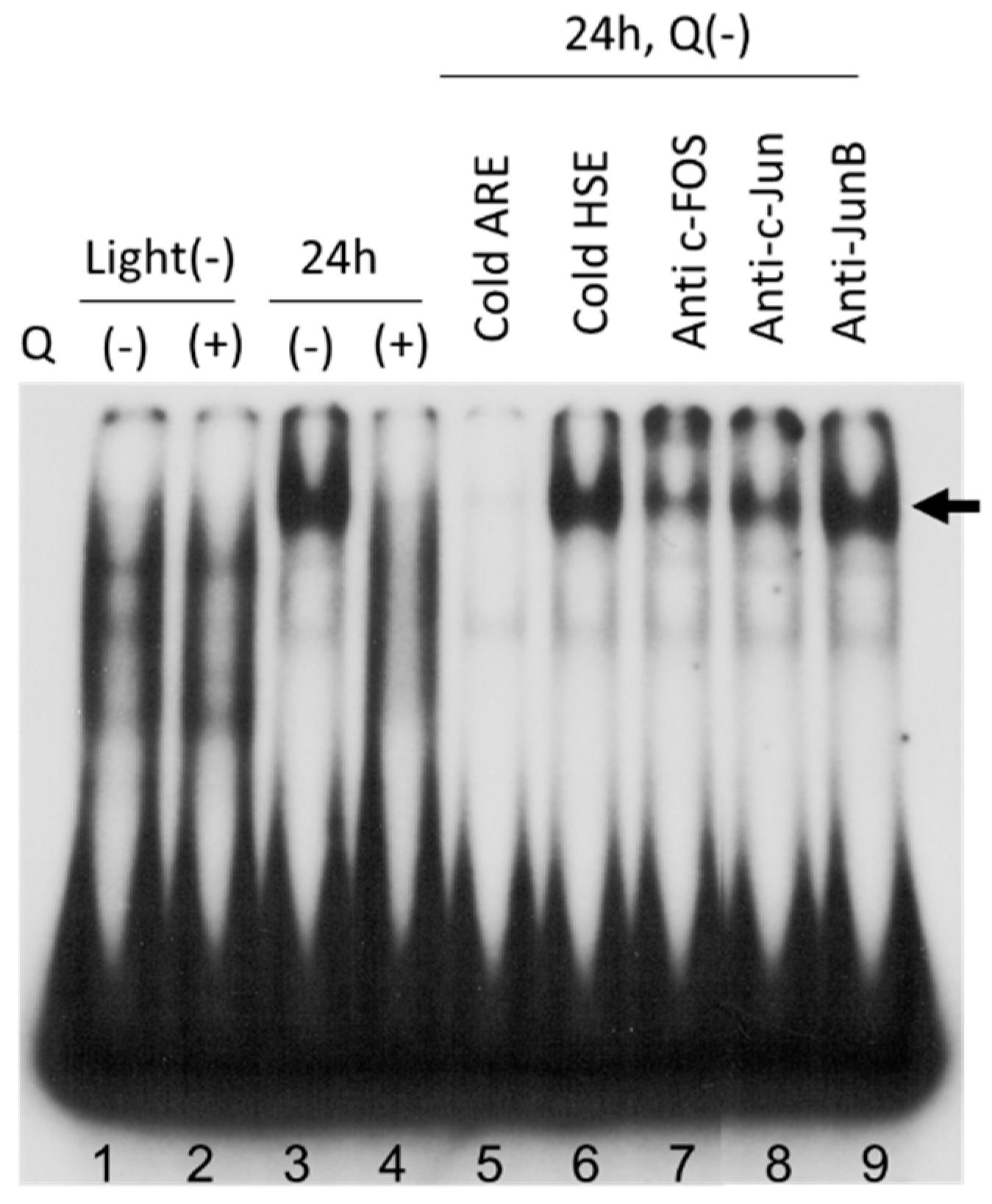
3.6. AP-1 Transcriptional Activity in Neural Retina
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Organisciak, D.T.; Darrow, R.A.; Barsalou, L.; Darrow, R.M.; Lininger, L.A. Light-induced damage in the retina: Differential effects of dimethylthiourea on photoreceptor survival, apoptosis and DNA oxidation. Photochem. Photobiol. 1999, 70, 261–268. [Google Scholar] [CrossRef]
- Contín, M.A.; Arietti, M.M.; Benedetto, M.M.; Bussi, C.; Guido, M.E. Photoreceptor damage induced by low-intensity light: Model of retinal degeneration in mammals. Mol. Vis. 2013, 19, 1614–1625. [Google Scholar] [PubMed]
- Hafezi, F.; Marti, A.; Grimm, C.; Wenzel, A.; Reme, C.E. Differential DNA binding activities of the transcription factors AP-1 and Oct-1 during light-induced apoptosis of photoreceptors. Vision Res. 1999, 39, 2511–2518. [Google Scholar] [CrossRef]
- LaVail, M.M.; Gorrin, G.M.; Yasumura, D.; Matthes, M.T. Increased susceptibility to constant light in nr and pcd mice with inherited retinal degenerations. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1020–1024. [Google Scholar]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Kortuem, K.; Geiger, L.K.; Levin, L.A. Differential susceptibility of retinal ganglion cells to reactive oxygen species. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3176–3182. [Google Scholar]
- Poon, H.K.; Tso, M.O.; Lam, T.T. c-Fos protein in photoreceptor cell death after photic injury in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2755–2758. [Google Scholar]
- Nir, I.; Agarwal, N. Diurnal expression of c-fos in the mouse retina. Brain Res. Mol. Brain Res. 1993, 19, 47–54. [Google Scholar] [CrossRef]
- Hafezi, F.; Steinbach, J.P.; Marti, A.; Munz, K.; Wang, Z.-Q.; Wagner, E.F.; Aguzzi, A.; Remé, C.E. The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat. Med. 1997, 3, 346–349. [Google Scholar] [CrossRef]
- Wenzel, A.; Grimm, C.; Marti, A.; Kueng-Hitz, N.; Hafezi, F.; Niemeyer, G.; Remé, C. c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J. Neurosci. 2000, 20, 81–88. [Google Scholar] [CrossRef]
- Erden Inal, M.; Kahraman, A.; Koken, T. Beneficial effects of quercetin on oxidative stress induced by ultraviolet, A. Clin. Exp. Dermatol. 2001, 26, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, A.; Erdeninal, M. Protective effects of quercetin on ultraviolet a light-induced oxidative stress in the blood of rat. J. Appl. Toxicol. 2002, 22, 303–309. [Google Scholar] [CrossRef]
