The Role of Isothiocyanates as Cancer Chemo-Preventive, Chemo-Therapeutic and Anti-Melanoma Agents

 , ,
, ,  ,
,  and
and 
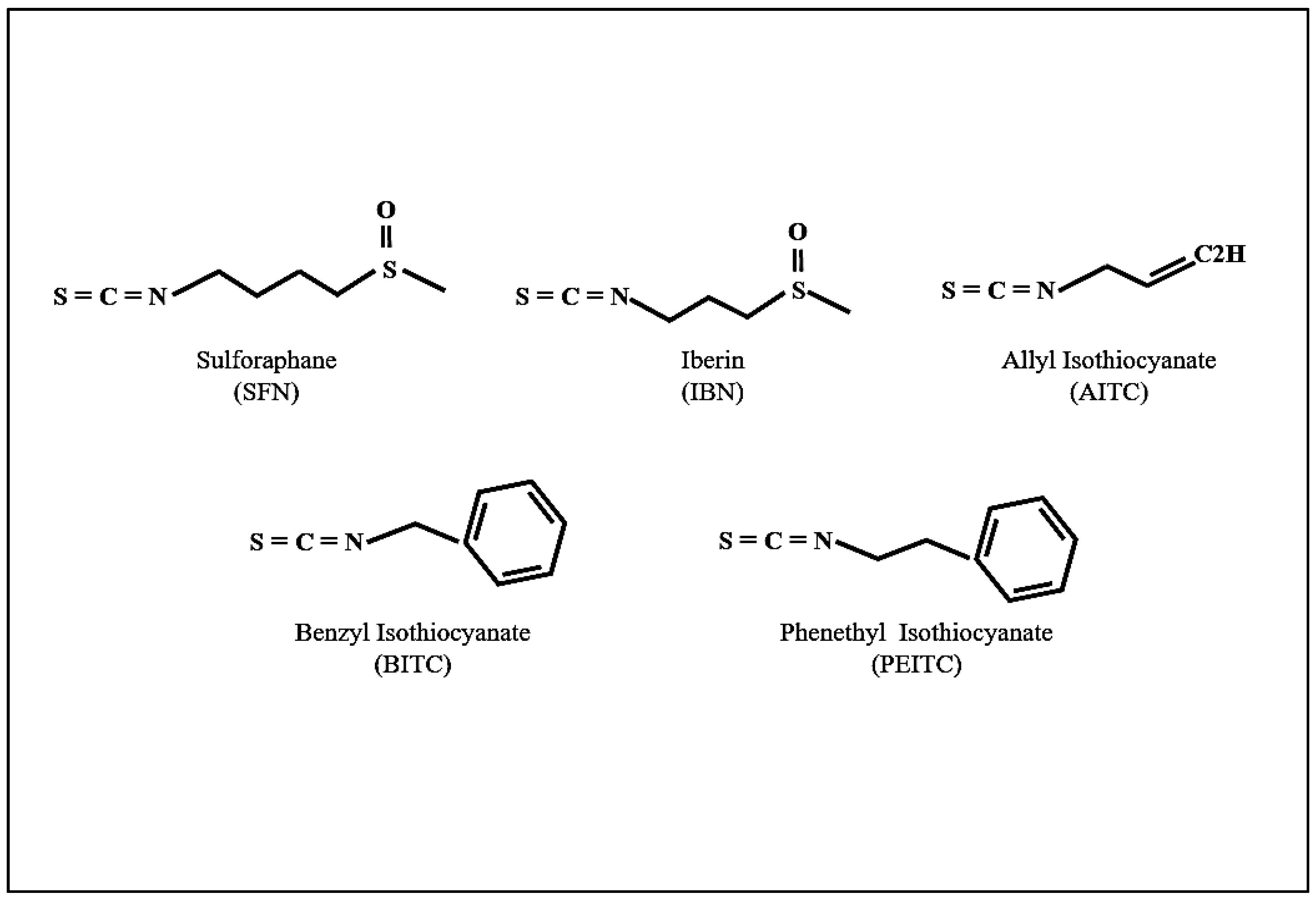{kind=link}
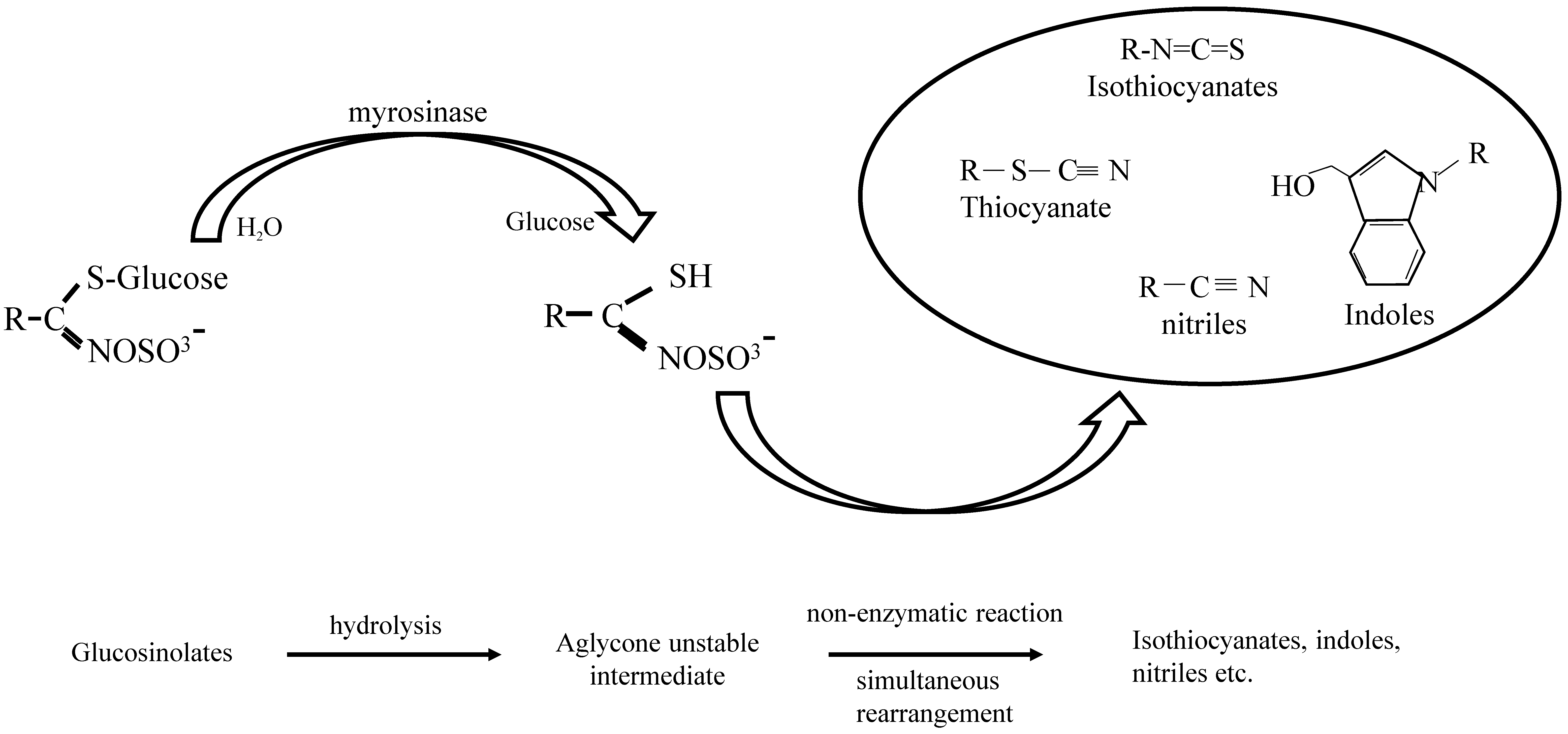{kind=link}
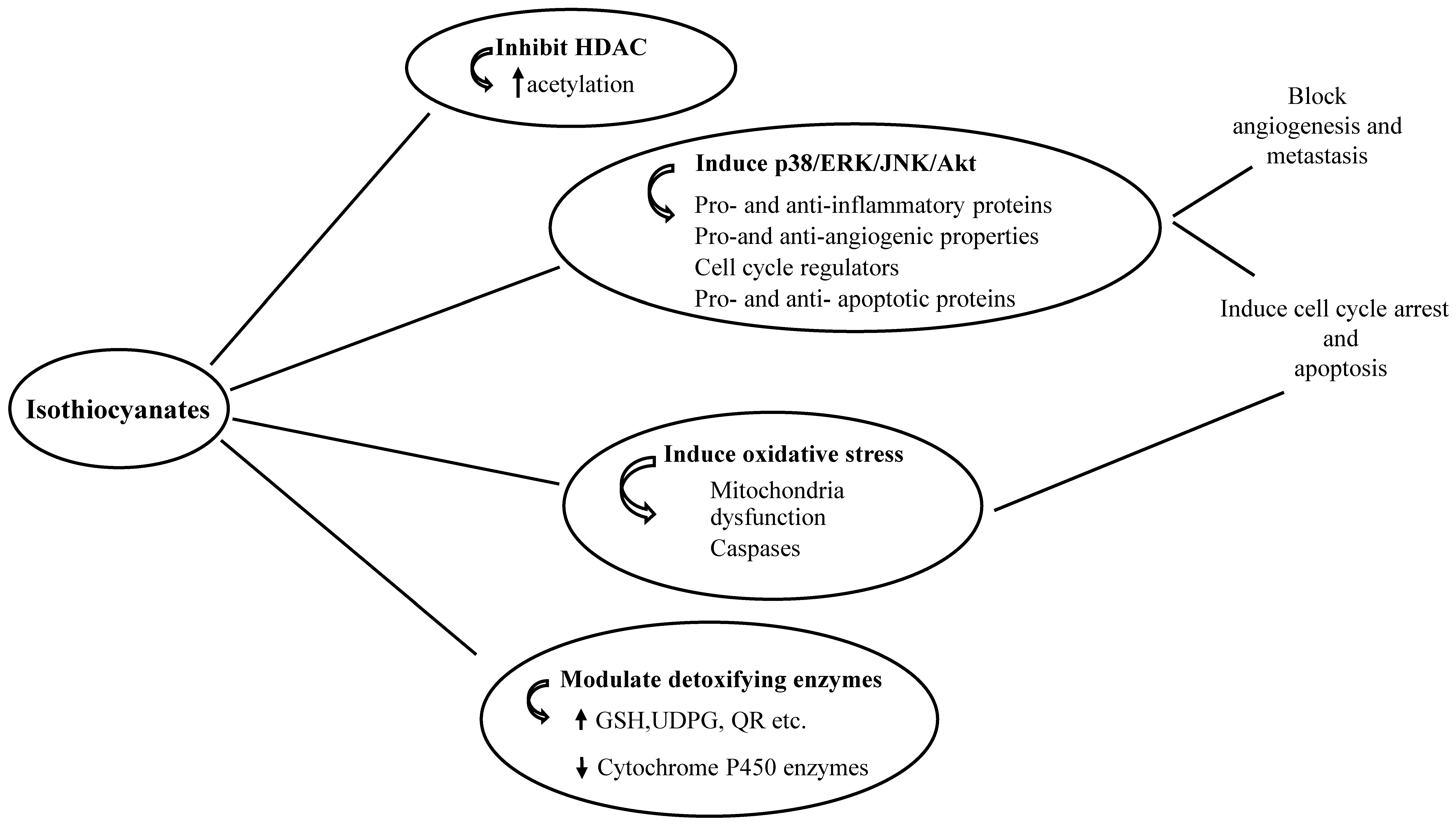{kind=link}
Abstract
1. Introduction
1.1. GLs-Myrosinase System
1.2. Anti-Cancer Properties
1.2.1. Inhibition of Phase I and Induction of Phase II Enzymes
1.2.2. Cell Cycle Arrest and Cell Death
1.2.3. Apoptosis
1.2.4. Oxidative Stress
1.2.5. Autophagy
1.2.6. Epigenetic Mechanisms
Inhibition of HDACs
Inhibition of DNMTs
Modulation of miRNAs
1.2.7. Anti-Angiogenic and Anti-Metastatic Properties
2. ITCs as Cancer Chemo-Preventive and Chemo-Therapeutic Agents
2.1. ITCs as Cancer Chemo-Preventive Agents: Human Studies
2.2. ITCs as Cancer Chemo-Therapeutic Agents: Animal Studies
2.2.1. In Vitro Studies Utilizing Various ITCs
2.2.2. In Vivo Studies Utilizing Various ITCs
3. ITCs as Anti-Melanoma Agents
3.1. In Vitro Studies Utilizing Various ITCs
3.2. In Vivo Studies Utilizing Various ITCs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slawson, D.L.; Fitzgerald, N.; Morgan, K.T. Position of the Academy of Nutrition and Dietetics: The role of nutrition in health promotion and chronic disease prevention. J. Acad. Nutr. Diet. 2013, 113, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L. Ins and outs of dietary phytochemicals in cancer chemoprevention. Biochem. Pharmacol. 2007, 74, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Fitsiou, E.; Anestopoulos, I.; Chlichlia, K.; Galanis, A.; Kourkoutas, I.; Panayiotidis, M.I.; Pappa, A. Antioxidant and Antiproliferative Properties of the Essential Oils of Satureja thymbra and Satureja parnassica and their Major Constituents. Anticancer Res. 2016, 36, 5757–5763. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.T. Phytochemicals and cancer. Proc. Nutr. Soc. 2007, 66, 207–215. [Google Scholar] [CrossRef]
- Li, A.-N.; Li, S.; Zhang, Y.-J.; Xu, X.-R.; Chen, Y.-M.; Li, H.-B. Resources and biological activities of natural polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef]
- Nohynek, L.J.; Alakomi, H.-L.; Kähkönen, M.P.; Heinonen, M.; Helander, I.M.; Oksman-Caldentey, K.-M.; Puupponen-Pimiä, R.H. Berry phenolics: Antimicrobial properties and mechanisms of action against severe human pathogens. Nutr. Cancer 2006, 54, 18–32. [Google Scholar] [CrossRef]
- Issa, A.Y.; Volate, S.R.; Wargovich, M.J. The role of phytochemicals in inhibition of cancer and inflammation: New directions and perspectives. J. Food Compost. Anal. 2006, 19, 405–419. [Google Scholar] [CrossRef]
- Rupasinghe, H.P.; Sekhon-Loodu, S.; Mantso, T.; Panayiotidis, M.I. Phytochemicals in regulating fatty acid beta-oxidation: Potential underlying mechanisms and their involvement in obesity and weight loss. Pharmacol. Ther. 2016, 165, 153–163. [Google Scholar] [CrossRef]
- Supic, G.; Jagodic, M.; Magic, Z. Epigenetics: A new link between nutrition and cancer. Nutr. Cancer 2013, 65, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, A.; Rodriguez-Rocha, H.; Madayiputhiya, N.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Biomarkers of protein oxidation in human disease. Curr. Mol. Med. 2012, 12, 681–697. [Google Scholar] [CrossRef]
- Panayiotidis, M. Reactive oxygen species (ROS) in multistage carcinogenesis. Cancer Lett. 2008, 266, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Wukovich, R.L.; Aziz, K.; Kalogerinis, P.T.; Richardson, C.C.; Panayiotidis, M.I.; Bonner, W.M.; Sedelnikova, O.A.; Georgakilas, A.G. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat. Res. 2009, 674, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Sanchez-Olea, R.; Reyes-Reyes, E.M.; Panayiotidis, M.I. Environmental toxicity, oxidative stress and apoptosis: Menage a trois. Mutat. Res. 2009, 674, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Foppoli, C.; Coccia, R.; Perluigi, M. Antioxidants in cervical cancer: Chemopreventive and chemotherapeutic effects of polyphenols. Biochim. Biophys. Acta 2012, 1822, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Anestopoulos, I.; Hanafi, R.; Voulgaridou, G.P.; Franco, R.; Georgakilas, A.G.; Pappa, A.; Panayiotidis, M.I. Pleiotrophic effects of natural products in ROS-induced carcinogenesis: The role of plant-derived natural products in oral cancer chemoprevention. Cancer Lett. 2012, 327, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-J.; Yen, G.-C. Chemopreventive effects of dietary phytochemicals against cancer invasion and metastasis: Phenolic acids, monophenol, polyphenol, and their derivatives. Cancer Treat. Rev. 2012, 38, 76–87. [Google Scholar] [CrossRef]
- Kang, N.J.; Shin, S.H.; Lee, H.J.; Lee, K.W. Polyphenols as small molecular inhibitors of signaling cascades in carcinogenesis. Pharmacol. Ther. 2011, 130, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H. Potential synergy of phytochemicals in cancer prevention: Mechanism of action. J. Nutr. 2004, 134, 3479S–3485S. [Google Scholar] [CrossRef]
- Verkerk, R.; Schreiner, M.; Krumbein, A.; Ciska, E.; Holst, B.; Rowland, I.; De Schrijver, R.; Hansen, M.; Gerhäuser, C.; Mithen, R. Glucosinolates in Brassica vegetables: The influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 2009, 53, S219. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; Kroymann, J.; Mitchell-Olds, T. The glucosinolate–myrosinase system in an ecological and evolutionary context. Curr. Opin. Plant Biol. 2005, 8, 264–271. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E. Glucosinolate structures in evolution. Phytochemistry 2012, 77, 16–45. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Clarke, D.B. Glucosinolates, structures and analysis in food. Anal. Methods 2010, 2, 310–325. [Google Scholar] [CrossRef]
- Śmiechowska, A.; Bartoszek, A.; Namieśnik, J. Determination of Glucosinolates and Their Decomposition Products—Indoles and Isothiocyanates in Cruciferous Vegetables. Crit. Rev. Anal. Chem. 2010, 40, 202–216. [Google Scholar] [CrossRef]
- Velasco, P.; Cartea, M.E.; González, C.; Vilar, M.; Ordás, A. Factors affecting the glucosinolate content of kale (Brassica oleracea acephala group). J. Agric. Food Chem. 2007, 55, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Charron, C.S.; Saxton, A.M.; Sams, C.E. Relationship of climate and genotype to seasonal variation in the glucosinolate–myrosinase system. I. Glucosinolate content in ten cultivars of Brassica oleracea grown in fall and spring seasons. J. Sci. Food Agric. 2005, 85, 671–681. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; Kroymann, J.; Brown, P.; Figuth, A.; Pedersen, D.; Gershenzon, J.; Mitchell-Olds, T. Genetic control of natural variation in Arabidopsis glucosinolate accumulation. Plant Physiol. 2001, 126, 811–825. [Google Scholar] [CrossRef]
- Fabre, N.; Poinsot, V.; Debrauwer, L.; Vigor, C.; Tulliez, J.; Fourasté, I.; Moulis, C. Characterisation of glucosinolates using electrospray ion trap and electrospray quadrupole time-of-flight mass spectrometry. Phytochem. Anal. 2007, 18, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.E.; Terschluesen, A.M.; Rimbach, G. Health promoting effects of brassica-derived phytochemicals: From chemopreventive and anti-inflammatory activities to epigenetic regulation. Oxid. Med. Cell. Longev. 2013, 2013, 964539. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef]
- Grubb, C.D.; Abel, S. Glucosinolate metabolism and its control. Trends Plant Sci. 2006, 11, 89–100. [Google Scholar] [CrossRef]
- Dufour, V.; Stahl, M.; Baysse, C. The antibacterial properties of isothiocyanates. Microbiology 2015, 161, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.J.; Mutters, N.T.; Blessing, B.; Günther, F. Natural isothiocyanates express antimicrobial activity against developing and mature biofilms of Pseudomonas aeruginosa. Fitoterapia 2017, 119, 57–63. [Google Scholar] [CrossRef]
- Ko, M.-O.; Kim, M.-B.; Lim, S.-B. Relationship between Chemical Structure and Antimicrobial Activities of Isothiocyanates from Cruciferous Vegetables against Oral Pathogens. J. Microbiol. Biotechnol. 2016, 26, 2036–2042. [Google Scholar] [CrossRef]
- Wilson, A.E.; Bergaentzlé, M.; Bindler, F.; Marchioni, E.; Lintz, A.; Ennahar, S. In vitro efficacies of various isothiocyanates from cruciferous vegetables as antimicrobial agents against foodborne pathogens and spoilage bacteria. Food Control 2013, 30, 318–324. [Google Scholar] [CrossRef]
- Borges, A.; Simões, L.C.; Saavedra, M.J.; Simões, M. The action of selected isothiocyanates on bacterial biofilm prevention and control. Int. Biodeterior. Biodegrad. 2014, 86, 25–33. [Google Scholar] [CrossRef]
- Manyes, L.; Luciano, F.B.; Mañes, J.; Meca, G. In vitro antifungal activity of allyl isothiocyanate (AITC) against Aspergillus parasiticus and Penicillium expansum and evaluation of the AITC estimated daily intake. Food Chem. Toxicol. 2015, 83, 293–299. [Google Scholar] [CrossRef]
- Freitas, E.; Aires, A.; Rosa, E.; Saavedra, M.J. Antibacterial activity and synergistic effect between watercress extracts, 2-phenylethyl isothiocyanate and antibiotics against 11 isolates of Escherichia coli from clinical and animal source. Lett. Appl. Microbiol. 2013, 57, 266–273. [Google Scholar]
