Mutual Influences between Nitric Oxide and Paraoxonase 1


Abstract
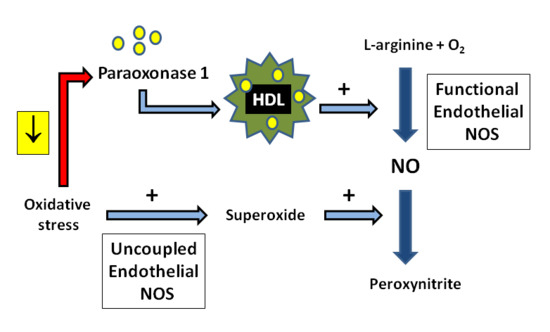
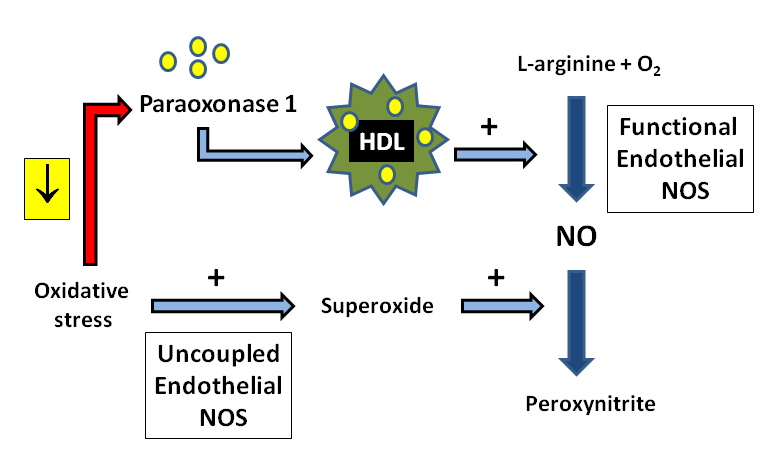{kind=link}
1. Introduction
2. On the Genesis of NO
3. PON1 as an Antioxidant Component of the Structure of HDL
4. Interplay between HDL and NOS
4.1. Mechanisms
4.2. HDL Changes in Disease
5. Vascular Effects of Gonadal Hormones
6. PON1 and Cholesterol Lowering Drugs
7. The Special Case of a Classical PON Substrate: Homocysteine Thiolactone
8. NO and Its Oxidized Metabolites as Inhibitors of PON1-Driven Effects
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar]
- Mineo, C.; Shaul, P.W. HDL stimulation of endothelial nitric oxide synthase: A novel mechanism of HDL action. Trends Cardiovasc. Med. 2003, 13, 226–231. [Google Scholar] [CrossRef]
- Abelló, D.; Sancho, E.; Camps, J.; Joven, J. Exploring the role of paraoxonases in the pathogenesis of coronary artery disease: A systematic review. Int. J. Mol. Sci. 2014, 15, 20997–21010. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Förstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Yilmaz, N.; Aydin, O. Functionally defective high-density lipoprotein and paraoxonase: A couple for endothelial dysfunction in atherosclerosis. Cholesterol 2013, 2013, 792090. [Google Scholar] [CrossRef]
- Yakovlev, V.A.; Barani, I.J.; Rabender, C.S.; Black, S.M.; Leach, J.K.; Graves, P.R.; Kellogg, G.E.; Mikkelsen, R.B. Tyrosine nitration of IκBα: A novel mechanism for NF-κB activation. Biochemistry 2007, 46, 11671–11683. [Google Scholar] [CrossRef]
- Mishra, B.B.; Rathinam, V.A.K.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef]
- Hernández-Cuéllar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M.G. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Liu, V.W.T.; Huang, P.L. Cardiovascular roles of nitric oxide: A review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc. Res. 2008, 77, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Upmacis, R.K.; Crabtree, M.J.; Deeb, R.S.; Shen, H.; Lane, P.B.; Benguigui, L.E.S.; Maeda, N.; Hajjar, D.P.; Gross, S.S. Profound biopterin oxidation and protein tyrosine nitration in tissues of ApoE-null mice on an atherogenic diet: Contribution of inducible nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2878–H2887. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajjar, R.; Picard, M.H.; Huang, P.L. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 2001, 104, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.; Donald, A.; Ferri, C.; Giannattasio, C.; Halcox, J.; Halligan, S.; Lerman, A.; Mancia, G.; Oliver, J.J.; Pessina, A.C.; et al. Endothelial function and dysfunction. Part I: Methodological issues for assessment in the different vascular beds: A statement by the working group on endothelin and endothelial factors of the European Society of Hypertension. J. Hypertens. 2005, 23, 7–17. [Google Scholar] [CrossRef]
- Mineo, C.; Yuhanna, I.S.; Quon, M.J.; Shaul, P.W. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J. Biol. Chem. 2003, 278, 9142–9149. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New therapeutic implications of endothelial nitric oxide synthase (eNOS) function/dysfunction in cardiovascular disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. PON-dering differences in HDL function in coronary artery disease. J. Clin. Investig. 2011, 121, 2545–2548. [Google Scholar] [CrossRef]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef]
