PCB11 Metabolite, 3,3’-Dichlorobiphenyl-4-ol, Exposure Alters the Expression of Genes Governing Fatty Acid Metabolism in the Absence of Functional Sirtuin 3: Examining the Contribution of MnSOD

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis and Authentication of 4OH-PCB11
2.2. Mouse Embryonic Fibroblast Isolation and Culture Conditions
2.3. Doubling Time
2.4. Western Blot Analysis
2.5. Mitosox and JC-1 Labeling for Mitochondrial ROS and Mitochondrial Membrane Potential
2.6. MnSOD Activity and Adenoviral Transduction of MnSOD
2.7. Cellular Bioenergetics Analysis and Fatty Acid Oxidation Assessment via Cellular Respirometry
2.8. Quantitative RT-PCR Analysis
2.9. Statistical Analysis
3. Results
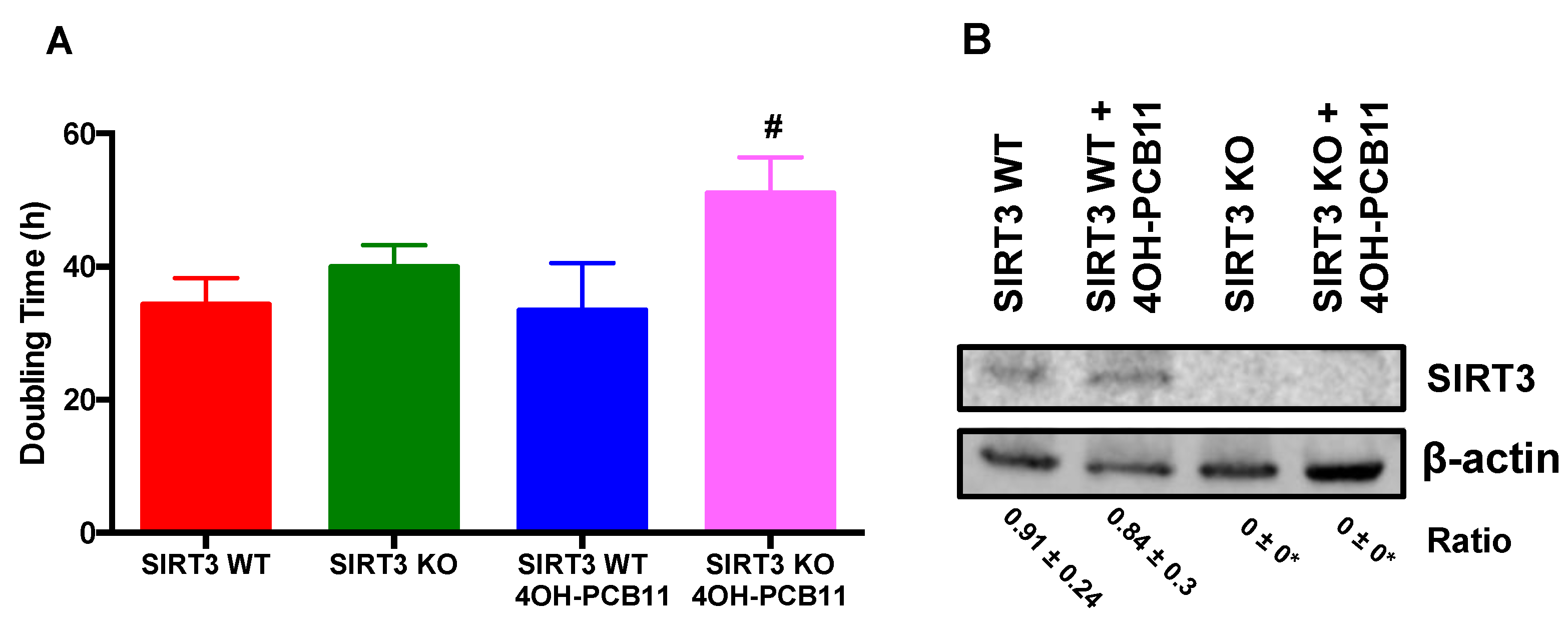
3.1. Effect of 4OH-PCB11 on the Growth of WT and SIRT3-KO MEFs
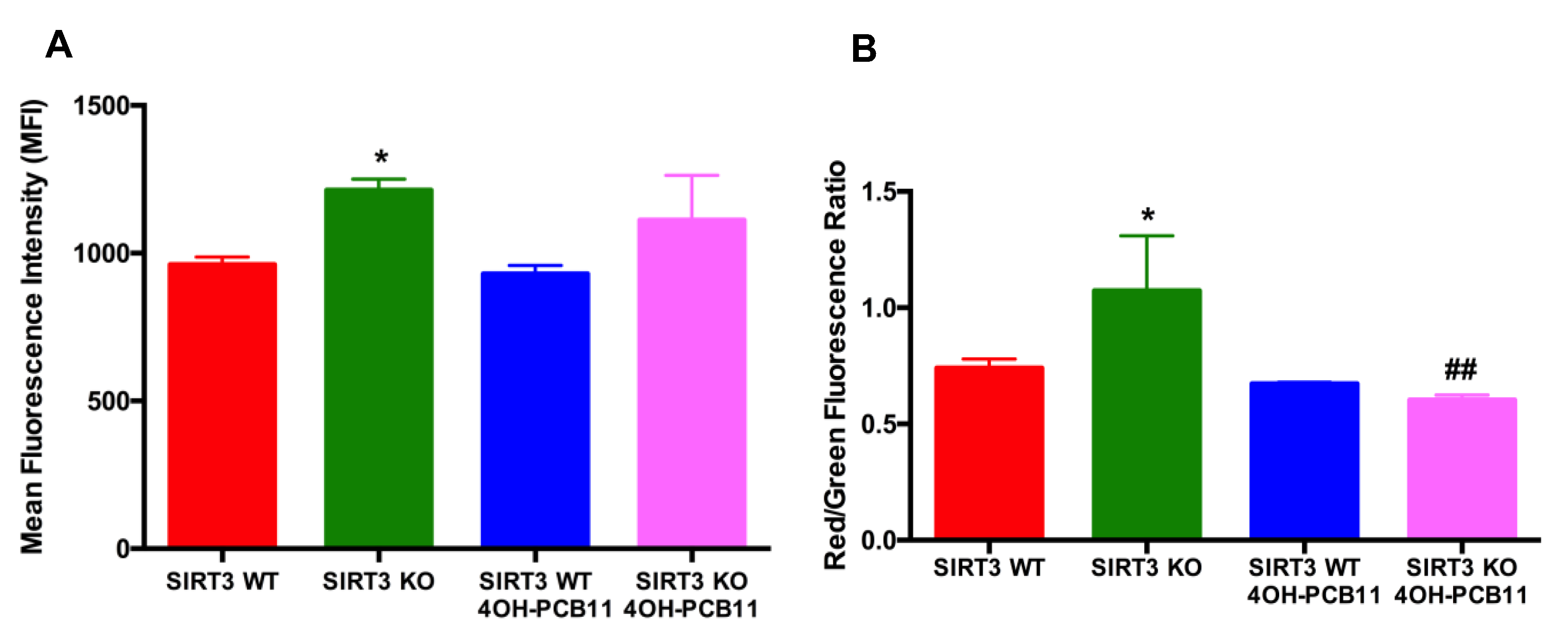
3.2. Effect of 4OH-PCB11 on Mitosox Oxidation and Mitochondrial Membrane Potential
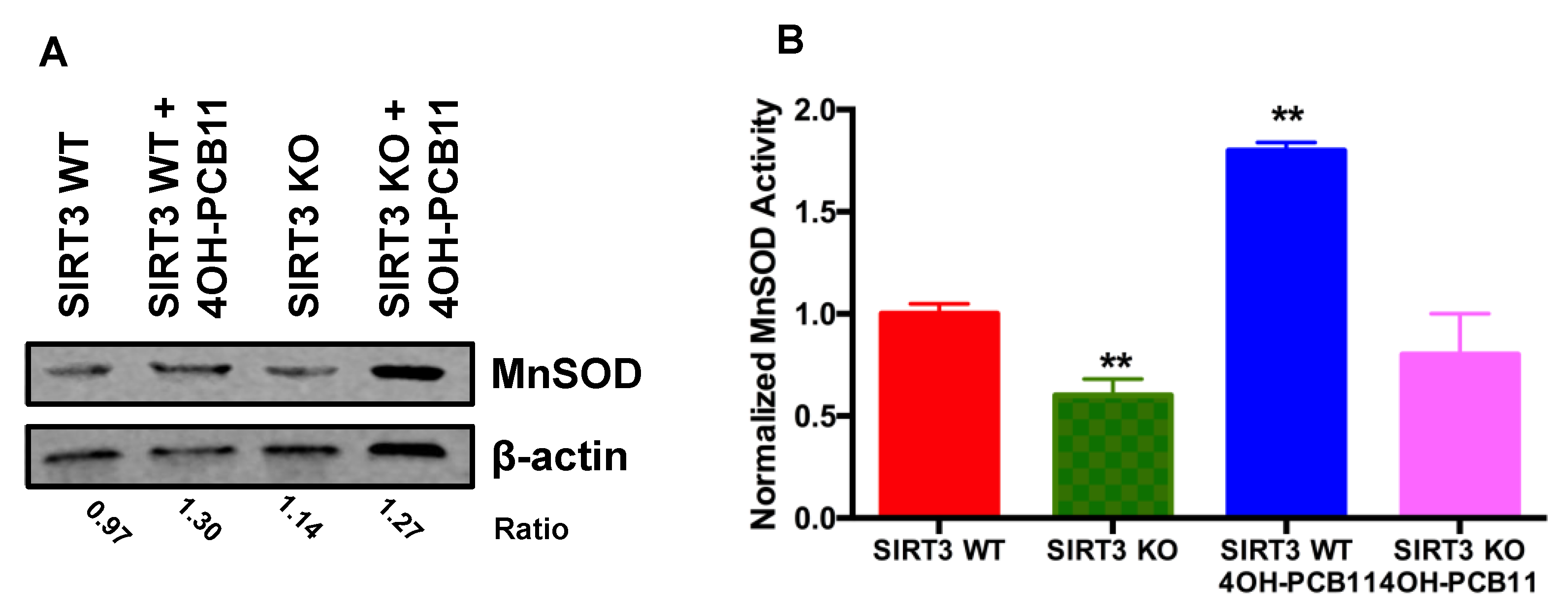
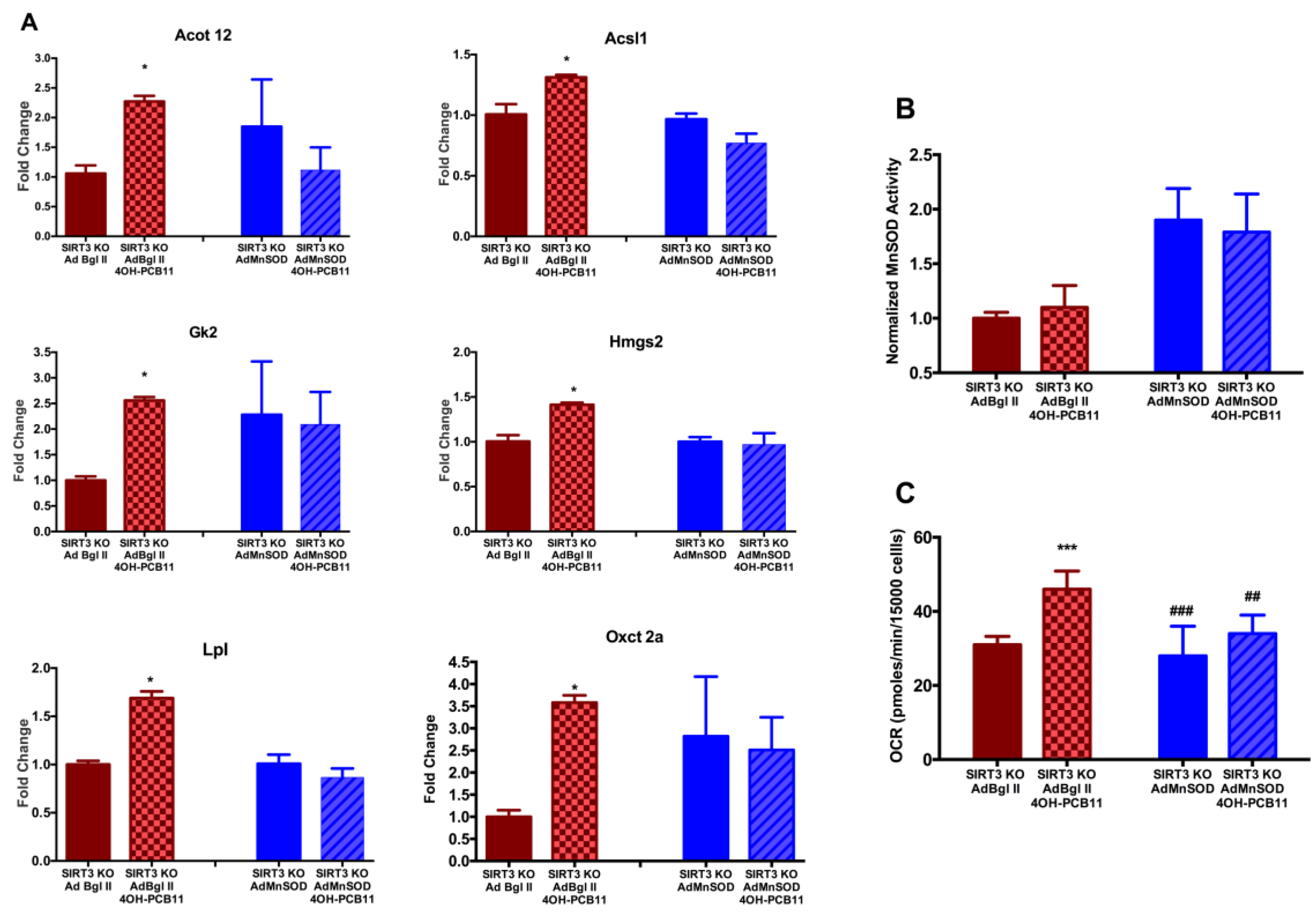
3.3. Effect of 4OH-PCB11 on MnSOD Abundance and Enzymatic Activity
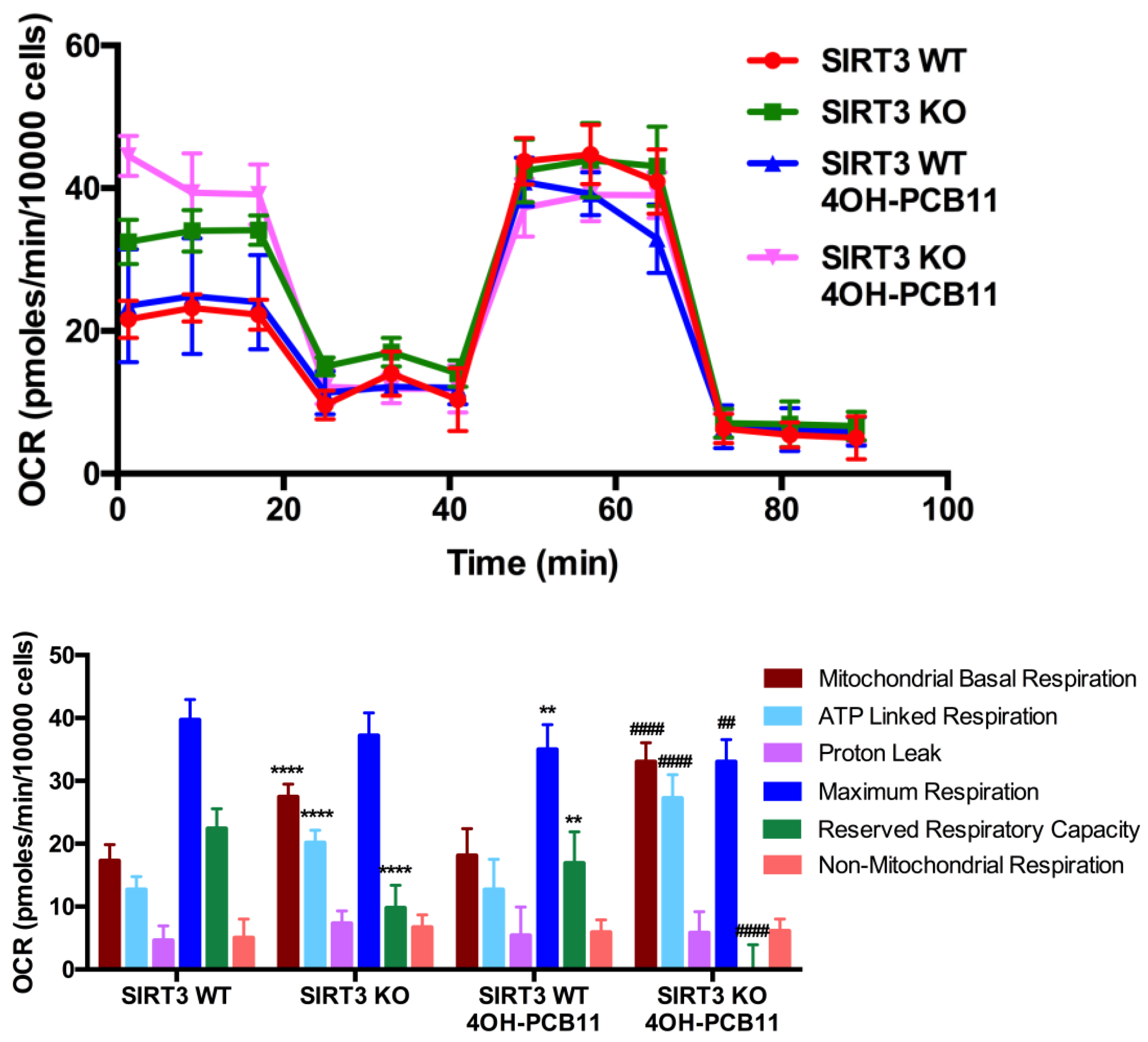
3.4. Mitochondrial Respiration Following 4OH-PCB11 Treatment in WT and SIRT3-KO MEFs
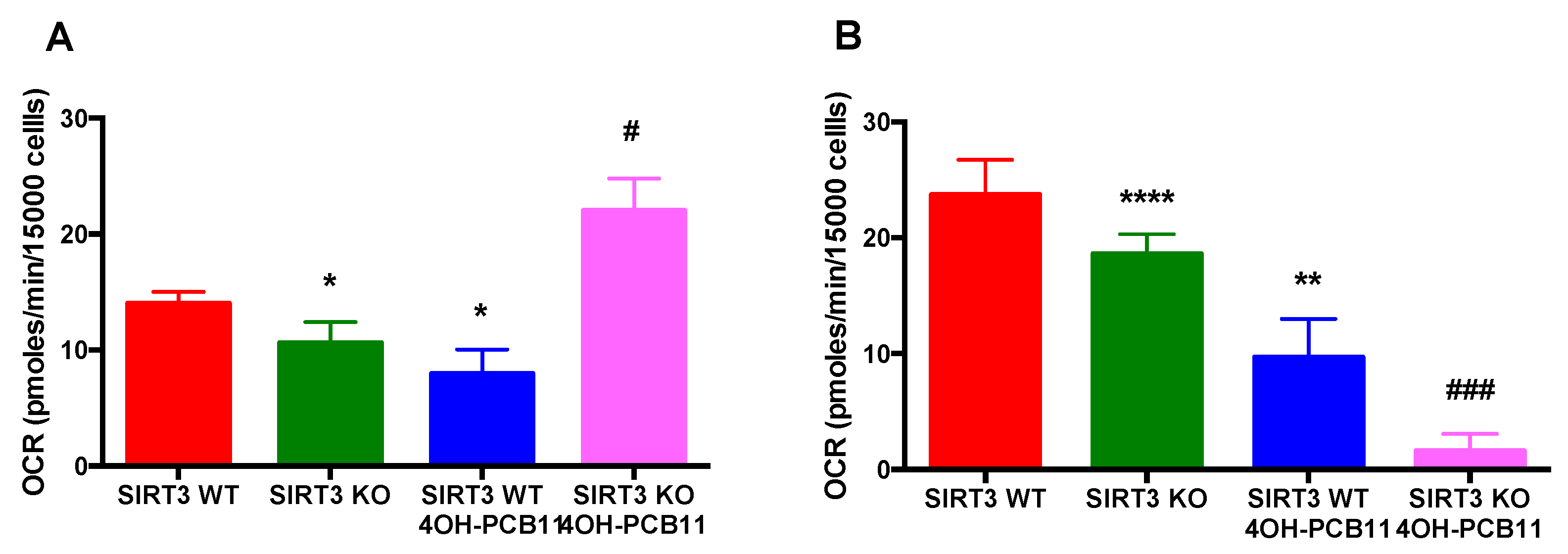
3.5. Effect of 4OH-PCB11 on Fatty Acid Oxidation-Associated Respiration in WT and SIRT3-KO MEFs
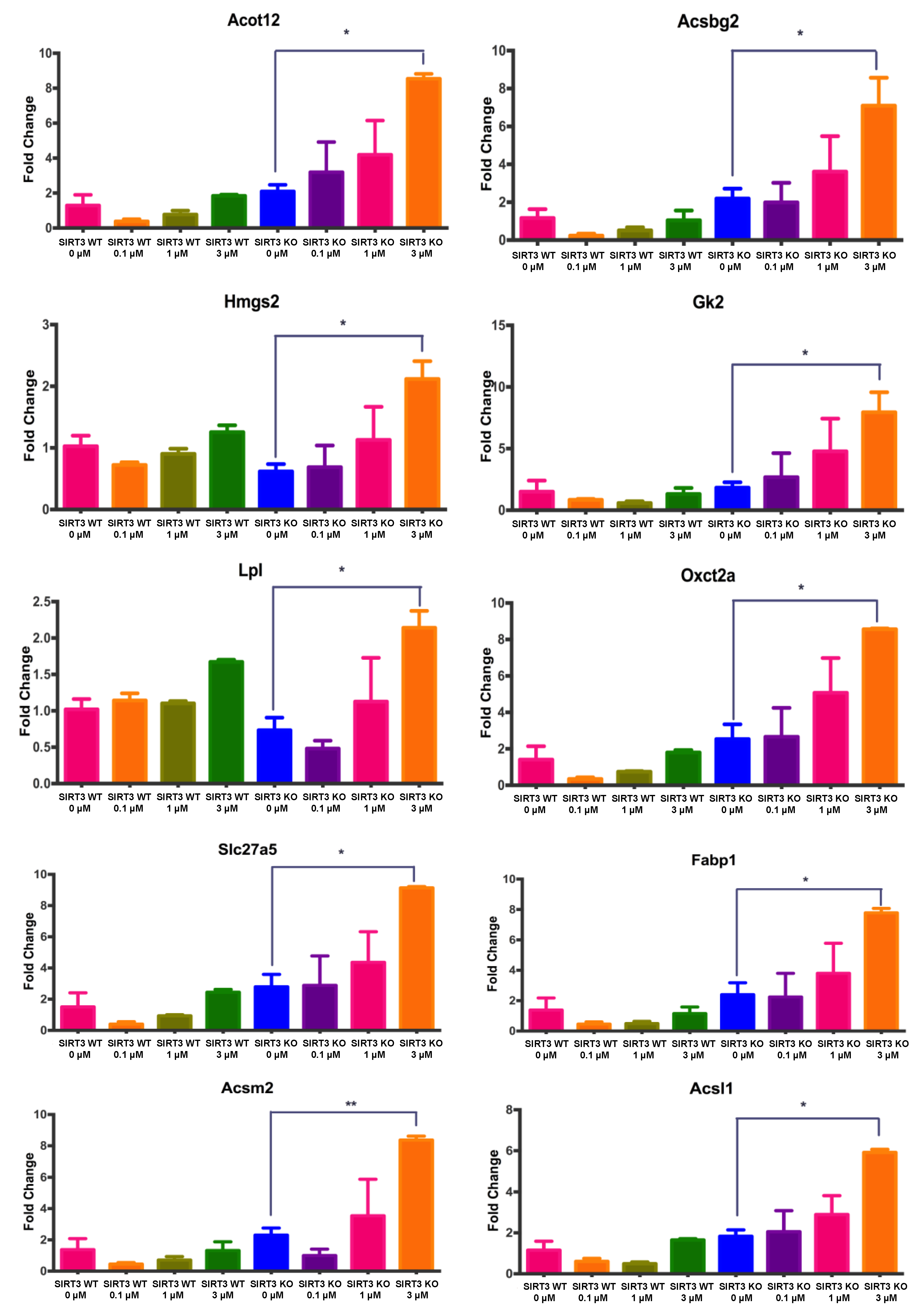
3.6. Changes in Gene Expression Associated with Fatty Acid Metabolism in WT and SIRT3-KO MEFs after 4OH-PCB11 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suvorov, A.; Takser, L. Facing the Challenge of Data Transfer from Animal Models to Humans: The Case of Persistent Organohalogens. Environ. Health 2008, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Ross, G. The public health implications of polychlorinated biphenyls (PCBs) in the environment. Ecotoxicol. Environ. Saf. 2004, 59, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Martinez, A.; Hornbuckle, K.C. Discovery of non-aroclor PCB (3,3’-dichlorobiphenyl) in Chicago air. Environ. Sci. Technol. 