- Afanas’ev, I.B.; Dorozhko, A.I.; Brodskii, A.V.; Kostyuk, V.A.; Potapovitch, A.I. Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin in lipid peroxidation. Biochem. Pharmacol. 1989, 38, 1763–1769. [Google Scholar] [CrossRef]
- da Silva, E.L.; Piskula, M.K.; Yamamoto, N.; Moon, J.H.; Terao, J. Quercetin metabolites inhibit copper ion-induced lipid peroxidation in rat plasma. FEBS Lett. 1998, 430, 405–408. [Google Scholar] [CrossRef]
- Sanderson, J.; McLauchlan, W.R.; Williamson, G. Quercetin inhibits hydrogen peroxide-induced oxidation of the rat lens. Free Radic. Biol. Med. 1999, 26, 639–645. [Google Scholar] [CrossRef]
- Areias, F.M.; Rego, A.C.; Oliveira, C.R.; Seabra, R.M. Antioxidant effect of flavonoids after ascorbate/Fe(2+)-induced oxidative stress in cultured retinal cells. Biochem. Pharmacol. 2001, 62, 111–118. [Google Scholar] [CrossRef]
- Kook, D.; Wolf, A.H.; Yu, A.L.; Neubauer, A.S.; Priglinger, S.G.; Kampik, A.; Welge-Lüssen, U.C. The protective effect of quercetin against oxidative stress in the human RPE in vitro. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1712–1720. [Google Scholar] [CrossRef]
- Cao, X.; Liu, M.; Tuo, J.; Shen, D.; Chan, C.C. The effects of quercetin in cultured human RPE cells under oxidative stress and in Ccl2/Cx3cr1 double deficient mice. Exp Eye Res. 2010, 91, 15–25. [Google Scholar] [CrossRef]
- Laabich, A.; Manmoto, C.C.; Kuksa, V.; Leung, D.W.; Vissvesvaran, G.P.; Karliga, I.; Kamat, M.; Scott, I.L.; Fawzi, A.; Kubota, R. Protective effects of myricetin and related flavonols against A2E and light mediated-cell death in bovine retinal primary cell culture. Exp. Eye Res. 2007, 85, 154–165. [Google Scholar] [CrossRef]
- Ueda, T.; Ueda, T.; Armstrong, D. Preventive effect of natural and synthetic antioxidants on lipid peroxidation in the mammalian eye. Ophthalmic Res. 1996, 28, 184–192. [Google Scholar] [CrossRef]
- Wu, B.Y.; Yu, A.C. Quercetin inhibits c-fos, heat shock protein, and glial fibrillary acidic protein expression in injured astrocytes. J. Neurosci. Res. 2000, 62, 730–736. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kitamura, M. Anti-apoptotic effect of quercetin: Intervention in the JNK- and ERK-mediated apoptotic pathways. Kidney Int. 2000, 58, 1078–1087. [Google Scholar] [CrossRef]
- Arikan, S.; Ersan, E.; Karaca, T.; Kara, S.; Gencer, B.; Karaboga, I.; Hasan Ali, T. Quercetin protects the retina by reducing apoptosis due to ischemia-reperfusion injury in a rat model. Arq. Bras. Oftalmol. 2014, 78, 100–104. [Google Scholar] [CrossRef] [PubMed]