- De Figueiredo, S.M.; Binda, N.S.; Nogueira-Machado, J.A.; Vieira-Filho, S.A.; Caligiorne, R.B. The antioxidant properties of organosulfur compounds (sulforaphane). Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 24–39. [Google Scholar] [CrossRef]
- Fuentes, F.; Paredes-Gonzalez, X.; Kong, A.-N.T. Dietary glucosinolates sulforaphane, phenethyl isothiocyanate, indole-3-carbinol/3, 3′-diindolylmethane: Antioxidative stress/inflammation, Nrf2, epigenetics/epigenomics and in vivo cancer chemopreventive efficacy. Curr. Pharmacol. Rep. 2015, 1, 179–196. [Google Scholar] [CrossRef]
- McWalter, G.K.; Higgins, L.G.; McLellan, L.I.; Henderson, C.J.; Song, L.; Thornalley, P.J.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 is essential for induction of NAD (P) H: Quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J. Nutr. 2004, 134, 3499S–3506S. [Google Scholar] [CrossRef]
- Trio, P.Z.; Kawahara, A.; Tanigawa, S.; Sakao, K.; Hou, D.-X. DNA Microarray Profiling Highlights Nrf2-Mediated Chemoprevention Targeted by Wasabi-Derived Isothiocyanates in HepG2 Cells. Nutr. Cancer 2017, 69, 105–116. [Google Scholar] [CrossRef]
- No, J.H.; Kim, Y.-B.; Song, Y.S. Targeting Nrf2 Signaling to Combat Chemoresistance. J. Cancer Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Furfaro, A.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.; Pronzato, M.; Nitti, M. The Nrf2/HO-1 axis in cancer cell growth and chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef]
- Catanzaro, E.; Calcabrini, C.; Turrini, E.; Sestili, P.; Fimognari, C. Nrf2: A potential therapeutic target for naturally occurring anticancer drugs? Expert Opin. Ther. Targets 2017, 21, 781–793. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martin-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Li, W.; Guo, Y.; Zhang, C.; Wu, R.; Yang, A.Y.; Gaspar, J.; Kong, A.-N.T. Dietary phytochemicals and cancer chemoprevention: A perspective on oxidative stress, inflammation, and epigenetics. Chem. Res. Toxicol. 2016, 29, 2071–2095. [Google Scholar] [CrossRef]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef]
- Surh, Y.-J.; Na, H.-K. NF-κB and Nrf2 as prime molecular targets for chemoprevention and cytoprotection with anti-inflammatory and antioxidant phytochemicals. Genes Nutr. 2008, 2, 313–317. [Google Scholar] [CrossRef]
- Shan, Y.; Lin, N.; Yang, X.; Tan, J.; Zhao, R.; Dong, S.; Wang, S. Sulphoraphane inhibited the expressions of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 through MyD88-dependent toll-like receptor-4 pathway in cultured endothelial cells. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 215–222. [Google Scholar] [CrossRef]
- Wagner, A.E.; Boesch-Saadatmandi, C.; Dose, J.; Schultheiss, G.; Rimbach, G. Anti-inflammatory potential of allyl-isothiocyanate–role of Nrf2, NF-κB and microRNA-155. J. Cell. Mol. Med. 2012, 16, 836–843. [Google Scholar] [CrossRef]
- Yang, Q.; Proll, M.J.; Salilew-Wondim, D.; Zhang, R.; Tesfaye, D.; Fan, H.; Cinar, M.U.; Grosse-Brinkhaus, C.; Tholen, E.; Islam, M.A.; et al. LPS-induced expression of CD14 in the TRIF pathway is epigenetically regulated by sulforaphane in porcine pulmonary alveolar macrophages. Innate Immun. 2016, 22, 682–695. [Google Scholar] [CrossRef]
- Qu, X.; Pröll, M.; Neuhoff, C.; Zhang, R.; Cinar, M.U.; Hossain, M.M.; Tesfaye, D.; Große-Brinkhaus, C.; Salilew-Wondim, D.; Tholen, E. Sulforaphane epigenetically regulates innate immune responses of porcine monocyte-derived dendritic cells induced with lipopolysaccharide. PLoS ONE 2015, 10, e0121574. [Google Scholar] [CrossRef]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef]
- Spencer, E.S.; Dale, E.J.; Gommans, A.L.; Rutledge, M.T.; Vo, C.T.; Nakatani, Y.; Gamble, A.B.; Smith, R.A.J.; Wilbanks, S.M.; Hampton, M.B.; et al. Multiple binding modes of isothiocyanates that inhibit macrophage migration inhibitory factor. Eur. J. Med. Chem. 2015, 93, 501–510. [Google Scholar] [CrossRef]
- Crichlow, G.V.; Fan, C.; Keeler, C.; Hodsdon, M.; Lolis, E.J. Structural interactions dictate the kinetics of macrophage migration inhibitory factor inhibition by different cancer-preventive isothiocyanates. Biochemistry 2012, 51, 7506–7514. [Google Scholar] [CrossRef]
- Tyndall, J.D.; Lue, H.; Rutledge, M.T.; Bernhagen, J.; Hampton, M.B.; Wilbanks, S.M. Macrophage migration inhibitory factor covalently complexed with phenethyl isothiocyanate. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 999–1002. [Google Scholar] [CrossRef]
- Gupta, P.; Kim, B.; Kim, S.H.; Srivastava, S.K. Molecular targets of isothiocyanates in cancer: Recent advances. Mol. Nutr. Food Res. 2014, 58, 1685–1707. [Google Scholar] [CrossRef]
- Navarro, S.L.; Li, F.; Lampe, J.W. Mechanisms of action of isothiocyanates in cancer chemoprevention: An update. Food Funct. 2011, 2, 579–587. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, Q.-H.; Xu, K. Are isothiocyanates potential anti-cancer drugs? Acta Pharmacol. Sin. 2009, 30, 501–512. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P. Anticarcinogenic activities of organic isothiocyanates: Chemistry and mechanisms. Cancer Res. 1994, 54 (Suppl. 7), 1976s–1981s. [Google Scholar] [PubMed]
- Mitsiogianni, M.; Amery, T.; Franco, R.; Zoumpourlis, V.; Pappa, A.; Panayiotidis, M.I. From chemo-prevention to epigenetic regulation: The role of isothiocyanates in skin cancer prevention. Pharmacol. Ther. 2018, 190, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, E.; Jørgensen, L.B. Chapter four Localization of plant myrosinases and glucosinolates. Recent Adv. Phytochem. 2003, 37, 79–99. [Google Scholar]
- Angelino, D.; Jeffery, E. Glucosinolate hydrolysis and bioavailability of resulting isothiocyanates: Focus on glucoraphanin. J. Funct. Foods 2014, 7, 67–76. [Google Scholar] [CrossRef]
- Andréasson, E.; Jørgensen, L.B.; Höglund, A.-S.; Rask, L.; Meijer, J. Different myrosinase and idioblast distribution in Arabidopsis and Brassica napus. Plant Physiol. 2001, 127, 1750–1763. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Martinez-Ballesta, M.; Carvajal, M. Myrosinase in Brassicaceae: The most important issue for glucosinolate turnover and food quality. Phytochem. Rev. 2015, 14, 1045–1051. [Google Scholar] [CrossRef]
- Koroleva, O.A.; Davies, A.; Deeken, R.; Thorpe, M.R.; Tomos, A.D.; Hedrich, R. Identification of a new glucosinolate-rich cell type in Arabidopsis flower stalk. Plant Physiol. 2000, 124, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Barba, F.J.; Nikmaram, N.; Roohinejad, S.; Khelfa, A.; Zhu, Z.; Koubaa, M. Bioavailability of glucosinolates and their breakdown products: Impact of processing. Front. Nutr. 2016, 3, 24. [Google Scholar] [CrossRef]
- Bones, A.M.; Rossiter, J.T. The enzymic and chemically induced decomposition of glucosinolates. Phytochemistry 2006, 67, 1053–1067. [Google Scholar] [CrossRef]
- Mithen, R.F.; Dekker, M.; Verkerk, R.; Rabot, S.; Johnson, I.T. The nutritional significance, biosynthesis and bioavailability of glucosinolates in human foods. J. Sci. Food Agric. 2000, 80, 967–984. [Google Scholar] [CrossRef]
- Thornalley, P. Cruciferous Vegetables, Isothiocyanates and Indoles (IARC Handbooks of Cancer Prevention); IARC Press: Lyon, France, 2004; pp. 1–261. [Google Scholar]
- Halkier, B.A.; Du, L. The biosynthesis of glucosinolates. Trends Plant Sci. 1997, 2, 425–431. [Google Scholar] [CrossRef]
- Kissen, R.; Rossiter, J.T.; Bones, A.M. The ‘mustard oil bomb’: Not so easy to assemble?! Localization, expression and distribution of the components of the myrosinase enzyme system. Phytochem. Rev. 2009, 8, 69–86. [Google Scholar] [CrossRef]
- Hanschen, F.S.; Platz, S.; Mewis, I.; Schreiner, M.; Rohn, S.; Kroh, L.W. Thermally induced degradation of sulfur-containing aliphatic glucosinolates in broccoli sprouts (Brassica oleracea var. italica) and model systems. J. Agric. Food Chem. 2012, 60, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Thornalley, P.J. Effect of storage, processing and cooking on glucosinolate content of Brassica vegetables. Food Chem. Toxicol. 2007, 45, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Ciska, E.; Martyniak-Przybyszewska, B.; Kozlowska, H. Content of glucosinolates in cruciferous vegetables grown at the same site for two years under different climatic conditions. J. Agric. Food Chem. 2000, 48, 2862–2867. [Google Scholar] [CrossRef]
- Deng, Q.; Zinoviadou, K.G.; Galanakis, C.M.; Orlien, V.; Grimi, N.; Vorobiev, E.; Lebovka, N.; Barba, F.J. The effects of conventional and non-conventional processing on glucosinolates and its derived forms, isothiocyanates: Extraction, degradation, and applications. Food Eng. Rev. 2015, 7, 357–381. [Google Scholar] [CrossRef]
- Angelino, D.; Dosz, E.B.; Sun, J.; Hoeflinger, J.L.; Van Tassell, M.L.; Chen, P.; Harnly, J.M.; Miller, M.J.; Jeffery, E.H. Myrosinase-dependent and–independent formation and control of isothiocyanate products of glucosinolate hydrolysis. Front. Plant Sci. 2015, 6, 831. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Kensler, T.W.; Chen, J.-G.; Gange, S.J.; Groopman, J.D.; Friesen, M.D. Quantification of sulforaphane mercapturic acid pathway conjugates in human urine by high-performance liquid chromatography and isotope-dilution tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 1991–1996. [Google Scholar] [CrossRef]