2008, 42, 7873–7877. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, L.A.; Guo, J.; Du, S.; Cavallo, G.J. Evidence for unique and ubiquitous environmental sources of 3,3’-dichlorobiphenyl (PCB 11). Environ. Sci. Technol. 2010, 44, 2816–2821. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Hornbuckle, K.C. Inadvertent polychlorinated biphenyls in commercial paint pigments. Environ. Sci. Technol. 2010, 44, 2822–2827. [Google Scholar] [CrossRef] [PubMed]
- Marek, R.F.; Thorne, P.S.; DeWall, J.; Hornbuckle, K.C. Variability in PCB and OH-PCB serum levels in children and their mothers in urban and rural U.S. communities. Environ. Sci. Technol. 2014, 48, 13459–13467. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Keil, K.P.; Chen, H.; Hayakawa, K.; Li, X.; Lin, Y.; Lehmler, H.J.; Puschner, B.; Lein, P.J. Detection of 3,3’-Dichlorobiphenyl in Human Maternal Plasma and Its Effects on Axonal and Dendritic Growth in Primary Rat Neurons. Toxicol. Sci. 2017, 158, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Mapuskar, K.A.; Marek, R.F.; Xu, W.; Lehmler, H.J.; Robertson, L.W.; Hornbuckle, K.C.; Spitz, D.R.; Aykin-Burns, N. A new player in environmentally induced oxidative stress: Polychlorinated biphenyl congener, 3,3’-dichlorobiphenyl (PCB11). Toxicol. Sci. 2013, 136, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, L.; Sarsour, E.H.; Kalen, A.L.; Aykin-Burns, N.; Spitz, D.R.; Goswami, P.C. Polychlorinated biphenyl induced ROS signaling delays the entry of quiescent human breast epithelial cells into the proliferative cycle. Free Radic. Biol. Med. 2010, 49, 40–49. [Google Scholar]