- van Kuijk, F.J.; Buck, P. Fatty acid composition of the human macula and peripheral retina. Investig. Ophthalmol. Vis. Sci. 1992, 33, 3493–3496. [Google Scholar]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD); associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 1–20. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and, E.; beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef]
- Dong, Y.S.; Wang, J.L.; Feng, D.Y.; Qin, H.Z.; Wen, H.; Yin, Z.M.; Gao, G.D.; Li, C. Protective effect of quercetin against oxidative stress and brain edema in an experimental rat model of subarachnoid hemorrhage. Int. J. Med. Sci. 2014, 11, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Lidia, K.; Gong, H.; Onitsuka, S.; Kotani, T.; Ohira, A. Changes in manganese superoxide dismutase expression after exposure of the retina to intense light. Histochem. J. 1999, 31, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Imamura, N.; Dake, Y.; Amemiya, T. Circadian rhythm in the retinal pigment epithelium related to vitamin B12. Life Sci. 1995, 57, 1317–1323. [Google Scholar] [CrossRef]
- Bobu, C.; Hicks, D. Regulation of retinal photoreceptor phagocytosis in a diurnal mammal by circadian clocks and ambient lighting. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3495–3502. [Google Scholar] [CrossRef]
- Ohira, A.; Tanito, M.; Kaidzu, S.; Kondo, T. Glutathione peroxidase induced in rat retinas to counteract photic injury. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1230–1236. [Google Scholar] [CrossRef]
- Tanito, M.; Nakamura, H.; Kwon, Y.W.; Teratani, A.; Masutani, H.; Shioji, K.; Kishimoto, C.; Ohira, A.; Horie, R.; Yodoi, J. Enhanced oxidative stress and impaired thioredoxin expression in spontaneously hypertensive rats. Antioxid. Redox Signal. 2004, 6, 89–97. [Google Scholar] [CrossRef]
- Tanito, M.; Nishiyama, A.; Tanaka, T.; Masutani, H.; Nakamura, H.; Yodoi, J.; Ohira, A. Change of redox status and modulation by thiol replenishment in retinal photooxidative damage. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2392–2400. [Google Scholar]
- Kim, J.H.; Yu, Y.S.; Chung, H.; Heo, J.W.; Seo, J.S. Effect of the absence of heat shock protein 70.1 (hsp70.1) on retinal photic injury. Korean J. Ophthalmol. 2003, 17, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; Silvia di Fazio de Escalante, M.; Careaga, M.M.; Bazan, H.E.; Giusto, N.M. High content of 22:6 (docosahexaenoate) and active [2-3H]glycerol metabolism of phosphatidic acid from photoreceptor membranes. Biochim. Biophys. Acta 1982, 712, 702–706. [Google Scholar] [CrossRef]