- Keum, Y.-S.; Jeong, W.-S.; Kong, A.T. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat. Res. 2004, 555, 191–202. [Google Scholar] [CrossRef]
- Talalay, P.; Fahey, J.W. Phytochemicals from cruciferous plants protect against cancer by modulating carcinogen metabolism. J. Nutr. 2001, 131, 3027S–3033S. [Google Scholar] [CrossRef] [PubMed]
- Yoxall, V.; Kentish, P.; Coldham, N.; Kuhnert, N.; Sauer, M.J.; Ioannides, C. Modulation of hepatic cytochromes P450 and phase II enzymes by dietary doses of sulforaphane in rats: Implications for its chemopreventive activity. Int. J. Cancer 2005, 117, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Telang, U.; Morris, M.E. Effect of orally administered phenethyl isothiocyanate on hepatic gene expression in rats. Mol. Nutr. Food Res. 2010, 54, 1802–1806. [Google Scholar] [CrossRef] [PubMed]
- Von Weymarn, L.B.; Chun, J.A.; Hollenberg, P.F. Effects of benzyl and phenethyl isothiocyanate on P450s 2A6 and 2A13: Potential for chemoprevention in smokers. Carcinogenesis 2006, 27, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.S.; Chen, X.Y.; Zhu, R.Z.; Choi, B.M.; Kim, B.R. Sulforaphane induces glutathione S-transferase isozymes which detoxify aflatoxin B1-8, 9-epoxide in AML 12 cells. BioFactors 2010, 36, 289–296. [Google Scholar] [CrossRef]
- Munday, C.M. Selective induction of phase II enzymes in the urinary bladder of rats by allyl isothiocyanate, a compound derived from Brassica vegetables. Nutr. Cancer 2002, 44, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.-L.; Shi, M.; Tang, H.; Han, W.; Spivack, S.D. Candidate dietary phytochemicals modulate expression of phase II enzymes GSTP1 and NQO1 in human lung cells. J. Nutr. 2010, 140, 1404–1410. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Howie, A.F.; Beckett, G.J.; Mithen, R.; Bao, Y. Sulforaphane, erucin, and iberin up-regulate thioredoxin reductase 1 expression in human MCF-7 cells. J. Agric. Food Chem. 2005, 53, 1417–1421. [Google Scholar] [CrossRef]
- Basten, G.P.; Bao, Y.; Williamson, G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis 2002, 23, 1399–1404. [Google Scholar] [CrossRef]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Parnaud, G.; Li, P.; Cassar, G.; Rouimi, P.; Tulliez, J.; Combaret, L.; Gamet-Payrastre, L. Mechanism of sulforaphane-induced cell cycle arrest and apoptosis in human colon cancer cells. Nutr. Cancer 2004, 48, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-M.; Tsai, C.-C.; Hsu, Y.-C. Sulforaphane, a dietary isothiocyanate, induces G2/M arrest in Cervical cancer cells through CyclinB1 downregulation and GADD45β/CDC2 association. Int. J. Mol. Sci. 2016, 17, 1530. [Google Scholar] [CrossRef]
- Shan, Y.; Sun, C.; Zhao, X.; Wu, K.; Cassidy, A.; Bao, Y. Effect of sulforaphane on cell growth, G0/G1 phase cell progression and apoptosis in human bladder cancer T24 cells. Int. J. Oncol. 2006, 29, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Lin, K.-C.; Lin, J.-P.; Tang, N.-Y.; Yang, J.-S.; Lu, K.-W.; Chung, J.-G. Phenethyl Isothiocyanate (PEITC) inhibits the growth of human oral squamous carcinoma HSC-3 cells through G0/G1 phase arrest and mitochondria-mediated apoptotic cell death. Evid. Based Complement. Alternat. Med. 2012, 2012, 718320. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Srivastava, S.K.; Lew, K.L.; Zeng, Y.; Hershberger, P.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003, 24, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Loganathan, S.; Humphreys, I.; Srivastava, S.K. Benzyl isothiocyanate-induced DNA damage causes G2/M cell cycle arrest and apoptosis in human pancreatic cancer cells. J. Nutr. 2006, 136, 2728–2734. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Uchida, K.; Osawa, T.; Nakamura, Y. Benzyl isothiocyanate modifies expression of the G2/M arrest-related genes. BioFactors 2004, 21, 23–26. [Google Scholar] [CrossRef]
- Cheung, K.L.; Khor, T.O.; Yu, S.; Kong, A.-N.T. PEITC induces G1 cell cycle arrest on HT-29 cells through the activation of p38 MAPK signaling pathway. AAPS J. 2008, 10, 277. [Google Scholar] [CrossRef]
- Jun-Hee, K.; Ki, H.K.; Jung, J.-Y.; Han, H.-S.; Shim, J.H.; Oh, S.; Choi, K.-H.; Choi, E.-S.; Shin, J.-A.; Leem, D.-H. Sulforaphane increases cyclin-dependent kinase inhibitor, p21 protein in human oral carcinoma cells and nude mouse animal model to induce G2/M cell cycle arrest. J. Clin. Biochem. Nutr. 2009, 46, 60–67. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Xiao, H.; Lew, K.L.; Singh, S.V. Induction of p21 protein protects against sulforaphane-induced mitotic arrest in LNCaP human prostate cancer cell line. Mol. Cancer Ther. 2007, 6, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.-A.; Murata, H.; Sakabe, T.; Sowa, Y.; Horie, N.; Nakanishi, R.; Sakai, T.; Kubo, T. Sulforaphane induces cell cycle arrest and apoptosis in murine osteosarcoma cells in vitro and inhibits tumor growth in vivo. Oncol. Rep. 2007, 18, 1263–1268. [Google Scholar] [CrossRef]
- Fimognari, C.; Nüsse, M.; Berti, F.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. A mixture of isothiocyanates induces cyclin B1-and p53-mediated cell-cycle arrest and apoptosis of human T lymphoblastoid cells. Mutat. Res. 2004, 554, 205–214. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Zörnig, M.; Hueber, A.-O.; Baum, W.; Evan, G. Apoptosis regulators and their role in tumorigenesis. Biochim. Biophys. Acta 2001, 1551, F1–F37. [Google Scholar] [CrossRef]
- Mi, L.; Di Pasqua, A.J.; Chung, F.L. Proteins as binding targets of isothiocyanates in cancer prevention. Carcinogenesis 2011, 32, 1405–1413. [Google Scholar] [CrossRef]
- Rudolf, E.; Andělová, H.; Červinka, M. Activation of several concurrent proapoptic pathways by sulforaphane in human colon cancer cells SW620. Food Chem. Toxicol. 2009, 47, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Mandlekar, S.; Harvey, K.J.; Ucker, D.S.; Kong, A.N. Chemopreventive isothiocyanates induce apoptosis and caspase-3-like protease activity. Cancer Res. 1998, 58, 402–408. [Google Scholar] [PubMed]
- Mondal, A.; Biswas, R.; Rhee, Y.-H.; Kim, J.; Ahn, J.-C. Sulforaphene promotes Bax/Bcl2, MAPK-dependent human gastric cancer AGS cells apoptosis and inhibits migration via EGFR, p-ERK1/2 downregulation. Gen. Physiol. Biophys. 2016, 35, 25–34. [Google Scholar] [PubMed]
- Tsai, S.-C.; Huang, W.-W.; Huang, W.-C.; Lu, C.-C.; Chiang, J.-H.; Peng, S.-F.; Chung, J.-G.; Lin, Y.-H.; Hsu, Y.-M.; Amagaya, S. ERK-modulated intrinsic signaling and G2/M phase arrest contribute to the induction of apoptotic death by allyl isothiocyanate in MDA-MB-468 human breast adenocarcinoma cells. Int. J. Oncol. 2012, 41, 2065–2072. [Google Scholar] [CrossRef]
- De Oliveira, J.M.P.F.; Costa, M.; Pedrosa, T.; Pinto, P.; Remédios, C.; Oliveira, H.; Pimentel, F.; Almeida, L.; Santos, C. Sulforaphane induces oxidative stress and death by p53-independent mechanism: Implication of impaired glutathione recycling. PLoS ONE 2014, 9, e92980. [Google Scholar] [CrossRef]
- Sarkar, R.; Mukherjee, S.; Biswas, J.; Roy, M. Sulphoraphane, a naturally occurring isothiocyanate induces apoptosis in breast cancer cells by targeting heat shock proteins. Biochem. Biophys. Res. Commun. 2012, 427, 80–85. [Google Scholar] [CrossRef]
- Sehrawat, A.; Croix, C.S.; Baty, C.J.; Watkins, S.; Tailor, D.; Singh, R.P.; Singh, S.V. Inhibition of mitochondrial fusion is an early and critical event in breast cancer cell apoptosis by dietary chemopreventative benzyl isothiocyanate. Mitochondrion 2016, 30, 67–77. [Google Scholar] [CrossRef]
- Xu, C.; Shen, G.; Yuan, X.; Kim, J.-H.; Gopalkrishnan, A.; Keum, Y.-S.; Nair, S.; Kong, A.-N.T. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis 2006, 27, 437–445. [Google Scholar] [CrossRef]
- Lin, F.; Lin, P.; Zhao, D.; Chen, Y.; Xiao, L.; Qin, W.; Li, D.; Chen, H.; Zhao, B.; Zou, H. Sox2 targets cyclinE, p27 and survivin to regulate androgen-independent human prostate cancer cell proliferation and apoptosis. Cell Prolif. 2012, 45, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Budhraja, A.; Cheng, S.; Liu, E.; Chen, J.; Yang, Z.; Chen, D.; Zhang, Z.; Shi, X. Phenethyl isothiocyanate exhibits antileukemic activity in vitro and in vivo by inactivation of Akt and activation of JNK pathways. Cell Death Dis. 2011, 2, e140. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-D.; Li, G.; Hu, H.; Jiang, C.; Kang, K.-S.; Lee, Y.-S.; Kim, S.-H.; Lu, J. Involvement of c-Jun N-terminal kinase in G2/M arrest and caspase-mediated apoptosis induced by sulforaphane in DU145 prostate cancer cells. Nutr. Cancer 2005, 52, 213–224. [Google Scholar] [CrossRef]
- Xiao, D.; Singh, S.V. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002, 62, 3615–3619. [Google Scholar]