- Glauert, H.P.; Tharappel, J.C.; Lu, Z.; Stemm, D.; Banerjee, S.; Chan, L.S.; Lee, E.Y.; Lehmler, H.J.; Robertson, L.W.; Spear, B.T. Role of oxidative stress in the promoting activities of pcbs. Environ. Toxicol. Pharmacol. 2008, 25, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Lai, I.; Chai, Y.; Simmons, D.; Luthe, G.; Coleman, M.C.; Spitz, D.; Haschek, W.M.; Ludewig, G.; Robertson, L.W. Acute toxicity of 3,3’,4,4’,5-pentachlorobiphenyl (PCB 126) in male Sprague-Dawley rats: Effects on hepatic oxidative stress, glutathione and metals status. Environ. Int. 2010, 36, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Kalen, A.L.; Li, L.; Lehmler, H.J.; Robertson, L.W.; Goswami, P.C.; Spitz, D.R.; Aykin-Burns, N. Polychlorinated-biphenyl-induced oxidative stress and cytotoxicity can be mitigated by antioxidants after exposure. Free Radic. Biol. Med. 2009, 47, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; Meer, R.V.D.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Scott, I.; Lu, Z.; Pang, L.; Dimond, C.C.; Gius, D.; Sack, M.N. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med. 2010, 49, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Richardson, S.J.; Kieswich, J.; Bugliani, M.; Holland, M.L.; Marchetti, P.; Morgan, N.G.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 2013, 56, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Pi, H.; Xu, S.; Reiter, R.J.; Guo, P.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy 2015, 11, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Li, L.; Chen, J.X. Loss of Sirt3 limits bone marrow cell-mediated angiogenesis and cardiac repair in post-myocardial infarction. PLoS ONE 2014, 9, e107011. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.N.; Vyas, S.M.; Duffel, M.W.; Parkin, S.; Lehmler, H.J. Synthesis of Sterically Hindered Polychlorinated Biphenyl Derivatives. Synthesis 2011, 7, 1045–1054. [Google Scholar] [PubMed]