- Blanks, J.C.; Pickford, M.S.; Organisciak, D.T. Ascorbate treatment prevents accumulation of phagosomes in RPE in light damage. Investig. Ophthalmol. Vis. Sci. 1992, 33, 2814–2821. [Google Scholar]
- Organisciak, D.T.; Darrow, R.M.; Jiang, Y.I.; Marak, G.E.; Blanks, J.C. Protection by dimethylthiourea against retinal light damage in rats. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1599–1609. [Google Scholar]
- Ranchon, I.; LaVail, M.M.; Kotake, Y.; Anderson, R.E. Free radical trap phenyl-N-tert-butylnitrone protects against light damage but does not rescue P23H and S334ter rhodopsin transgenic rats from inherited retinal degeneration. J Neurosci. 2003, 23, 6050–6057. [Google Scholar] [CrossRef]
- Tomita, H.; Kotake, Y.; Anderson, R.E. Mechanism of protection from light-induced retinal degeneration by the synthetic antioxidant phenyl-N-tert-butylnitrone. Investig. Ophthalmol. Vis. Sci. 2005, 46, 427–434. [Google Scholar] [CrossRef]
- Lewden, O.; Garcher, C.; Assem, M.; Morales, C.; Rochette, L.; Bron, A.M. Changes of the inducible heat shock protein 70 mRNA level in rat retina after ischemia and reperfusion. Ophthalmic Res. 1998, 30, 291–294. [Google Scholar] [CrossRef]
- Kim, Y.; Yamaguchi, Y.; Kondo, N.; Masutani, H.; Yodoi, J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene 2003, 22, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Barbe, M.F.; Tytell, M.; Gower, D.J.; Welch, W.J. Hyperthermia protects against light damage in the rat retina. Science 1988, 241, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Kent, C.R.; Tytell, M. Retinal uptake of intravitreally injected Hsc/Hsp70 and its effect on susceptibility to light damage. Mol. Vis. 2001, 7, 48–56. [Google Scholar] [PubMed]
- Roca, A.; Shin, K.J.; Liu, X.; Simon, M.I.; Chen, J. Comparative analysis of transcriptional profiles between two apoptotic pathways of light-induced retinal degeneration. Neuroscience 2004, 129, 779–790. [Google Scholar] [CrossRef]
- Grimm, C.; Wenzel, A.; Behrens, A.; Hafezi, F.; Wagner, E.F.; Reme, C.E. AP-1 mediated retinal photoreceptor apoptosis is independent of N-terminal phosphorylation of c-Jun. Cell Death Differ. 2001, 8, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, J.D.; Wang, B.; Lv, Y.J.; Jiang, H.; Liu, G.L.; Qiao, Y.; Ren, M.; Guo, X.F. Quercetin inhibits left ventricular hypertrophy in spontaneously hypertensive rats and inhibits angiotensin II-induce H9C2 cells hypertrophy by enhancing PPAR-gamma expression and suppressing AP-1 activity. PLoS ONE 2013, 8, e72548. [Google Scholar]