- Stan, S.D.; Singh, S.V.; Whitcomb, D.C.; Brand, R.E. Phenethyl isothiocyanate inhibits proliferation and induces apoptosis in pancreatic cancer cells in vitro and in a MIAPaca2 xenograft animal model. Nutr. Cancer 2014, 66, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Nüsse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liu, X.; Chang, K.; Liu, X.; Xiong, J. Allyl Isothiocyanate Inhibits the Proliferation of Renal Carcinoma Cell Line GRC-1 by Inducing an Imbalance Between Bcl2 and Bax. Med. Sci. Monit. 2016, 22, 4283. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Lee, S.-H. Sulforaphane induces antioxidative and antiproliferative responses by generating reactive oxygen species in human bronchial epithelial BEAS-2B cells. J. Korean Med. Sci. 2011, 26, 1474–1482. [Google Scholar] [CrossRef]
- Yeh, Y.-T.; Hsu, Y.-N.; Huang, S.-Y.; Lin, J.-S.; Chen, Z.-F.; Chow, N.-H.; Su, S.-H.; Shyu, H.-W.; Lin, C.-C.; Huang, W.-T.; et al. Benzyl isothiocyanate promotes apoptosis of oral cancer cells via an acute redox stress-mediated DNA damage response. Food Chem. Toxicol. 2016, 97, 336–345. [Google Scholar] [CrossRef]
- Yeh, Y.T.; Yeh, H.; Su, S.H.; Lin, J.S.; Lee, K.J.; Shyu, H.W.; Chen, Z.F.; Huang, S.Y.; Su, S.J. Phenethyl isothiocyanate induces DNA damage-associated G2/M arrest and subsequent apoptosis in oral cancer cells with varying p53 mutations. Free Radic. Biol. Med. 2014, 74, 1–13. [Google Scholar] [CrossRef]
- Safe, S.; Kasiappan, R. Natural products as mechanism-based anticancer agents: Sp transcription factors as targets. Phytother. Res. 2016, 30, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Cheng, Y.; Jin, U.H.; Kim, K.; Safe, S. Specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4 are non-oncogene addiction genes in cancer cells. Oncotarget 2016, 7, 22245–22256. [Google Scholar] [CrossRef] [PubMed]
- Jutooru, I.; Guthrie, A.S.; Chadalapaka, G.; Pathi, S.; Kim, K.; Burghardt, R.; Jin, U.-H.; Safe, S. Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol. Cell. Biol. 2014, 34, 2382–2395. [Google Scholar] [CrossRef]
- Kasiappan, R.; Jutooru, I.; Karki, K.; Hedrick, E.; Safe, S. Benzyl Isothiocyanate (BITC) Induces Reactive Oxygen Species-dependent Repression of STAT3 Protein by Downregulation of Specificity Proteins in Pancreatic Cancer. J. Biol. Chem. 2016, 291, 27122–27133. [Google Scholar] [CrossRef] [PubMed]
- Chadalapaka, G.; Jutooru, I.; Safe, S. Celastrol decreases specificity proteins (Sp) and fibroblast growth factor receptor-3 (FGFR3) in bladder cancer cells. Carcinogenesis 2012, 33, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Crose, L.; Linardic, C.M.; Safe, S. Histone Deacetylase Inhibitors Inhibit Rhabdomyosarcoma by Reactive Oxygen Species-Dependent Targeting of Specificity Protein Transcription Factors. Mol. Cancer Ther. 2015, 14, 2143–2153. [Google Scholar] [CrossRef]
- Hedrick, E.; Li, X.; Safe, S. Penfluridol represses integrin expression in breast cancer through induction of reactive oxygen species and downregulation of Sp transcription factors. Mol. Cancer Ther. 2017, 16, 205–216. [Google Scholar] [CrossRef]
- Karki, K.; Hedrick, E.; Kasiappan, R.; Jin, U.-H.; Safe, S. Piperlongumine induces reactive oxygen species (ROS)-dependent downregulation of specificity protein transcription factors. Cancer Prev. Res. (Phila) 2017, 10, 467–477. [Google Scholar] [CrossRef]
- Gandhy, S.U.; Kim, K.; Larsen, L.; Rosengren, R.J.; Safe, S. Curcumin and synthetic analogs induce reactive oxygen species and decreases specificity protein (Sp) transcription factors by targeting microRNAs. BMC Cancer 2012, 12, 564. [Google Scholar] [CrossRef] [PubMed]
- Noratto, G.D.; Jutooru, I.; Safe, S.; Angel-Morales, G.; Mertens-Talcott, S.U. The drug resistance suppression induced by curcuminoids in colon cancer SW-480 cells is mediated by reactive oxygen species-induced disruption of the microRNA-27a-ZBTB10-Sp axis. Mol. Nutr. Food Res. 2013, 57, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Taoka, R.; Mmeje, C.O.; Jinesh, G.G.; Safe, S.; Kamat, A.M. CDODA-Me decreases specificity protein transcription factors and induces apoptosis in bladder cancer cells through induction of reactive oxygen species. Urol Oncol. 2016, 34, e11–e337. [Google Scholar] [CrossRef] [PubMed]
- Taoka, R.; Jinesh, G.G.; Xue, W.; Safe, S.; Kamat, A.M. CF3DODA-Me induces apoptosis, degrades Sp1, and blocks the transformation phase of the blebbishield emergency program. Apoptosis 2017, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.S.; Jutooru, I.; Chadalapaka, G.; Sreevalsan, S.; Anand, S.; Thatcher, G.R.; Safe, S. GT-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol. Cancer Res. 2011, 9, 195–202. [Google Scholar] [CrossRef]
- Safe, S.; Abbruzzese, J.; Abdelrahim, M.; Hedrick, E. Specificity Protein Transcription Factors and Cancer: Opportunities for Drug Development. Cancer Prev. Res. (Phila) 2018, 11, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Levine, B. Autophagy and cancer. Nature 2007, 446, 745. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Maiuri, M.C.; Madeo, F.; Kroemer, G. Autophagy induction for the treatment of cancer. Autophagy 2016, 12, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006, 66, 5828–5835. [Google Scholar] [CrossRef]
- Zhang, Q.-C.; Pan, Z.-H.; Liu, B.-N.; Meng, Z.-W.; Wu, X.; Zhou, Q.-H.; Xu, K. Benzyl isothiocyanate induces protective autophagy in human lung cancer cells through an endoplasmic reticulum stress-mediated mechanism. Acta Pharmacol. Sin. 2017, 38, 539. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-F.; Liao, P.-C.; Lin, Y.-H.; Chen, H.-E.; Lin, Y.-C.; Chou, K.-Y.; Tsai, T.-F.; Hwang, T.I.-S. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis 2012, 34, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-F.; Tsai, T.-F.; Yang, S.-C.; Lin, Y.-C.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.S. Benzyl isothiocyanate induces reactive oxygen species-initiated autophagy and apoptosis in human prostate cancer cells. Oncotarget 2017, 8, 20220–20234. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Cao, L.; Zhang, Q.; Song, Q.; Meng, Z.; Wu, X.; Xu, K. Inhibition of autophagy potentiates the anti-metastasis effect of phenethyl isothiocyanate through JAK2/STAT3 pathway in lung cancer cells. Mol. Carcinog. 2018, 57, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-E.; Lin, J.-F.; Tsai, T.-F.; Lin, Y.-C.; Chou, K.-Y.; Hwang, T.I.-S. Allyl Isothiocyanate Induces Autophagy through the Up-Regulation of Beclin-1 in Human Prostate Cancer Cells. Am. J. Chin. Med. 2018, 46, 1625–1643. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, A.; Wała, M.; Hać, A.; Felczykowska, A.; Herman-Antosiewicz, A. Sulforaphene, an isothiocyanate present in radish plants, inhibits proliferation of human breast cancer cells. Phytomedicine 2017, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, S.H. Pro-oxidant activity of sulforaphane and cisplatin potentiates apoptosis and simultaneously promotes autophagy in malignant mesothelioma cells. Mol. Med. Rep. 2017, 16, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Hee Kim, Y.; Kim, K.Y.; Jun do, Y.; Kim, J.S.; Kim, Y.H. Inhibition of autophagy enhances dynamin inhibitor-induced apoptosis via promoting Bak activation and mitochondrial damage in human Jurkat T cells. Biochem. Biophys. Res. Commun. 2016, 478, 1609–1616. [Google Scholar] [CrossRef]
- Horwacik, I.; Gaik, M.; Durbas, M.; Boratyn, E.; Zajac, G.; Szychowska, K.; Szczodrak, M.; Koloczek, H.; Rokita, H. Inhibition of autophagy by 3-methyladenine potentiates sulforaphane-induced cell death of BE(2)-C human neuroblastoma cells. Mol. Med. Rep. 2015, 12, 535–542. [Google Scholar] [CrossRef]
- Xiao, D.; Bommareddy, A.; Kim, S.H.; Sehrawat, A.; Hahm, E.R.; Singh, S.V. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS ONE 2012, 7, e32597. [Google Scholar] [CrossRef]
- Bommareddy, A.; Hahm, E.-R.; Xiao, D.; Powolny, A.A.; Fisher, A.L.; Jiang, Y.; Singh, S.V. Atg5 Regulates Phenethyl Isothiocyanate–Induced Autophagic and Apoptotic Cell Death in Human Prostate Cancer Cells. Cancer Res. 2009, 69, 3704–3712. [Google Scholar] [CrossRef]
- Liu, H.J.; Wang, L.; Kang, L.; Du, J.; Li, S.; Cui, H.X. Sulforaphane-N-Acetyl-Cysteine Induces Autophagy Through Activation of ERK1/2 in U87MG and U373MG Cells. Cell. Physiol. Biochem. 2018, 51, 528–542. [Google Scholar] [CrossRef]
- Yang, F.; Wang, F.; Liu, Y.; Wang, S.; Li, X.; Huang, Y.; Xia, Y.; Cao, C. Sulforaphane induces autophagy by inhibition of HDAC6-mediated PTEN activation in triple negative breast cancer cells. Life Sci. 2018, 213, 149–157. [Google Scholar] [CrossRef]
- Milczarek, M.; Wiktorska, K.; Mielczarek, L.; Koronkiewicz, M.; Dąbrowska, A.; Lubelska, K.; Matosiuk, D.; Chilmonczyk, Z. Autophagic cell death and premature senescence: New mechanism of 5-fluorouracil and sulforaphane synergistic anticancer effect in MDA-MB-231 triple negative breast cancer cell line. Food Chem. Toxicol. 2018, 111, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C.; Surani, M.A. Imprinting and the epigenetic asymmetry between parental genomes. Science 2001, 293, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef]
- Avner, P.; Heard, E. X-chromosome inactivation: Counting, choice and initiation. Nat. Rev. Genet. 2001, 2, 59–67. [Google Scholar] [CrossRef]