- Bauer, U.; Amaro, A.R.; Robertson, L.W. A new strategy for the synthesis of polychlorinated biphenyl metabolites. Chem. Res. Toxicol. 1995, 8, 92–95. [Google Scholar] [CrossRef]
- Li, X.; Holland, E.B.; Feng, W.; Zheng, J.; Dong, Y.; Pessah, I.N.; Duffel, M.W.; Robertson, L.W.; Lehmler, H.J. Authentication of synthetic environmental contaminants and their (bio)transformation products in toxicology: Polychlorinated biphenyls as an example. Environ. Sci. Pollut. Res. Int. 2018, 25, 16508–16521. [Google Scholar] [CrossRef] [PubMed]
- Dayal, D.; Martin, S.M.; Owens, K.M.; Aykin-Burns, N.; Zhu, Y.; Boominathan, A.; Pain, D.; Limoli, C.L.; Goswami, P.C.; Domann, F.E.; et al. Mitochondrial complex II dysfunction can contribute significantly to genomic instability after exposure to ionizing radiation. Radiat. Res. 2009, 172, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Oberley, L.W. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal. Biochem. 1989, 179, 8–18. [Google Scholar] [CrossRef]
- Banerjee, S.; Melnyk, S.B.; Krager, K.J.; Aykin-Burns, N.; Letzig, L.G.; James, L.P.; Hinson, J.A. The neuronal nitric oxide synthase inhibitor NANT blocks acetaminophen toxicity and protein nitration in freshly isolated hepatocytes. Free Radic. Biol. Med. 2015, 89, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Melnyk, S.B.; Krager, K.J.; Aykin-Burns, N.; McCullough, S.S.; James, L.P.; Hinson, J.A. Trifluoperazine inhibits acetaminophen-induced hepatotoxicity and hepatic reactive nitrogen formation in mice and in freshly isolated hepatocytes. Toxicol. Rep. 2017, 4, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Krager, K.J.; Pineda, E.N.; Kharade, S.V.; Kordsmeier, M.; Howard, L.; Breen, P.J.; Compadre, C.M.; Hauer-Jensen, M.; Aykin-Burns, N. Tocotrienol-Rich Fraction from Rice Bran Demonstrates Potent Radiation Protection Activity. Evid. Based Complement Alternat. Med. 2015, 2015, 148791. [Google Scholar] [CrossRef] [PubMed]