- Sung, K.C.; Ryu, S.; Wild, S.H.; Byrne, C.D. An Increased high-density lipoprotein cholesterol/apolipoprotein A-I ratio is associated with increased cardiovascular and all-cause mortality. Heart 2015, 101, 553–558. [Google Scholar] [CrossRef]
- James, R.W.; Deakin, S.P. The contribution of high density lipoprotein apolipoproteins and derivatives to serum paraoxonase-1 activity and function. Adv. Exp. Med. Biol. 2010, 660, 173–181. [Google Scholar]
- Moya, C.; Máñez, S. Paraoxonases: Metabolic role and pharmacological projection. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry. 2005, 44, 6371–6382. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, Z.; Riwanto, M.; Gao, S.; Levison, B.S.; Gu, X.; Fu, X.; Wagner, M.A.; Besler, C.; Gerstenecker, G.; et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J. Clin. Invest. 2013, 123, 3815–3828. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Huang, Y.; Levison, B.S.; Gerstenecker, G.; Di Donato, A.J.; Hazen, L.B.; Lee, J.; Gogonea, V.; Di Donato, J.A.; Hazen, S.L. Identification of critical paraoxonase 1 residues involved in high density lipoprotein interaction. J. Biol. Chem. 2016, 291, 1890–1904. [Google Scholar] [CrossRef]
- Sozer, V.; Uzun, H.; Gelisgen, R.; Kaya, M.; Kalayci, R.; Tabak, O.; Arican, N.; Konukoglu, D. The effects of atorvastatin on oxidative stress in L-NAME-treated rats. Scan. J. Clin. Lab. Invest. 2013, 73, 591–597. [Google Scholar] [CrossRef]
- Viktorinova, A.; Jurkovicova, I.; Fabryova, L.; Kinova, S.; Koren, M.; Stecova, A.; Svitekova, K. Abnormalities in the relationship of paraoxonase 1 with HDL and apolipoprotein A1 and their possible connection to HDL dysfunctionality in type 2 diabetes. Diabetes Res. Clin. Pract. 2018, 140, 174–182. [Google Scholar] [CrossRef]
- Han, C.Y.; Chiba, T.; Campbell, J.S.; Fausto, N.; Chaisson, M.; Orasanu, G.; Plutzky, J.; Chait, A. Reciprocal and coordinate regulation of serum amyloid A versus apolipoprotein A-I and paraoxonase-1 by inflammation in murine hepatocytes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1806–1813. [Google Scholar] [CrossRef]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef]
- Kratzer, A.; Giral, H.; Landmesser, U. High-density lipoproteins as modulators of endothelial cell functions: Alterations in patients with coronary artery disease. Cardiovasc. Res. 2014, 103, 350–361. [Google Scholar] [CrossRef]
- Sugiura, T.; Dohi, Y.; Yamashita, S.; Yamamoto, K.; Tanaka, S.; Wakamatsu, Y.; Kimura, G. Malondialdehyde-modified LDL to HDL-cholesterol ratio reflects endothelial damage. Int. J. Cardiol. 2011, 147, 461–463. [Google Scholar] [CrossRef]
- Wu, A.; Hinds, C.J.; Thiemermann, C. High-density lipoproteins in sepsis and septic shock: Metabolism, actions, and therapeutic applications. Shock 2004, 21, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Gomaraschi, M.; Ossoli, A.; Favari, E.; Adorni, M.P.; Sinagra, G.; Cattin, L.; Veglia, F.; Bernini, F.; Franceschini, G.; Calabresi, L. Inflammation impairs eNOS activation by HDL in patients with acute coronary syndrome. Cardiovasc. Res. 2013, 100, 36–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Givvimani, S.; Kundu, S.; Pushpakumar, S.; Doyle, V.; Narayanan, N.; Winchester, L.J.; Veeranki, S.; Metreveli, N.; Tyagi, S.C. Hyperhomocysteinemia: A missing link to dysfunctional HDL via paraoxanase-1. Can. J. Physiol. Pharmacol. 2015, 93, 755–763. [Google Scholar] [CrossRef]