- Panning, B.; Jaenisch, R. RNA and the epigenetic regulation of X chromosome inactivation. Cell 1998, 93, 305–308. [Google Scholar] [CrossRef]
- Maltepe, E.; Bakardjiev, A.I.; Fisher, S.J. The placenta: Transcriptional, epigenetic, and physiological integration during development. J. Clin. Investig. 2010, 120, 1016–1025. [Google Scholar] [CrossRef]
- Hemberger, M. Epigenetic landscape required for placental development. Cell. Mol. Life Sci. 2007, 64, 2422–2436. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar] [CrossRef]
- Toyota, M.; Suzuki, H.; Sasaki, Y.; Maruyama, R.; Imai, K.; Shinomura, Y.; Tokino, T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008, 68, 4123–4132. [Google Scholar] [CrossRef]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Müller, M.; Römermann, D.; Länger, F.; Kreipe, H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J. Pathol. 2008, 214, 17–24. [Google Scholar] [CrossRef]
- Zhang, C.; Li, H.; Zhou, G.; Zhang, Q.; Zhang, T.; Li, J.; Zhang, J.; Hou, J.; Liew, C.; Yin, D. Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J. Pathol. 2007, 212, 134–142. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Franco, R.; Pappa, A.; Malamou-Mitsi, V.; Georgakila, S.; Georgakilas, A.G.; Panayiotidis, M.I. The role of epigenetics in environmental and occupational carcinogenesis. Chem.-Biol. Interact. 2010, 188, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)--induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Yuanfeng, W.; Gongnian, X.; Jianwei, M.; Shiwang, L.; Jun, H.; Lehe, M. Dietary sulforaphane inhibits histone deacetylase activity in B16 melanoma cells. J. Funct. Foods 2015, 18, 182–189. [Google Scholar] [CrossRef]
- Rajendran, P.; Kidane, A.I.; Yu, T.-W.; Dashwood, W.-M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef]
- Su, Z.-Y.; Zhang, C.; Lee, J.H.; Shu, L.; Wu, T.-Y.; Khor, T.O.; Conney, A.H.; Lu, Y.-P.; Kong, A.-N.T. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev. Res. (Phila) 2014, 7, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Liu, X.M.; Fang, Y.; Dai, W.; Chiao, F.B.; Puccio, G.M.; Feng, J.; Liu, D.; Chiao, J.W. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int. J. Oncol. 2008, 33, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2005, 27, 811–819. [Google Scholar] [CrossRef]
- Abbaoui, B.; Telu, K.H.; Lucas, C.R.; Thomas-Ahner, J.M.; Schwartz, S.J.; Clinton, S.K.; Freitas, M.A.; Mortazavi, A. The impact of cruciferous vegetable isothiocyanates on histone acetylation and histone phosphorylation in bladder cancer. J. Proteom. 2017, 156, 94–103. [Google Scholar] [CrossRef]
- Batra, S.; Sahu, R.P.; Kandala, P.K.; Srivastava, S.K. Benzyl isothiocyanate–mediated inhibition of histone deacetylase leads to NF-κB turnoff in human pancreatic carcinoma cells. Mol. Cancer Ther. 2010, 9, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Beklemisheva, A.A.; Fang, Y.; Feng, J.; Ma, X.; Dai, W.; Chiao, J.W. Epigenetic mechanism of growth inhibition induced by phenylhexyl isothiocyanate in prostate cancer cells. Anticancer Res. 2006, 26, 1225–1230. [Google Scholar]
- Jiang, L.-L.; Zhou, S.-J.; Zhang, X.-M.; Chen, H.-Q.; Liu, W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed. Pharmacother. 2016, 78, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.-L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Myzak, M.C.; Ho, E. Dietary HDAC inhibitors: Time to rethink weak ligands in cancer chemoprevention? Carcinogenesis 2005, 27, 344–349. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Ho, E. Dietary agents as histone deacetylase inhibitors: Sulforaphane and structurally related isothiocyanates. Nutr. Rev. 2008, 66 (Suppl. 1), S36–S38. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Baylin, S.B. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Akhavan-Niaki, H.; Samadani, A.A. DNA methylation and cancer development: Molecular mechanism. Cell Biochem. Biophys. 2013, 67, 501–513. [Google Scholar] [CrossRef]
- Kaufman-Szymczyk, A.; Majewski, G.; Lubecka-Pietruszewska, K.; Fabianowska-Majewska, K. The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation. Int. J. Mol. Sci. 2015, 16, 29732–29743. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Hsu, A.; Buchanan, A.; Palomera-Sanchez, Z.; Beaver, L.M.; Houseman, E.A.; Williams, D.E.; Dashwood, R.H.; Ho, E. Effects of sulforaphane and 3,3’-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE 2014, 9, e86787. [Google Scholar] [CrossRef]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenet. 2011, 3, 3. [Google Scholar] [CrossRef]
- Jiang, S.; Ma, X.; Huang, Y.; Xu, Y.; Zheng, R.; Chiao, J.-W. Reactivating aberrantly hypermethylated p15 gene in leukemic T cells by a phenylhexyl isothiocyanate mediated inter-active mechanism on DNA and chromatin. J. Hematol. Oncol. 2010, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Li, Y.; Shukla, S.; Tollefsbol, T.O. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS ONE 2012, 7, e37748. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef]
- Boyanapalli, S.S.; Li, W.; Fuentes, F.; Guo, Y.; Ramirez, C.N.; Gonzalez, X.-P.; Pung, D.; Kong, A.-N.T. Epigenetic reactivation of RASSF1A by phenethyl isothiocyanate (PEITC) and promotion of apoptosis in LNCaP cells. Pharmacol. Res. 2016, 114, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Slaby, O.; Sachlova, M.; Brezkova, V.; Hezova, R.; Kovarikova, A.; Bischofová, S.; Sevcikova, S.; Bienertova-Vasku, J.; Vasku, A.; Svoboda, M. Identification of microRNAs regulated by isothiocyanates and association of polymorphisms inside their target sites with risk of sporadic colorectal cancer. Nutr. Cancer 2013, 65, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shu, L.; Kim, H.; Khor, T.O.; Wu, R.; Li, W.; Kong, A.N.T. Phenethyl isothiocyanate (PEITC) suppresses prostate cancer cell invasion epigenetically through regulating microRNA-194. Mol. Nutr. Food Res. 2016, 60, 1427–1436. [Google Scholar] [CrossRef]
- Wang, D.X.; Zou, Y.J.; Zhuang, X.B.; Chen, S.X.; Lin, Y.; Li, W.L.; Lin, J.J.; Lin, Z.Q. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3beta/beta-catenin signaling pathways. Acta Pharmacol. Sin. 2017, 38, 241–251. [Google Scholar] [CrossRef]
- Lan, F.; Pan, Q.; Yu, H.; Yue, X. Sulforaphane enhances temozolomide-induced apoptosis because of downregulation of miR-21 via Wnt/beta-catenin signaling in glioblastoma. J. Neurochem. 2015, 134, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Zhang, L.; Bao, Y.; Li, B.; He, C.; Gao, M.; Feng, X.; Xu, W.; Zhang, X.; Wang, S. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J. Nutr. Biochem. 2013, 24, 1062–1069. [Google Scholar] [CrossRef]
- Xiao, J.; Gong, A.Y.; Eischeid, A.N.; Chen, D.; Deng, C.; Young, C.Y.; Chen, X.M. miR-141 modulates androgen receptor transcriptional activity in human prostate cancer cells through targeting the small heterodimer partner protein. Prostate 2012, 72, 1514–1522. [Google Scholar] [CrossRef]
- Yu, C.; Gong, A.Y.; Chen, D.; Solelo Leon, D.; Young, C.Y.; Chen, X.M. Phenethyl isothiocyanate inhibits androgen receptor-regulated transcriptional activity in prostate cancer cells through suppressing PCAF. Mol. Nutr. Food Res. 2013, 57, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Moserle, L.; Casanovas, O. Anti-angiogenesis and metastasis: A tumour and stromal cell alliance. J. Intern. Med. 2013, 273, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Rudek, M.A.; Venitz, J.; Figg, W.D. Matrix metalloproteinase inhibitors: Do they have a place in anticancer therapy? Pharmacother. J. Hum. Pharmacol. Drug Ther. 2002, 22, 705–720. [Google Scholar] [CrossRef]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. Molecular mechanism of angiogenesis Transcription factors and their therapeutic relevance. Pharmacol. Ther. 2000, 87, 51–60. [Google Scholar] [CrossRef]
- Thejass, P.; Kuttan, G. Inhibition of endothelial cell differentiation and proinflammatory cytokine production during angiogenesis by allyl isothiocyanate and phenyl isothiocyanate. Integr. Cancer Ther. 2007, 6, 389–399. [Google Scholar] [CrossRef]
- Lai, K.-C.; Lu, C.-C.; Tang, Y.-J.; Chiang, J.-H.; Kuo, D.-H.; Chen, F.-A.; Chen, I.-L.; Yang, J.-S. Allyl isothiocyanate inhibits cell metastasis through suppression of the MAPK pathways in epidermal growth factor-stimulated HT29 human colorectal adenocarcinoma cells. Oncol. Rep. 2014, 31, 189–196. [Google Scholar] [CrossRef]
- Xiao, D.; Singh, S.V. Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res. 2007, 67, 2239–2246. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, L.; Zhang, G.-D.; Wang, H.-O.; Liu, M.-Y.; Jiang, Y.; Qi, L.-S.; Li, Q.; Yang, P. Potential mechanisms of benzyl isothiocyanate suppression of invasion and angiogenesis by the U87MG human glioma cell line. Asian Pac. J. Cancer Prev. 2014, 15, 8225–8228. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.; Wang, L.; Jiao, B. HIFs, angiogenesis, and cancer. J. Cell. Biochem. 2013, 114, 967–974. [Google Scholar] [CrossRef]
- Kim, D.H.; Sung, B.; Kang, Y.J.; Hwang, S.Y.; Kim, M.J.; Yoon, J.-H.; Im, E.; Kim, N.D. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int. J. Oncol. 2015, 47, 2226–2232. [Google Scholar] [CrossRef]