- Marine, A.; Krager, K.J.; Aykin-Burns, N.; Macmillan-Crow, L.A. Peroxynitrite induced mitochondrial biogenesis following MnSOD knockdown in normal rat kidney (NRK) cells. Redox Biol. 2014, 2, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta. 2011, 1807, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012, 287, 42436–42443. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Gallant, T.L.; Singh, A.; Chu, I. PCB 118 induces ultrastructural alterations in the rat liver. Toxicology 2000, 145, 127–134. [Google Scholar] [CrossRef]
- Johansson, C.; Tofighi, R.; Tamm, C.; Goldoni, M.; Mutti, A.; Ceccatelli, S. Cell death mechanisms in AtT20 pituitary cells exposed to polychlorinated biphenyls (PCB 126 and PCB 153) and methylmercury. Toxicol. Lett. 2006, 167, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.P.; Roelandt, L.; Goffinet, G.; Stouvenakers, N.; Kremers, P. Cytotoxic effects of Aroclor 1254 on ultrastructure and biochemical parameters in cultured foetal rat hepatocytes. Toxicology 1995, 98, 83–94. [Google Scholar] [CrossRef]
- Nishihara, Y.; Robertson, L.W.; Oesch, F.; Utsumi, K. The effects of tetrachlorobiphenyls on the electron transfer reaction of isolated rat liver mitochondria. Life Sci. 1986, 38, 627–635. [Google Scholar] [CrossRef]
- Nishihara, Y.; Utsumi, K. 4-Chloro-4’-biphenylol as an uncoupler and an inhibitor of mitochondrial oxidative phosphorylation. Biochem. Pharmacol. 1987, 36, 3453–3457. [Google Scholar] [CrossRef]
- Sadeghi-Aliabadi, H.; Chan, K.; Lehmler, H.J.; Robertson, L.W.; O’Brien, P.J. Molecular cytotoxic mechanisms of catecholic polychlorinated biphenyl metabolites in isolated rat hepatocytes. Chem. Biol. Interact. 