Modulation of the Oxidative Stress and Lipid Peroxidation by Endocannabinoids and Their Lipid Analogues

 , , , ,
, , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
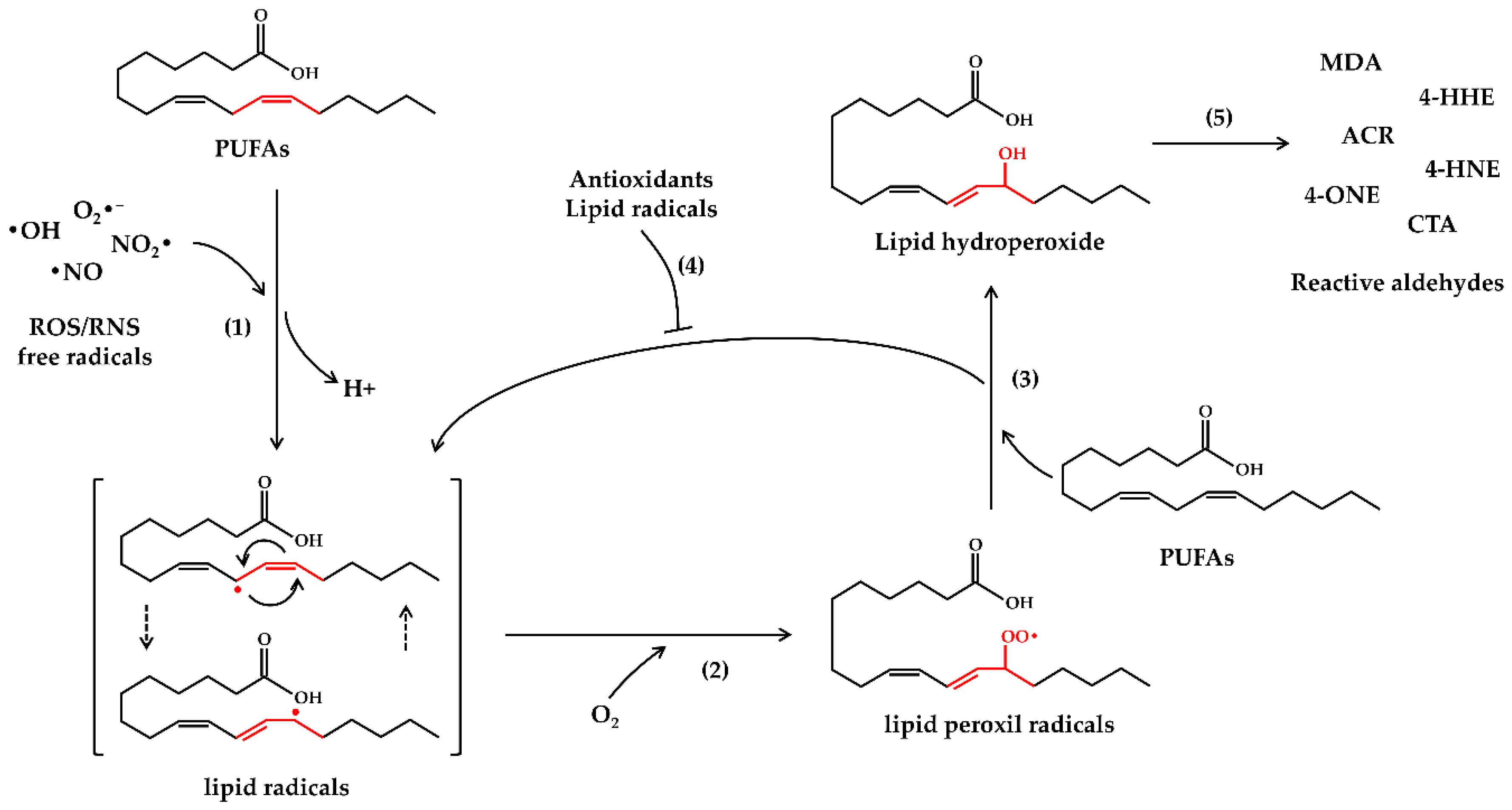
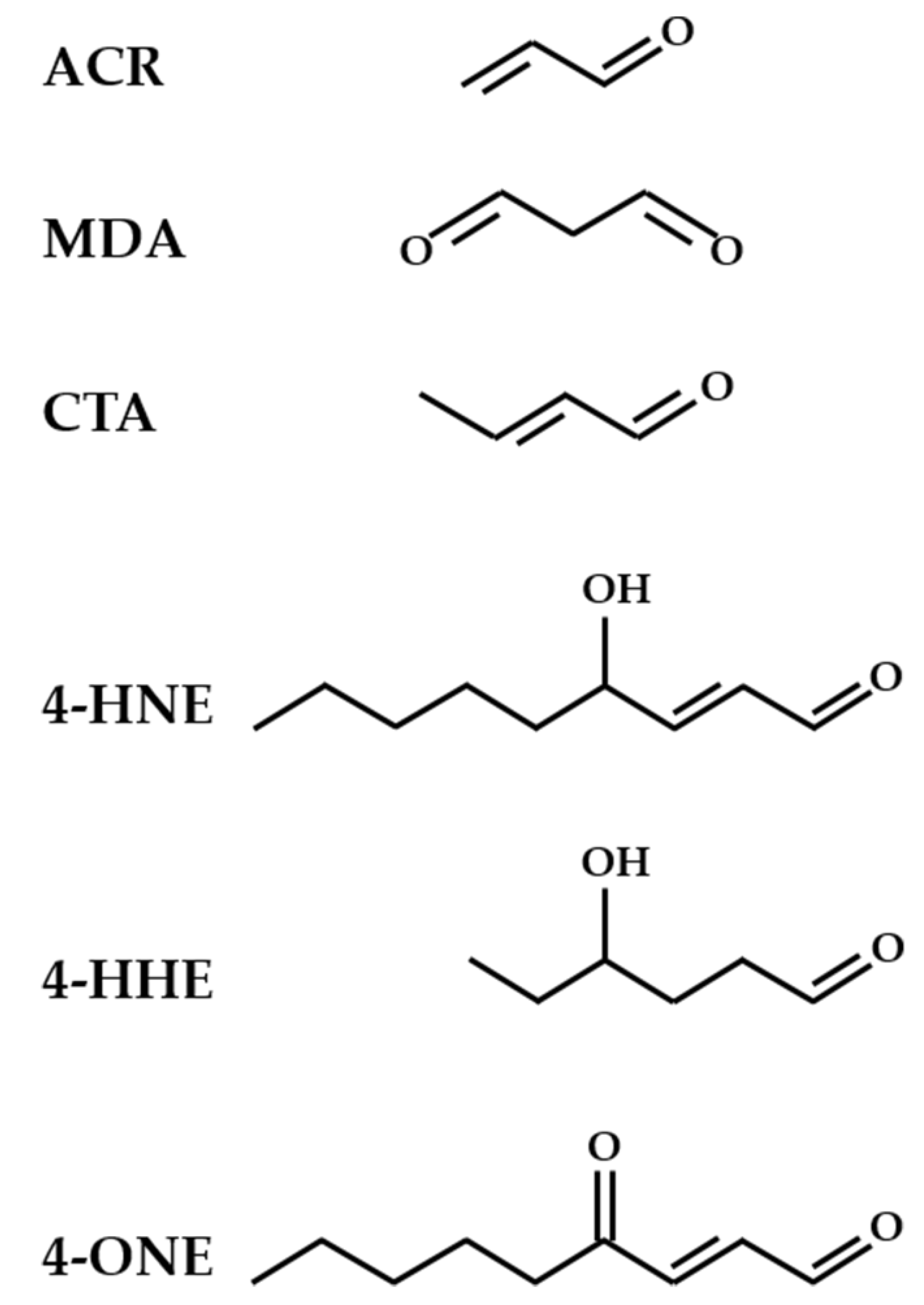
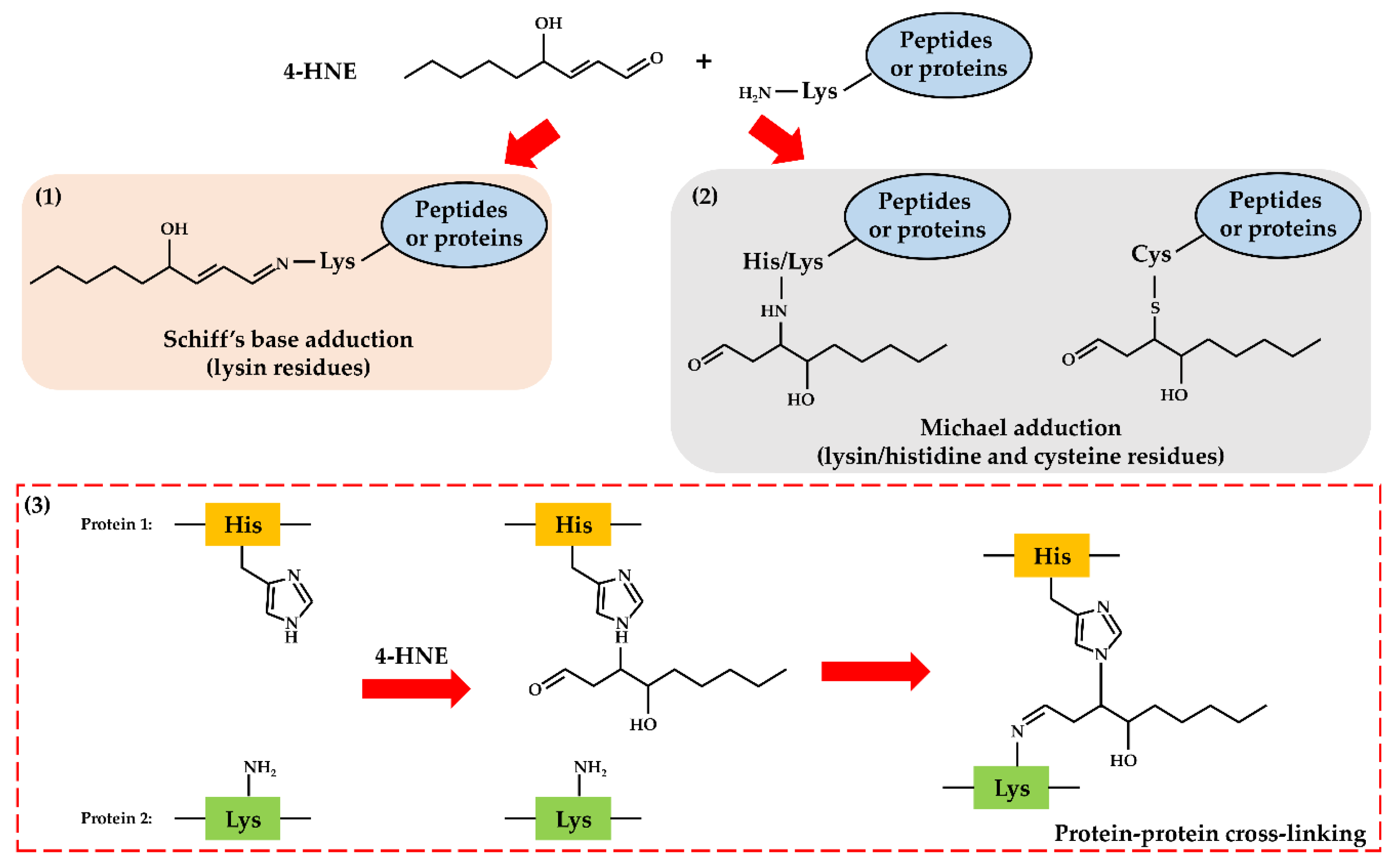
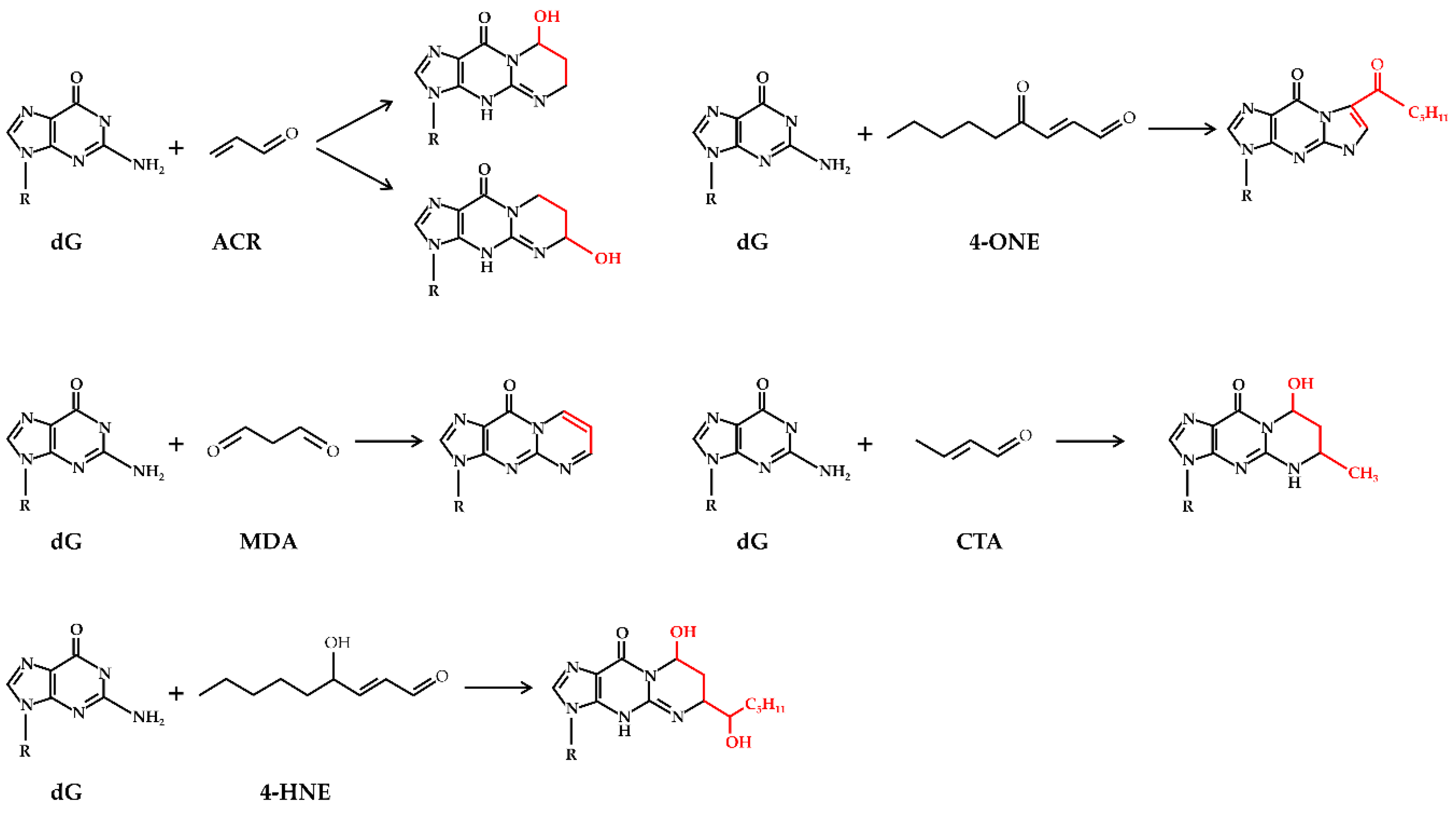
2. Oxidative Stress and Lipid Peroxidation
3. The Endocannabinoid System: Endocannabinoids, Their Lipid Analogues, and the Receptors
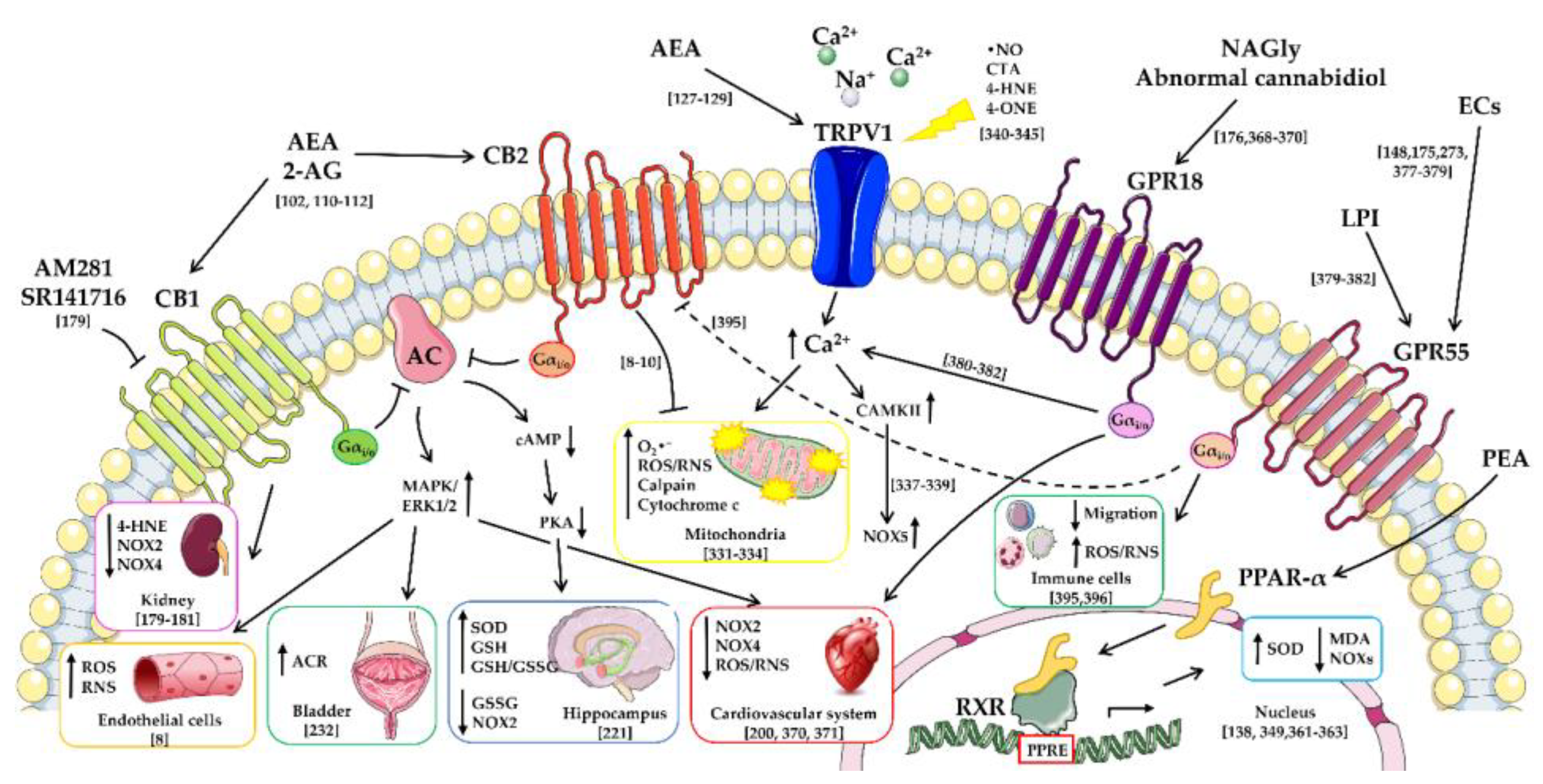
4. Modulation of Oxidative Stress and Lipid Peroxidation through Cannabinoid Receptors by Endocannabinoids and Their Lipid Analogues
5. Modulation of Oxidative Stress and Lipid Peroxidation through the Transient Receptor Potential Vanilloid Channels by Endocannabinoids and Their Lipid Analogues
6. Modulation of Oxidative Stress and Lipid Peroxidation through the Peroxisome Proliferator-Activated Receptors-Alpha by Endocannabinoids and Their Lipid Analogues
7. Modulation of Oxidative Stress and Lipid Peroxidation through Other Receptors by Endocannabinoids and Their Lipid Analogues
8. The Role of Antioxidant System as Scavenger of ROS/RNS and Reactive Aldehydes