- Jia, H.; Chen, W.; Yu, X.; Wu, X.; Li, S.; Liu, H.; Liao, J.; Liu, W.; Mi, M.; Liu, L.; et al. Black rice anthocyanidins prevent retinal photochemical damage via involvement of the AP-1/NF-kB/Caspase-1 pathway in Sprague-Dawley rats. J. Vet. Sci. 2013, 14, 345–353. [Google Scholar] [CrossRef]
- Chu, K.O.; Chan, K.P.; Yang, Y.P.; Qin, Y.J.; Li, W.Y.; Chan, S.O.; Wang, C.C.; Pang, C.P. Effects of EGCG content in green tea extract on pharmacokinetics, oxidative status and expression of inflammatory and apoptotic genes in the rat ocular tissues. J. Nutr. Biochem. 2015, 26, 1357–1367. [Google Scholar] [CrossRef]
- Kumar, B.; Gupta, S.K.; Nag, T.C.; Srivastava, S.; Saxena, R.; Jha, K.A.; Srinivasan, B.P. Retinal neuroprotective effects of quercetin in streptozotocin-induced diabetic rats. Exp. Eye Res. 2014, 125, 193–202. [Google Scholar] [CrossRef]
- Zhu, X.; Li, N.; Wang, Y.; Ding, L.; Chen, H.; Yu, Y.; Shi, X. Protective effects of quercetin on UVB irradiation-induced cytotoxicity through ROS clearance in keratinocyte cells. Oncol. Rep. 2017, 37, 209–218. [Google Scholar] [CrossRef]
- Jomova, K.; Lawson, M.; Drostinova, L.; Lauro, P.; Poprac, P.; Brezova, V.; Michalik, M.; Lukes, V.; Valko, M. Protective role of quercetin against copper(II)-induced oxidative stress: A spectroscopic, theoretical and DNA damage study. Food Chem. Toxicol. 2017, 110, 340–350. [Google Scholar] [CrossRef]
- Li, F.; Bai, Y.; Zhao, M.; Huang, L.; Li, S.; Li, X.; Chen, Y. Quercetin inhibits vascular endothelial growth factor-induced choroidal and retinal angiogenesis in vitro. Ophthalmic Res. 2015, 53, 109–116. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koyama, Y.; Kaidzu, S.; Kim, Y.-C.; Matsuoka, Y.; Ishihara, T.; Ohira, A.; Tanito, M. Suppression of Light-Induced Retinal Degeneration by Quercetin via the AP-1 Pathway in Rats. Antioxidants 2019, 8, 79. https://doi.org/10.3390/antiox8040079
Koyama Y, Kaidzu S, Kim Y-C, Matsuoka Y, Ishihara T, Ohira A, Tanito M. Suppression of Light-Induced Retinal Degeneration by Quercetin via the AP-1 Pathway in Rats. Antioxidants. 2019; 8(4):79. https://doi.org/10.3390/antiox8040079
Chicago/Turabian StyleKoyama, Yasurou, Sachiko Kaidzu, Yong-Chul Kim, Yotaro Matsuoka, Tomoe Ishihara, Akihiro Ohira, and Masaki Tanito. 2019. "Suppression of Light-Induced Retinal Degeneration by Quercetin via the AP-1 Pathway in Rats" Antioxidants 8, no. 4: 79. https://doi.org/10.3390/antiox8040079
APA StyleKoyama, Y., Kaidzu, S., Kim, Y.-C., Matsuoka, Y., Ishihara, T., Ohira, A., & Tanito, M. (2019). Suppression of Light-Induced Retinal Degeneration by Quercetin via the AP-1 Pathway in Rats. Antioxidants, 8(4), 79. https://doi.org/10.3390/antiox8040079