2007, 167, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chen, C.H.; Lawrence, D.; Carpenter, D.O. Ortho-substituted PCBs kill cells by altering membrane structure. Toxicol. Sci. 2004, 80, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Martinez, A.; Hornbuckle, K.C. Sedimentary Records of Non-Aroclor and Aroclor PCB mixtures in the Great Lakes. J. Great Lakes Res. 2011, 37, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Adamcakova-Dodd, A.; Lehmler, H.J.; Gibson-Corley, K.; Thorne, P.S. Toxicity Evaluation of Exposure to an Atmospheric Mixture of Polychlorinated Biphenyls by Nose-Only and Whole-Body Inhalation Regimens. Environ. Sci. Technol. 2015, 49, 11875–11883. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Adamcakova-Dodd, A.; Thorne, P.S. The fate of inhaled (14)C-labeled PCB11 and its metabolites in vivo. Environ. Int. 2014, 63, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Brace, L.E.; Vose, S.C.; Stanya, K.; Gathungu, R.M.; Marur, V.R.; Longchamp, A.; Treviño-Villarreal, H.; Mejia, P.; Vargas, D.; Inouye, K.; et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Alrob, O.A.; Sankaralingam, S.; Ma, C.; Wagg, C.S.; Fillmore, N.; Jaswal, J.S.; Sack, M.N.; Lehner, R.; Gupta, M.P.; Michelakis, E.D.; et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res. 2014, 103, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Weddle, A.; Kinter, C.S.; Humphries, K.M.; Mather, T.; Szweda, L.I.; Kinter, M. Lysine Acetylation Activates Mitochondrial Aconitase in the Heart. Biochemistry 2015, 54, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A. Fatty acid activation. Prog Lipid Res. 1997, 36, 55–83. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Kosinski, C.; Gavino, B.; Horton, J.D.; Skarnes, W.C.; Young, S.G. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 2004, 279, 9557–9564. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Yamamoto, J.; Okamura, M.; Fujino, T.; Takahashi, S.; Takeuchi, K.; Osborne, T.F.; Yamamoto, T.T.; Ito, S.; Sakai, J. Transcriptional regulation of the murine acetyl-CoA synthetase 1 gene through multiple clustered binding sites for sterol regulatory element-binding proteins and a single neighboring site for Sp1. J. Biol. Chem. 2001, 276, 34259–34269. [Google Scholar] [CrossRef] [PubMed]