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Pomara, C.; Cassano, T.; D’Errico, S.; Bello, S.; Romano, A.D.; Riezzo, I.; Serviddio, G. Data available on the extent of cocaine use and dependence: Biochemistry, pharmacologic effects and global burden of disease of cocaine abusers. Curr. Med. Chem. 2012, 19, 5647–5657. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.T.; Ross, M.K. Oxyradical Stress, Endocannabinoids, and Atherosclerosis. Toxics 2015, 3, 481–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, T.; Serviddio, G.; Gaetani, S.; Romano, A.; Dipasquale, P.; Cianci, S.; Bellanti, F.; Laconca, L.; Romano, A.D.; Padalino, I.; et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 2012, 33, 1121-e1. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Pace, L.; Bedse, G.; Lavecchia, A.M.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and Mitochondria: Two Prominent Players in the Oxidative Stress-Induced Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Romano, A.D.; Cassano, T.; Bellanti, F.; Altomare, E.; Vendemiale, G. Principles and therapeutic relevance for targeting mitochondria in aging and neurodegenerative diseases. Curr. Pharm. Des. 2011, 17, 2036–2055. [Google Scholar] [CrossRef] [PubMed]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Patel, V.; Kashiwaya, Y.; Liaudet, L.; Evgenov, O.V.; Mackie, K.; Haskó, G.; Pacher, P. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 2010, 85, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Lim, S.; Ryu, J.; Lee, C.W.; Kim, Y.; Kang, J.H.; Kang, S.S.; Ahn, Y.K.; Park, C.S.; Kim, J.J. CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc. Res. 2009, 84, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, X.; Chen, J.; Luo, Z.; He, H.; Yu, H.; Ma, L.; Ma, S.; Zhu, T.; Liu, D.; Zhu, Z. TRPV1 activation prevents high-salt diet-induced nocturnal hypertension in mice. Pflügers Arch. Eur. J. Physiol. 2011, 461, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, J.; Lambeth, J.D. Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic. Biol. Med. 2010, 49, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Sevier, C.S.; Kaiser, C.A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta 2008, 1783, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Yan, L.; Madara, P.; Del