- Nofer, J.R.; van der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef]
- Seetharam, D.; Mineo, C.; Gormley, A.K.; Gibson, L.L.; Vongpatanasin, W.; Chambliss, K.L.; Hahner, L.D.; Cummings, M.L.; Kitchens, R.L.; Marcel, Y.L.; et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B Type I. Circ. Res. 2006, 98, 63–72. [Google Scholar] [CrossRef]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Lüscher, T.F. Oxidized low-density lipoprotein activates P66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2090–2097. [Google Scholar] [CrossRef]
- Xu, X.; Gao, X.; Potter, B.J.; Cao, J.M.; Zhang, C. Anti-LOX-1 rescues endothelial function in coronary arterioles in atherosclerotic ApoE knockout mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 871–877. [Google Scholar] [CrossRef]
- Cominacini, L.; Rigoni, A.; Pasini, A.F.; Garbin, U.; Davoli, A.; Campagnola, M.; Pastorino, A.M.; Lo Cascio, V.; Sawamura, T. The binding of oxidized low density lipoprotein (Ox-LDL) to Ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J. Biol. Chem. 2001, 276, 13750–13755. [Google Scholar] [CrossRef]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef]
- Li, H.; Horke, S.; Förstermann, U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Charakida, M.; Besler, C.; Batuca, J.R.; Sangle, S.; Marques, S.; Sousa, M.; Wang, G.; Tousoulis, D.; Delgado Alves, J.; Loukogeorgakis, S.P.; et al. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA 2009, 302, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Kränkel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef]
- O’Neill, F.; Riwanto, M.; Charakida, M.; Colin, S.; Manz, J.; McLoughlin, E.; Khan, T.; Klein, N.; Kay, C.W.M.; Patel, K.; et al. Structural and functional changes in HDL with low grade and chronic inflammation. Int. J. Cardiol. 2015, 188, 111–116. [Google Scholar] [CrossRef]
- Sang, H.; Yao, S.; Zhang, L.; Li, X.; Yang, N.; Zhao, J.; Zhao, L.; Si, Y.; Zhang, Y.; Lv, X.; et al. Walk-run training improves the anti-inflammation properties of high-density lipoprotein in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 2015, 100, 870–879. [Google Scholar] [CrossRef]
- Gamliel-Lazarovich, A.; Abassi, Z.; Khatib, S.; Tavori, H.; Vaya, J.; Aviram, M.; Keidar, S. Paraoxonase1 deficiency in mice is associated with hypotension and increased levels of 5,6-epoxyeicosatrienoic acid. Atherosclerosis 2012, 222, 92–98. [Google Scholar] [CrossRef]
- Eryanni-Levin, S.; Khatib, S.; Levy-Rosenzvig, R.; Tamir, S.; Szuchman-Sapir, A. 5,6-δ-DHTL, a stable metabolite of arachidonic acid, is a potential substrate for paraoxonase 1. Biochim. Biophys. Acta 2015, 1851, 1118–1122. [Google Scholar] [CrossRef]
- Gilad, D.; Atiya, S.; Mozes-Autmazgin, Z.; Ben-Shushan, R.S.; Ben-David, R.; Amram, E.; Tamir, S.; Chuyun, D.; Szuchman-Sapir, A. Paraoxonase 1 in endothelial cells impairs vasodilation induced by arachidonic acid lactone metabolite. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids 2019, 1864, 386–393. [Google Scholar] [CrossRef]