- Wang, X.-H.; Cavell, B.E.; Syed Alwi, S.S.; Packham, G. Inhibition of hypoxia inducible factor by phenethyl isothiocyanate. Biochem. Pharmacol. 2009, 78, 261–272. [Google Scholar] [CrossRef]
- Gupta, B.; Chiang, L.; Chae, K.; Lee, D.-H. Phenethyl isothiocyanate inhibits hypoxia-induced accumulation of HIF-1α and VEGF expression in human glioma cells. Food Chem. 2013, 141, 1841–1846. [Google Scholar] [CrossRef]
- Chaw, S.; Majeed, A.A.; Dalley, A.; Chan, A.; Stein, S.; Farah, C. Epithelial to mesenchymal transition (EMT) biomarkers–E-cadherin, beta-catenin, APC and Vimentin–in oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef]
- Shih, J.-Y.; Yang, P.-C. The EMT regulator slug and lung carcinogenesis. Carcinogenesis 2011, 32, 1299–1304. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Z.; Yang, Q.; Li, H.; Guan, H.; Shi, B.; Hou, P.; Ji, M. Sulforaphane inhibits thyroid cancer cell growth and invasiveness through the reactive oxygen species-dependent pathway. Oncotarget 2015, 6, 25917–25931. [Google Scholar] [CrossRef]
- Sehrawat, A.; Kim, S.-H.; Vogt, A.; Singh, S.V. Suppression of FOXQ1 in benzyl isothiocyanate-mediated inhibition of epithelial–mesenchymal transition in human breast cancer cells. Carcinogenesis 2012, 34, 864–873. [Google Scholar] [CrossRef]
- Milkiewicz, M.; Roudier, E.; Doyle, J.L.; Trifonova, A.; Birot, O.; Haas, T.L. Identification of a Mechanism Underlying Regulation of the Anti-Angiogenic Forkhead Transcription Factor FoxO1 in Cultured Endothelial Cells and Ischemic Muscle. Am. J. Pathol. 2011, 178, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-C.; Huang, A.-C.; Hsu, S.-C.; Kuo, C.-L.; Yang, J.-S.; Wu, S.-H.; Chung, J.-G. Benzyl isothiocyanate (BITC) inhibits migration and invasion of human colon cancer HT29 cells by inhibiting matrix metalloproteinase-2/-9 and urokinase plasminogen (uPA) through PKC and MAPK signaling pathway. J. Agric. Food Chem. 2010, 58, 2935–2942. [Google Scholar] [CrossRef]
- Jackson, S.J.T.; Singletary, K.W.; Venema, R.C. Sulforaphane suppresses angiogenesis and disrupts endothelial mitotic progression and microtubule polymerization. Vascul. Pharmacol. 2007, 46, 77–84. [Google Scholar] [CrossRef]
- Bertl, E.; Bartsch, H.; Gerhauser, C. Inhibition of angiogenesis and endothelial cell functions are novel sulforaphane-mediated mechanisms in chemoprevention. Mol. Cancer Ther. 2006, 5, 575–585. [Google Scholar] [CrossRef]
- Abdull, R.; Ahmad, F.; Noor, N.M. Cruciferous vegetables: Dietary phytochemicals for cancer prevention. Asian Pac. J. Cancer Prev. 2013, 14, 1565–1570. [Google Scholar] [CrossRef]
- Sahu, R.P.; Srivastava, S.K. The role of STAT-3 in the induction of apoptosis in pancreatic cancer cells by benzyl isothiocyanate. J. Natl. Cancer Inst. 2009, 101, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Murillo, G.; Mehta, R.G. Cruciferous vegetables and cancer prevention. Nutr. Cancer 2001, 41, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; Zhang, Y. Chemoprotection against Cancer by Isothiocyanates and Glucosinolates; Portland Press Limited: London, UK, 1996. [Google Scholar]
- Fahey, J.W.; Zhang, Y.; Talalay, P. Broccoli sprouts: An exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. USA 1997, 94, 10367–10372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef]
- Brennan, P.; Hsu, C.C.; Moullan, N.; Szeszenia-Dabrowska, N.; Lissowska, J.; Zaridze, D.; Rudnai, P.; Fabianova, E.; Mates, D.; Bencko, V. Effect of cruciferous vegetables on lung cancer in patients stratified by genetic status: A mendelian randomisation approach. Lancet 2005, 366, 1558–1560. [Google Scholar] [CrossRef]
- Zhao, H.; Lin, J.; Grossman, H.B.; Hernandez, L.M.; Dinney, C.P.; Wu, X. Dietary isothiocyanates, GSTM1, GSTT1, NAT2 polymorphisms and bladder cancer risk. Int. J. Cancer 2007, 120, 2208–2213. [Google Scholar] [CrossRef]
- Seow, A.; Yuan, J.-M.; Sun, C.-L.; Van Den Berg, D.; Lee, H.-P.; Yu, M.C. Dietary isothiocyanates, glutathione S-transferase polymorphisms and colorectal cancer risk in the Singapore Chinese Health Study. Carcinogenesis 2002, 23, 2055–2061. [Google Scholar] [CrossRef]
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef]
- Seow, A.; Vainio, H.; Mimi, C.Y. Effect of glutathione-S-transferase polymorphisms on the cancer preventive potential of isothiocyanates: An epidemiological perspective. Mutat. Res. 2005, 592, 58–67. [Google Scholar] [CrossRef]
- Zhao, B.; Seow, A.; Lee, E.J.; Poh, W.-T.; Teh, M.; Eng, P.; Wang, Y.-T.; Tan, W.-C.; Mimi, C.Y.; Lee, H.-P. Dietary isothiocyanates, glutathione S-transferase-M1,-T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol. Biomark. Prev. 2001, 10, 1063–1067. [Google Scholar]
- Cai, X.; Yang, L.; Chen, H.; Wang, C. An updated meta-analysis of the association between GSTM1 polymorphism and colorectal cancer in Asians. Tumor Biol. 2014, 35, 949–953. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, B.; Hu, K.; Wang, J.; Lu, S.; Zhang, Y.; Lu, W.; Zhao, E.; Yuan, L. Glutathione S-transferase θ1 polymorphism contributes to lung cancer susceptibility: A meta-analysis of 26 case-control studies. Oncol. Lett. 2015, 9, 1947–1953. [Google Scholar] [CrossRef]
- Yang, H.; Yang, S.; Liu, J.; Shao, F.; Wang, H.; Wang, Y. The association of GSTM1 deletion polymorphism with lung cancer risk in Chinese population: Evidence from an updated meta-analysis. Sci. Rep. 2015, 5, 9392. [Google Scholar] [CrossRef] [PubMed]
- Vogtmann, E.; Xiang, Y.-B.; Li, H.-L.; Cai, Q.; Wu, Q.-J.; Xie, L.; Li, G.-L.; Yang, G.; Waterbor, J.W.; Levitan, E.B.; et al. Cruciferous vegetables, glutathione S-transferase polymorphisms, and the risk of colorectal cancer among Chinese men. Ann. Epidemiol. 2014, 24, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Feng, M.; Luo, Y.; Sun, C.; Fan, Z.; Tan, Y.; Fu, B.; Lang, J. GSTP1 Ile105Val polymorphism and colorectal cancer risk: An updated analysis. Gene 2013, 527, 275–282. [Google Scholar] [CrossRef]
- Wang, J.; Han, L.; Zhang, W.; Wang, J.; Ni, Q.; Shen, M.; Gao, Y. [A case-control study on the association between urinary levels of isothiocyanates and the risk of pancreatic cancer]. Zhonghua Yu Fang Yi Xue Za Zhi. 2014, 48, 172–176. [Google Scholar] [PubMed]
- Yang, G.; Gao, Y.T.; Shu, X.O.; Cai, Q.; Li, G.L.; Li, H.L.; Ji, B.T.; Rothman, N.; Dyba, M.; Xiang, Y.B.; et al. Isothiocyanate exposure, glutathione S-transferase polymorphisms, and colorectal cancer risk. Am. J. Clin. Nutr. 2010, 91, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Kolonel, L.N.; Hankin, J.H.; Whittemore, A.S.; Wu, A.H.; Gallagher, R.P.; Wilkens, L.R.; John, E.M.; Howe, G.R.; Dreon, D.M.; West, D.W. Vegetables, fruits, legumes and prostate cancer: A multiethnic case-control study. Cancer Epidemiol. Biomark. Prev. 2000, 9, 795–804. [Google Scholar]
- Tang, L.; Zirpoli, G.R.; Jayaprakash, V.; Reid, M.E.; McCann, S.E.; Nwogu, C.E.; Zhang, Y.; Ambrosone, C.B.; Moysich, K.B. Cruciferous vegetable intake is inversely associated with lung cancer risk among smokers: A case-control study. BMC Cancer 2010, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Neuhouser, M.L.; Patterson, R.E.; Thornquist, M.D.; Omenn, G.S.; King, I.B.; Goodman, G.E. Fruits and vegetables are associated with lower lung cancer risk only in the placebo arm of the β-carotene and retinol efficacy trial (CARET). Cancer Epidemiol. Biomark. Prev. 2003, 12, 350–358. [Google Scholar]
- Fowke, J.H.; Chung, F.-L.; Jin, F.; Qi, D.; Cai, Q.; Conaway, C.; Cheng, J.-R.; Shu, X.-O.; Gao, Y.-T.; Zheng, W. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003, 63, 3980–3986. [Google Scholar] [PubMed]
- Ambrosone, C.B.; McCann, S.E.; Freudenheim, J.L.; Marshall, J.R.; Zhang, Y.; Shields, P.G. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J. Nutr. 2004, 134, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.G.; Hislop, G.T.; Howe, G.R.; Ghadirian, P. Plant foods, antioxidants, and prostate cancer risk: Findings from case-control studies in Canada. Nutr. Cancer 1999, 34, 173–184. [Google Scholar] [CrossRef]
- Cohen, J.H.; Kristal, A.R.; Stanford, J.L. Fruit and vegetable intakes and prostate cancer risk. J. Natl. Cancer Inst. 2000, 92, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Romagnolo, D.F.; Selmin, O.I. Flavonoids and cancer prevention: A review of the evidence. J. Nutr. Gerontol. Geriatr. 2012, 31, 206–238. [Google Scholar] [CrossRef]
- Wu, Q.J.; Wang, J.; Gao, J.; Zhang, W.; Han, L.H.; Gao, S.; Gao, Y.T.; Ji, B.T.; Zheng, W.; Shu, X.O.; et al. Urinary isothiocyanates level and liver cancer risk: A nested case-control study in Shanghai, China. Nutr. Cancer 2014, 66, 1023–1029. [Google Scholar] [CrossRef]
- Fowke, J.H.; Gao, Y.-T.; Chow, W.-H.; Cai, Q.; Shu, X.-O.; Li, H.-l.; Ji, B.-T.; Rothman, N.; Yang, G.; Chung, F.-L.; et al. Urinary isothiocyanate levels and lung cancer risk among non-smoking women: A prospective investigation. Lung Cancer 2011, 73, 18–24. [Google Scholar] [CrossRef]