- Luong, A.; Hannah, V.C.; Brown, M.S.; Goldstein, J.L. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2000, 275, 26458–26466. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yasunaga, T.; Tanaka, H.; Nishimune, Y.; Nozaki, M. Gene structure and evolution of testicular haploid germ cell-specific genes, Oxct2a and Oxct2b. Genomics 2004, 83, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Theisen, M.J.; Misra, I.; Saadat, D.; Campobasso, N.; Miziorko, H.M.; Harrison, D.H. 3-hydroxy-3-methylglutaryl-CoA synthase intermediate complex observed in “real-time”. Proc. Natl. Acad. Sci. USA. 2004, 101, 16442–16447. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Eckel, R.H.; Abumrad, N.A. Regulation of fatty acid uptake into tissues: Lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res. 2009, 50, S86–S90. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Storch, J.; Thumser, A.E.A. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1486, 28–44. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.E.L.; Landrigan, P.J.; Glueck, C.J.; Zack, J.M.M.; Liddle, J.A.; Burse, V.W.; Jere Housworth, W.; Needham, L.L. Metabolic consequences of exposure to polychlorinated biphenyls (PCB) in sewage sludge. Am. J. Epidemiol. 1980, 112, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Chase, K.H.; Wong, O.; Thomas, D.; Berney, B.W.; Simon, R.K. Clinical and metabolic abnormalities associated with occupational exposure to polychlorinated biphenyls (PCBs). J. Occup Med. 1982, 24, 109–114. [Google Scholar] [PubMed]