- Duckles, S.P.; Miller, V.M. Hormonal modulation of endothelial NO production. Pflugers Arch. 2010, 459, 841–851. [Google Scholar] [CrossRef]
- Nuedling, S.; Kahlert, S.; Loebbert, K.; Doevendans, P.A.; Meyer, R.; Vetter, H.; Grohé, C. 17-Beta-estradiol stimulates expression of endothelial and inducible NO synthase in rat myocardium in-vitro and in-vivo. Cardiovasc. Res. 1999, 43, 666–674. [Google Scholar] [CrossRef]
- Vincent-Viry, M.; Sass, C.; Bastien, S.; Aguillon, D.; Siest, G.; Visvikis, S. PON1-192 phenotype and genotype assessments in 918 subjects of the Stanislas Cohort Study. Clin. Chem. Lab. Med. 2003, 41, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Kumru, S.; Aydin, S.; Aras, A.; Gursu, M.F.; Gulcu, F. Effects of surgical menopause and estrogen replacement therapy on serum paraoxonase activity and plasma malondialdehyde concentration. Gynecol. Obstet. Investig. 2005, 59, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kiranoglu, S.; Sinan, S.; Gencer, N.; Köckar, F.; Arslan, O. In vivo effects of oral contraceptives on paraoxonase, catalase and carbonic anhydrase enzyme activities on mouse. Biol. Pharm. Bull. 2007, 30, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Scott, J.E. Estradiol enhances cell-associated paraoxonase 1 (PON1) activity in vitro without altering PON1 expression. Biochem. Biophys. Res. Commun. 2010, 397, 441–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castardo-de-Paula, J.C.; de Campos, B.H.; Amorim, E.D.T.; da Silva, R.V.; de Farias, C.C.; Higachi, L.; Pinge-Filho, P.; Barbosa, D.S.; Martins-Pinge, M.C. Cardiovascular risk and the effect of nitric oxide synthase inhibition in female rats: The role of estrogen. Exp. Gerontol. 2017, 97, 38–48. [Google Scholar] [CrossRef]
- Nathan, L.; Shi, W.; Dinh, H.; Mukherjee, T.K.; Wang, X.; Lusis, A.J.; Chaudhuri, G. Testosterone inhibits early atherogenesis by conversion to estradiol: Critical role of aromatase. Proc. Natl. Acad. Sci. USA 2001, 98, 3589–3593. [Google Scholar] [CrossRef]
- Orio, F.; Palomba, S.; Cascella, T.; De Simone, B.; Di Biase, S.; Russo, T.; Labella, D.; Zullo, F.; Lombardi, G.; Colao, A. Early impairment of endothelial structure and function in young normal-weight women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 4588–4593. [Google Scholar] [CrossRef]
- Bayram, F.; Kocer, D.; Ozsan, M.; Muhtaroglu, S. Evaluation of endothelial dysfunction, lipid metabolism in women with polycystic ovary syndrome: Relationship of paraoxonase 1 activity, malondialdehyde levels, low-Density lipoprotein subfractions, and endothelial dysfunction. Gynecol. Endocrinol. 2012, 28, 497–501. [Google Scholar] [CrossRef]
- Bin Ali, A.; Zhang, Q.; Lim, Y.K.; Fang, D.; Retnam, L.; Lim, S.K. Expression of major HDL-associated antioxidant PON-1 is gender dependent and regulated during inflammation. Free Radic. Biol. Med. 2003, 34, 824–829. [Google Scholar] [CrossRef]