- Wu, C.L.; Huang, A.C.; Yang, J.S.; Liao, C.L.; Lu, H.F.; Chou, S.T.; Ma, C.Y.; Hsia, T.C.; Ko, Y.C.; Chung, J.G. Benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells. J. Orth. Res. 2011, 29, 1199–1209. [Google Scholar]
- Xiao, D.; Vogel, V.; Singh, S.V. Benzyl isothiocyanate–induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol. Cancer Ther. 2006, 5, 2931–2945. [Google Scholar] [CrossRef]
- Tripathi, K.; Hussein, U.K.; Anupalli, R.; Barnett, R.; Bachaboina, L.; Scalici, J.; Rocconi, R.P.; Owen, L.B.; Piazza, G.A.; Palle, K. Allyl isothiocyanate induces replication-associated DNA damage response in NSCLC cells and sensitizes to ionizing radiation. Oncotarget 2015, 6, 5237. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.-S.; Lee, H.J. Effects of phenylethyl isothiocyanate and its metabolite on cell-cycle arrest and apoptosis in LNCaP human prostate cancer cells. Int. J. Food Sci. Nutr. 2010, 61, 324–336. [Google Scholar] [CrossRef]
- Pappa, G.; Lichtenberg, M.; Iori, R.; Barillari, J.; Bartsch, H.; Gerhäuser, C. Comparison of growth inhibition profiles and mechanisms of apoptosis induction in human colon cancer cell lines by isothiocyanates and indoles from Brassicaceae. Mutat. Res. 2006, 599, 76–87. [Google Scholar] [CrossRef]
- Ma, X.; Fang, Y.; Beklemisheva, A.; Dai, W.; Feng, J.; Ahmed, T.; Liu, D.; Chiao, J. Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int. J. Oncol. 2006, 28, 1287–1293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.-W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Beklemisheva, A.; Liu, X.M.; Ferrari, A.C.; Feng, J.; Chiao, J.W. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol. Carcinog. 2007, 46, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shen, G.; Chen, C.; Gélinas, C.; Ah-Ng, T.K. Suppression of NF-[kappa] B and NF-[kappa] B-regulated gene expression by sulforaphane and PEITC through I [kappa] B [alpha], IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005, 24, 4486. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.R.; Sahu, R.P.; Srivastava, S.K. Benzyl isothiocyanate suppresses pancreatic tumor angiogenesis and invasion by inhibiting HIF-α/VEGF/Rho-GTPases: Pivotal role of STAT-3. PLoS ONE 2011, 6, e25799. [Google Scholar] [CrossRef] [PubMed]
- Lawson, A.P.; Long, M.J.C.; Coffey, R.T.; Qian, Y.; Weerapana, E.; El Oualid, F.; Hedstrom, L. Naturally Occurring Isothiocyanates Exert Anticancer Effects by Inhibiting Deubiquitinating Enzymes. Cancer Res. 2015, 75, 5130–5142. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Xiao, Z.; Hood, B.L.; Dakshanamurthy, S.; Wang, X.; Govind, S.; Conrads, T.P.; Veenstra, T.D.; Chung, F.-L. Covalent binding to tubulin by isothiocyanates A mechanism of cell growth arrest and apoptosis. J. Biol. Chem. 2008, 283, 22136–22146. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.K.; Lund, E.K.; Parker, M.L.; Clarke, R.G.; Johnson, I.T. Allyl-isothiocyanate causes mitotic block, loss of cell adhesion and disrupted cytoskeletal structure in HT29 cells. Carcinogenesis 2004, 25, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Singletary, K.W. Sulforaphane inhibits human MCF-7 mammary cancer cell mitotic progression and tubulin polymerization. J. Nutr. 2004, 134, 2229–2236. [Google Scholar] [CrossRef]
- Bryant, C.S.; Kumar, S.; Chamala, S.; Shah, J.; Pal, J.; Haider, M.; Seward, S.; Qazi, A.M.; Morris, R.; Semaan, A. Sulforaphane induces cell cycle arrest by protecting RB-E2F-1 complex in epithelial ovarian cancer cells. Mol. Cancer 2010, 9, 47. [Google Scholar] [CrossRef]
- Lee, C.-S.; Cho, H.-J.; Jeong, Y.-J.; Shin, J.-M.; Park, K.-K.; Park, Y.-Y.; Bae, Y.-S.; Chung, I.-K.; Kim, M.; Kim, C.-H. Isothiocyanates inhibit the invasion and migration of C6 glioma cells by blocking FAK/JNK-mediated MMP-9 expression. Oncol. Rep. 2015, 34, 2901–2908. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y. Dietary isothiocyanates inhibit the growth of human bladder carcinoma cells. J. Nutr. 2004, 134, 2004–2010. [Google Scholar] [CrossRef]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N.E. Sulforaphane induces cell type–specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef]
- Xiao, D.; Powolny, A.A.; Singh, S.V. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger reactive oxygen species-dependent apoptosis in human breast cancer cells. J. Biol. Chem. 2008, 283, 30151–30163. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lin, X.; Feng, J.; Zhao, X.; Gallagher, R.; Lee, M.Y.; Chiao, J.W.; Liu, D. Phenylhexyl isothiocyanate has dual function as histone deacetylase inhibitor and hypomethylating agent and can inhibit myeloma cell growth by targeting critical pathways. J. Hematol. Oncol. 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, J.; Lin, Y.; Tsai, T.; Chou, K.; Hwang, T. 44 Allyl isothiocyanate induces reactive oxygen species-mediated autophagy through beclin-1 in human prostate cancer cells. Eur. Urol. 2016, 15, e44. [Google Scholar] [CrossRef]
- Singh, S.V.; Srivastava, S.K.; Choi, S.; Lew, K.L.; Antosiewicz, J.; Xiao, D.; Zeng, Y.; Watkins, S.C.; Johnson, C.S.; Trump, D.L. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem. 2005, 280, 19911–19924. [Google Scholar] [CrossRef]
- Kim, J.-H.; Xu, C.; Keum, Y.-S.; Reddy, B.; Conney, A.; Kong, A.-N.T. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with β-phenylethyl isothiocyanate and curcumin. Carcinogenesis 2006, 27, 475–482. [Google Scholar] [CrossRef]
- Gamet-Payrastre, L.; Li, P.; Lumeau, S.; Cassar, G.; Dupont, M.-A.; Chevolleau, S.; Gasc, N.; Tulliez, J.; Tercé, F. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000, 60, 1426–1433. [Google Scholar] [PubMed]
- Bhattacharya, A.; Li, Y.; Wade, K.L.; Paonessa, J.D.; Fahey, J.W.; Zhang, Y. Allyl isothiocyanate-rich mustard seed powder inhibits bladder cancer growth and muscle invasion. Carcinogenesis 2010, 31, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lim, D.Y.; Kwon, G.T.; Kim, J.H.; Huang, Z.; Song, H.; Oh, Y.S.; Kang, Y.-H.; Lee, K.W.; Dong, Z. Benzyl isothiocyanate inhibits prostate cancer development in the transgenic adenocarcinoma mouse prostate (TRAMP) model, which is associated with the induction of cell cycle G1 arrest. Int. J. Mol. Sci. 2016, 17, 264. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Xiao, D.; Lew, K.L.; Hershberger, P.; Kokkinakis, D.M.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits growth of PC-3 human prostate cancer xenografts in vivo. Carcinogenesis 2003, 24, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Tong, P.; Dashwood, W.-M.; Dashwood, R.H.; Ho, E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. 2007, 232, 227–234. [Google Scholar]
- Liang, H.; Lai, B.; Yuan, Q. Sulforaphane induces cell-cycle arrest and apoptosis in cultured human lung adenocarcinoma LTEP-A2 cells and retards growth of LTEP-A2 xenografts in vivo. J. Nat. Prod. 2008, 71, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.-L.; Conaway, C.C.; Rao, C.; Reddy, B.S. Chemoprevention of colonic aberrant crypt foci in Fischer rats by sulforaphane and phenethyl isothiocyanate. Carcinogenesis 2000, 21, 2287–2291. [Google Scholar] [CrossRef]
- Boreddy, S.R.; Pramanik, K.C.; Srivastava, S.K. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin. Cancer Res. 2011, 17, 1784–1795. [Google Scholar] [CrossRef]
- Xu, C.; Huang, M.T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.N. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef]
- Hu, R.; Xu, C.; Shen, G.; Jain, M.R.; Khor, T.O.; Gopalkrishnan, A.; Lin, W.; Reddy, B.; Chan, J.Y.; Kong, A.-N.T. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 2006, 243, 170–192. [Google Scholar] [CrossRef]
- Gupta, P.; Adkins, C.; Lockman, P.; Srivastava, S.K. Metastasis of breast tumor cells to brain is suppressed by phenethyl isothiocyanate in a novel in vivo metastasis model. PLoS ONE 2013, 8, e67278. [Google Scholar] [CrossRef] [PubMed]
- Aras, U.; Gandhi, Y.A.; Masso-Welch, P.A.; Morris, M.E. Chemopreventive and anti-angiogenic effects of dietary phenethyl isothiocyanate in an N-methyl nitrosourea-induced breast cancer animal model. Biopharm. Drug Dispos. 2013, 34, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Stahr, S.; Kadlubar, S.A.; Chiang, T.-C.; Wong, H.K. Tanning bed use, risk of melanoma and opportunity for prevention with sulforaphane. Transl. Cancer Res. 2016, 5, S944–S950. [Google Scholar] [CrossRef]
- Ko, J.M.; Fisher, D.E. A new era: Melanoma genetics and therapeutics. J. Pathol. 2011, 223, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Davies, M.Q.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef]
- Hocker, T.L.; Singh, M.K.; Tsao, H. Melanoma Genetics and Therapeutic Approaches in the 21st Century: Moving from the Benchside to the Bedside. J. Investig. Dermatol. 2008, 128, 2575–2595. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Covre, A.; Fratta, E.; Parisi, G.; Colizzi, F.; Rizzo, A.; Danielli, R.; Nicolay, H.J.; Coral, S.; Maio, M. Epigenetics of human cutaneous melanoma: Setting the stage for new therapeutic strategies. J. Transl. Med. 2010, 8, 56. [Google Scholar] [CrossRef]