- Lee, D.H.; Steffes, M.W.; Sjodin, A.; Jones, R.S.; Needham, L.L.; Jacobs, J.D.R. Low dose of some persistent organic pollutants predicts type 2 diabetes: A nested case-control study. Environ. Health Perspect. 2010, 118, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Steffes, M.W.; Sjodin, A.; Jones, R.S.; Needham, L.L.; Jacobs, J.D.R. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE 2011, 6, e15977. [Google Scholar] [CrossRef] [PubMed]
- Stehr-Green, P.A.; Welty, E.; Steele, G.; Steinberg, K. Evaluation of potential health effects associated with serum polychlorinated biphenyl levels. Environ. Health Perspect. 1986, 70, 255–259. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, S.; Carter, G.S.; Krager, K.J.; Li, X.; Lehmler, H.-J.; Aykin-Burns, N. PCB11 Metabolite, 3,3’-Dichlorobiphenyl-4-ol, Exposure Alters the Expression of Genes Governing Fatty Acid Metabolism in the Absence of Functional Sirtuin 3: Examining the Contribution of MnSOD. Antioxidants 2018, 7, 121. https://doi.org/10.3390/antiox7090121
Alam S, Carter GS, Krager KJ, Li X, Lehmler H-J, Aykin-Burns N. PCB11 Metabolite, 3,3’-Dichlorobiphenyl-4-ol, Exposure Alters the Expression of Genes Governing Fatty Acid Metabolism in the Absence of Functional Sirtuin 3: Examining the Contribution of MnSOD. Antioxidants. 2018; 7(9):121. https://doi.org/10.3390/antiox7090121
Chicago/Turabian StyleAlam, Sinthia, Gwendolyn S. Carter, Kimberly J. Krager, Xueshu Li, Hans-Joachim Lehmler, and Nukhet Aykin-Burns. 2018. "PCB11 Metabolite, 3,3’-Dichlorobiphenyl-4-ol, Exposure Alters the Expression of Genes Governing Fatty Acid Metabolism in the Absence of Functional Sirtuin 3: Examining the Contribution of MnSOD" Antioxidants 7, no. 9: 121. https://doi.org/10.3390/antiox7090121
APA StyleAlam, S., Carter, G. S., Krager, K. J., Li, X., Lehmler, H.-J., & Aykin-Burns, N. (2018). PCB11 Metabolite, 3,3’-Dichlorobiphenyl-4-ol, Exposure Alters the Expression of Genes Governing Fatty Acid Metabolism in the Absence of Functional Sirtuin 3: Examining the Contribution of MnSOD. Antioxidants, 7(9), 121. https://doi.org/10.3390/antiox7090121