- Bayrak, T.; Dursun, P.; Bayrak, A.; Gültekin, M.; Kolusarı, A.; Cakır, E.; Ozyurt, M.; Zeyneloğlu, H.B. Paraoxonase lactonase activity (PON-HTLase), asymmetric dimethylarginine (ADMA) and platelet activating factor-acetylhydrolase (PAF-AH) activity in non-obese women with PCOS. Gynecol. Endocrinol. 2012, 28, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Harangi, M.; Seres, I.; Varga, Z.; Emri, G.; Szilvássy, Z.; Paragh, G.; Remenyik, E. Atorvastatin effect on high-density lipoprotein-associated paraoxonase activity and oxidative DNA damage. Eur. J. Clin. Pharmacol. 2004, 60, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Kural, B.V.; Orem, C.; Uydu, H.A.; Alver, A.; Orem, A. The effects of lipid-lowering therapy on paraoxonase activities and their relationships with the oxidant-antioxidant system in patients with dyslipidemia. Coron. Artery Dis. 2004, 15, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Costa, L.G.; Guizzetti, M. Assessment of cholesterol homeostasis in astrocytes and neurons. Methods Mol. Biol. 2011, 758, 403–414. [Google Scholar]
- Akalin Çiftçi, G.; Ertorun, İ.; Akalin, A.; Alataş, İ.Ö.; Musmul, A. The effects of atorvastatin on antioxidant/antiinflammatory properties of HDLs in hypercholesterolemics. Turk. J. Med. Sci. 2015, 45, 345–351. [Google Scholar] [CrossRef]
- Deakin, S.P.; James, R.W. Genetic and environmental factors modulating serum concentrations and activities of the antioxidant enzyme paraoxonase-1. Clin. Sci. 2004, 107, 435–447. [Google Scholar] [CrossRef]
- Laufs, U. Beyond lipid-lowering: Effects of statins on endothelial nitric oxide. Eur. J. Clin. Pharmacol. 2003, 58, 719–731. [Google Scholar] [CrossRef]
- Bełtowski, J.; Kedra, A. Asymmetric dimethylarginine (ADMA) as a target for pharmacotherapy. Pharmacol. Rep. 2006, 58, 159–178. [Google Scholar]
- Bolayirli, I.M.; Aslan, M.; Balci, H.; Altug, T.; Hacibekiroglu, M.; Seven, A. Effects of atorvastatin therapy on hypercholesterolemic rabbits with respect to oxidative stress, nitric oxide pathway and homocysteine. Life Sci. 2007, 81, 121–127. [Google Scholar] [CrossRef]
- Hernáez, Á.; Castañer, O.; Elosúa, R.; Pintó, X.; Estruch, R.; Salas-Salvadó, J.; Corella, D.; Arós, F.; Serra-Majem, L.; Fiol, M.; et al. Mediterranean diet improves high-density lipoprotein function in high-cardiovascular-risk individuals: A randomized controlled trial. Circulation 2017, 135, 633–643. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Kumar, M.; Sen, U.; Mishra, P.K.; Tyagi, N.; Metreveli, N.; Lominadze, D.; Rodriguez, W.; Tyagi, S.C. Nitrotyrosinylation, remodeling and endothelial-myocyte uncoupling in iNOS, cystathionine beta synthase (CBS) knockouts and iNOS/CBS double knockout mice. J. Cell. Biochem. 2009, 106, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wu, C.; Lu, G. Atorvastatin inhibits homocysteine-induced dysfunction and apoptosis in endothelial progenitor cells. Acta Pharmacol. Sin. 2010, 31, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hernando, C.; Ackah, E.; Yu, J.; Suárez, Y.; Murata, T.; Iwakiri, Y.; Prendergast, J.; Miao, R.Q.; Birnbaum, M.J.; Sessa, W.C. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007, 6, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Jakubowski, H. Paraoxonase 1 and homocysteine metabolism. Amino Acids 2012, 43, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Yang, X.; Wang, H. Hyperhomocysteinemia and high-density lipoprotein metabolism in cardiovascular disease. Clin. Chem. Lab. Med. 2007, 45, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Weijun, G.; Juming, L.; Guoqing, Y.; Jingtao, D.; Qinghua, G.; Yiming, M.; Changyu, P. Effects of plasma homocysteine levels on serum HTase/PON activity in patients with type 2 diabetes. Adv. Ther. 2008, 25, 884–893. [Google Scholar] [CrossRef]