- Huang, S.; Hsu, M.; Hsu, S.; Yang, J.; Huang, W.; Huang, A.; Hsiao, Y.; Yu, C.; Chung, J. Phenethyl isothiocyanate triggers apoptosis in human malignant melanoma A375. S2 cells through reactive oxygen species and the mitochondria-dependent pathways. Hum. Exp. Toxicol. 2014, 33, 270–283. [Google Scholar] [CrossRef]
- Fuke, Y.; Shinoda, S.; Nagata, I.; Sawaki, S.; Murata, M.; Ryoyama, K.; Koizumi, K.; Saiki, I.; Nomura, T. Preventive effect of oral administration of 6-(methylsulfinyl) hexyl isothiocyanate derived from wasabi (Wasabia japonica Matsum) against pulmonary metastasis of B16-BL6 mouse melanoma cells. Cancer Detect. Prev. 2006, 30, 174–179. [Google Scholar] [CrossRef]
- Sasaki, T.; Kudoh, K.; Uda, Y.; Ozawa, Y.; Shimizu, J.; Kanke, Y.; Takita, T. Effects of isothiocyanates on growth and metastaticity of B16-F10 melanoma cells. Nutr. Cancer 1999, 33, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Hamsa, T.P.; Thejass, P.; Kuttan, G. Induction of apoptosis by sulforaphane in highly metastatic B16F-10 melanoma cells. Drug Chem. Toxicol. 2011, 34, 332–340. [Google Scholar] [CrossRef]
- Thejass, P.; Kuttan, G. Allyl isothiocyanate (AITC) and phenyl isothiocyanate (PITC) inhibit tumour-specific angiogenesis by downregulating nitric oxide (NO) and tumour necrosis factor-α (TNF-α) production. Nitric Oxide 2007, 16, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.P. Expression of the platelet-activating factor receptor enhances benzyl isothiocyanate-induced apoptosis in murine and human melanoma cells. Mol. Med. Rep. 2015, 12, 394–400. [Google Scholar] [CrossRef]
- Lai, K.C.; Hsiao, Y.T.; Yang, J.L.; Ma, Y.S.; Huang, Y.P.; Chiang, T.A.; Chung, J.G. Benzyl isothiocyanate and phenethyl isothiocyanate inhibit murine melanoma B16F10 cell migration and invasion in vitro. Int. J. Oncol. 2017, 51, 832–840. [Google Scholar] [CrossRef]
- Ma, Y.S.; Hsiao, Y.T.; Lin, J.J.; Liao, C.L.; Lin, C.C.; Chung, J.G. Phenethyl Isothiocyanate (PEITC) and Benzyl Isothiocyanate (BITC) Inhibit Human Melanoma A375.S2 Cell Migration and Invasion by Affecting MAPK Signaling Pathway In Vitro. Anticancer Res. 2017, 37, 6223–6234. [Google Scholar] [PubMed]
- Rudolf, K.; Cervinka, M.; Rudolf, E. Sulforaphane-induced apoptosis involves p53 and p38 in melanoma cells. Apoptosis 2014, 19, 734–747. [Google Scholar] [CrossRef]
- Mantso, T.; Sfakianos, A.P.; Atkinson, A.; Anestopoulos, I.; Mitsiogianni, M.; Botaitis, S.; Perente, S.; Simopoulos, C.; Vasileiadis, S.; Franco, R.; et al. Development of a Novel Experimental In Vitro Model of Isothiocyanate-induced Apoptosis in Human Malignant Melanoma Cells. Anticancer Res. 2016, 36, 6303–6309. [Google Scholar] [CrossRef]
- Huang, S.-H.; Wu, L.-W.; Huang, A.-C.; Yu, C.-C.; Lien, J.-C.; Huang, Y.-P.; Yang, J.-S.; Yang, J.-H.; Hsiao, Y.-P.; Wood, W.G. Benzyl isothiocyanate (BITC) induces G2/M phase arrest and apoptosis in human melanoma A375. S2 cells through reactive oxygen species (ROS) and both mitochondria-dependent and death receptor-mediated multiple signaling pathways. J. Agric. Food Chem. 2012, 60, 665–675. [Google Scholar] [CrossRef]
- Mantso, T.; Anestopoulos, I.; Lamprianidou, E.; Kotsianidis, I.; Pappa, A.; Panayiotidis, M.I. Isothiocyanate-induced Cell Cycle Arrest in a Novel In Vitro Exposure Protocol of Human Malignant Melanoma (A375) Cells. Anticancer Res. 2019, 39, 591–596. [Google Scholar] [CrossRef]
- Mitsiogianni, M.; Mantso, T.; Trafalis, D.T.; Vasantha Rupasinghe, H.P.; Zoumpourlis, V.; Franco, R.; Botaitis, S.; Pappa, A.; Panayiotidis, M.I. Allyl isothiocyanate regulates lysine acetylation and methylation marks in an experimental model of malignant melanoma. Eur. J. Nutr. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Pang, Z. TRP channels in skin biology and pathophysiology. Pharmaceuticals 2016, 9, 77. [Google Scholar] [CrossRef]
- Oehler, B.; Scholze, A.; Schaefer, M.; Hill, K. TRPA1 is functionally expressed in melanoma cells but is not critical for impaired proliferation caused by allyl isothiocyanate or cinnamaldehyde. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 555–563. [Google Scholar] [CrossRef]
- Khoobchandani, M.; Ganesh, N.; Gabbanini, S.; Valgimigli, L.; Srivastava, M. Phytochemical potential of Eruca sativa for inhibition of melanoma tumor growth. Fitoterapia 2011, 82, 647–653. [Google Scholar] [CrossRef]
- Bansal, P.; Medhe, S.; Ganesh, N.; Srivastava, M. In vitro anticancer activity of dietary bioagent (isothiocyanates) on HepG2 and B16F10 cell lines: A comparative study. Annals Plant Sci. 2013, 2, 234–237. [Google Scholar]
- Fisher, M.L.; Adhikary, G.; Grun, D.; Kaetzel, D.M.; Eckert, R.L. The Ezh2 polycomb group protein drives an aggressive phenotype in melanoma cancer stem cells and is a target of diet derived sulforaphane. Mol. Carcinog. 2016, 55, 2024–2036. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, P.; Stabile, A.M.; Ragonese, F.; Pistilli, A.; Calvieri, S.; Bottoni, U.; Crisanti, A.; Spaccapelo, R.; Rende, M. Anticarcinogenic activities of sulforaphane are influenced by Nerve Growth Factor in human melanoma A375 cells. Food Chem. Toxicol. 2018, 113, 154–161. [Google Scholar] [CrossRef]
- Bansal, P.; Medhe, S.; Ganesh, N.; Srivastava, M.M. Antimelanoma Potential of Eruca sativa Seed Oil and its Bioactive Principles. Indian J. Pharm. Sci. 2015, 77, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.Y.; Lu, H.F.; Hsu, S.C.; Hsiao, Y.P.; Liu, K.C.; Liu, J.Y.; Ji, B.C.; Hsueh, S.C.; Hung, F.M.; Shang, H.S.; Chung, J.G. Phenethyl isothiocyanate inhibits in vivo growth of subcutaneous xenograft tumors of human malignant melanoma A375.S2 cells. In Vivo 2014, 28, 891–894. [Google Scholar]
- Ni, W.-Y.; Hsiao, Y.-P.; Hsu, S.-C.; Hsueh, S.-C.; Chang, C.-H.; Ji, B.-C.; Yang, J.-S.; Lu, H.-F.; Chung, J.-G. Oral administration of benzyl-isothiocyanate inhibits in vivo growth of subcutaneous xenograft tumors of human malignant melanoma A375. S2 cells. In Vivo 2013, 27, 623–626. [Google Scholar]
- Manesh, C.; Kuttan, G. Effect of naturally occurring allyl and phenyl isothiocyanates in the inhibition of experimental pulmonary metastasis induced by B16F-10 melanoma cells. Fitoterapia 2003, 74, 355–363. [Google Scholar] [CrossRef]
- Thejass, P.; Kuttan, G. Modulation of cell-mediated immune response in B16F-10 melanoma-induced metastatic tumor-bearing C57BL/6 mice by sulforaphane. Immunopharmacol. Immunotoxicol. 2007, 29, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.J.; Mishra, R.; Sharma, P.; Kundu, G.C. Quercetin and sulforaphane in combination suppress the progression of melanoma through the downregulation of matrix metalloproteinase-9. Exp. Ther. Med. 2010, 1, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Do, D.; Pai, S.; Rizvi, S.A.; D’Souza, M.J. Development of sulforaphane-encapsulated microspheres for cancer epigenetic therapy. Int. J. Pharm. 2010, 386, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Sharma, A.; Nguyen, N.; Sharma, A.K.; Desai, D.; Huh, S.J.; Amin, S.; Meyers, C.; Robertson, G.P. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev. Res. 2011, 4, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Sharma, A.; Desai, D.; Madhunapantula, S.V.; Huh, S.J.; Robertson, G.P.; Amin, S. Synthesis and anticancer activity comparison of phenylalkyl isoselenocyanates with corresponding naturally occurring and synthetic isothiocyanates. J. Med. Chem. 2008, 51, 7820–7826. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, A.K.; Madhunapantula, S.V.; Desai, D.; Huh, S.J.; Mosca, P.; Amin, S.; Robertson, G.P. Targeting Akt3 signaling in malignant melanoma using isoselenocyanates. Clin. Cancer Res. 2009, 15, 1674–1685. [Google Scholar] [CrossRef]
- Sk, U.H.; Gowda, A.P.; Crampsie, M.A.; Yun, J.K.; Spratt, T.E.; Amin, S.; Sharma, A.K. Development of novel naphthalimide derivatives and their evaluation as potential melanoma therapeutics. Eur. J. Med. Chem. 2011, 46, 3331–3338. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsiogianni, M.; Koutsidis, G.; Mavroudis, N.; Trafalis, D.T.; Botaitis, S.; Franco, R.; Zoumpourlis, V.; Amery, T.; Galanis, A.; Pappa, A.; et al. The Role of Isothiocyanates as Cancer Chemo-Preventive, Chemo-Therapeutic and Anti-Melanoma Agents. Antioxidants 2019, 8, 106. https://doi.org/10.3390/antiox8040106
Mitsiogianni M, Koutsidis G, Mavroudis N, Trafalis DT, Botaitis S, Franco R, Zoumpourlis V, Amery T, Galanis A, Pappa A, et al. The Role of Isothiocyanates as Cancer Chemo-Preventive, Chemo-Therapeutic and Anti-Melanoma Agents. Antioxidants. 2019; 8(4):106. https://doi.org/10.3390/antiox8040106
Chicago/Turabian StyleMitsiogianni, Melina, Georgios Koutsidis, Nikos Mavroudis, Dimitrios T. Trafalis, Sotiris Botaitis, Rodrigo Franco, Vasilis Zoumpourlis, Tom Amery, Alex Galanis, Aglaia Pappa, and et al. 2019. "The Role of Isothiocyanates as Cancer Chemo-Preventive, Chemo-Therapeutic and Anti-Melanoma Agents" Antioxidants 8, no. 4: 106. https://doi.org/10.3390/antiox8040106
APA StyleMitsiogianni, M., Koutsidis, G., Mavroudis, N., Trafalis, D. T., Botaitis, S., Franco, R., Zoumpourlis, V., Amery, T., Galanis, A., Pappa, A., & Panayiotidis, M. I. (2019). The Role of Isothiocyanates as Cancer Chemo-Preventive, Chemo-Therapeutic and Anti-Melanoma Agents. Antioxidants, 8(4), 106. https://doi.org/10.3390/antiox8040106