- Liu, L.H.; Guo, Z.; Feng, M.; Wu, Z.Z.; He, Z.-M.; Xiong, Y. Protection of DDAH2 overexpression against homocysteine-induced impairments of DDAH/ADMA/NOS/NO pathway in endothelial cells. Cell. Physiol. Biochem. 2012, 30, 1413–1422. [Google Scholar] [CrossRef]
- Holven, K.B.; Scholz, H.; Halvorsen, B.; Aukrust, P.; Ose, L.; Nenseter, M.S. Hyperhomocysteinemic subjects have enhanced expression of lectin-like oxidized LDL receptor-1 in mononuclear cells. J. Nutr. 2003, 133, 3588–3591. [Google Scholar] [CrossRef][Green Version]
- Yilmaz, N. Relationship between paraoxonase and homocysteine: Crossroads of oxidative diseases. Arch. Med. Sci. 2012, 8, 138–153. [Google Scholar] [CrossRef]
- Maron, B.A.; Michel, T. Subcellular localization of oxidants and redox modulation of endothelial nitric oxide synthase. Circ. J. 2012, 76, 2497–2512. [Google Scholar] [CrossRef] [PubMed]
- Khatib, S.; Artoul, F.; Gershko, M.; Markman, G.; Vaya, J. The synthesis and analysis of S-nitrosylated paraoxonase 1. Biochem. Biophys. Res. Commun. 2014, 444, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahhab, K.G.; Fawzi, H.; Mannaa, F.A. Paraoxonase-1 (PON1) inhibition by tienilic acid produces hepatic injury: Antioxidant protection by fennel extract and whey protein concentrate. Pathophysiology 2016, 23, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ames, P.R.J.; Guardascione, M.; Batuca, J.R.; Arcaro, A.; Gentile, F.; Amitrano, L. Nitric oxide metabolites, nitrative stress, and paraoxonase activity in hepatopulmonary syndrome. Scand. J. Gastroenterol. 2016, 51, 73–77. [Google Scholar] [CrossRef]
- Rodríguez-Carrio, J.; Alperi-López, M.; López-Mejías, R.; López, P.; Ballina-García, F.J.; Abal, F.; González-Gay, M.A.; Suárez, A. Antibodies to paraoxonase 1 are associated with oxidant status and endothelial activation in rheumatoid arthritis. Clin. Sci. (Lond.) 2016, 130, 1889–1899. [Google Scholar] [CrossRef]
- Maes, M.; Bonifacio, K.L.; Morelli, N.R.; Vargas, H.O.; Moreira, E.G.; St Stoyanov, D.; Barbosa, D.S.; Carvalho, A.F.; Nunes, S.O.V. Generalized anxiety disorder (GAD) and comorbid major depression with GAD are characterized by enhanced nitro-oxidative stress, increased lipid peroxidation, and lowered lipid-associated antioxidant defenses. Neurotox. Res. 2018, 34, 489–510. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marín, M.; Moya, C.; Máñez, S. Mutual Influences between Nitric Oxide and Paraoxonase 1. Antioxidants 2019, 8, 619. https://doi.org/10.3390/antiox8120619
Marín M, Moya C, Máñez S. Mutual Influences between Nitric Oxide and Paraoxonase 1. Antioxidants. 2019; 8(12):619. https://doi.org/10.3390/antiox8120619
Chicago/Turabian StyleMarín, Marta, Carlos Moya, and Salvador Máñez. 2019. "Mutual Influences between Nitric Oxide and Paraoxonase 1" Antioxidants 8, no. 12: 619. https://doi.org/10.3390/antiox8120619
APA StyleMarín, M., Moya, C., & Máñez, S. (2019). Mutual Influences between Nitric Oxide and Paraoxonase 1. Antioxidants, 8(12), 619. https://doi.org/10.3390/antiox8120619