Pilar Cintron, A.; Wesley, R.A.; Danner, R.L. Superoxide production and reactive oxygen species signaling by endothelial nitric-oxide synthase. J. Biol. Chem. 2000, 275, 16899–16903. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Vergeade, A.; Mulder, P.; Vendeville, C.; Ventura-Clapier, R.; Thuillez, C.; Monteil, C. Xanthine oxidase contributes to mitochondrial ROS generation in an experimental model of cocaine-induced diastolic dysfunction. J. Cardiovasc. Pharmacol. 2012, 60, 538–543. [Google Scholar] [CrossRef] [PubMed]
- McGrath, A.P.; Hilmer, K.M.; Collyer, C.A.; Shepard, E.M.; Elmore, B.O.; Brown, D.E.; Dooley, D.M.; Guss, J.M. Structure and inhibition of human diamine oxidase. Biochemistry 2009, 48, 9810–9822. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Prostaglandin synthase-mediated metabolism of carcinogens and a potential role for peroxyl radicals as reactive intermediates. Environ. Health Perspect. 1990, 88, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Schröder, P.; Krutmann, J. Environmental Oxidative Stress—Environmental Sources of ROS. In Reactions, Processes; Grune, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 2, ISBN 3540235876. [Google Scholar]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef]
- Quie, P.G.; White, J.G.; Holmes, B.; Good, R.A. In vitro bactericidal capacity of human polymorphonuclear leukocytes: Diminished activity in chronic granulomatous disease of childhood. J. Clin. Investig. 1967, 46, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Quie, P.G.; Windhorst, D.B.; Good, R.A. Fatal granulomatous disease of childhood. An inborn abnormality of phagocytic function. Lancet 1966, 1, 1225–1228. [Google Scholar] [CrossRef]
- Bylund, J.; Goldblatt, D.; Speert, D.P. Chronic granulomatous disease: From genetic defect to clinical presentation. Adv. Exp. Med. Biol. 2005, 568, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Ammons, M.C.; Deleo, F.R. The expanding role of NADPH oxidases in health and disease: No longer just agents of death and destruction. Clin. Sci. 2006, 111, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.E.; Pagano, P.J. Targeting reactive oxygen species in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Redox regulation of the afferent arteriole and tubuloglomerular feedback. Acta Physiol. Scand. 2003, 179, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.P.; Cowley, A.W., Jr. Reactive oxygen species and molecular regulation of renal oxygenation. Acta Physiol. Scand. 2003, 179, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Juncos, R.; Hong, N.J.; Garvin, J.L. Differential effects of superoxide on luminal and basolateral Na+/H+ exchange in the thick ascending limb. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R79–R83. [Google Scholar] [CrossRef] [PubMed]
- Hoidal, J.R.; Brar, S.S.; Sturrock, A.B.; Sanders, K.A.; Dinger, B.; Fidone, S.; Kennedy, T.P. The role of endogenous NADPH oxidases in airway and pulmonary vascular smooth muscle function. Antioxid. Redox Signal. 2003, 5, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Kennedy, T.P.; Sturrock, A.B.; Huecksteadt, T.P.; Quinn, M.T.; Murphy, T.M.; Chitano, P.; Hoidal, J.R. NADPH oxidase promotes NF-kappaB activation and proliferation in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L782L795. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.J.; Seo, Y.H.; Hong, F.; Kim, J.H.; Kim, Y.J.; Kang, M.H.; Kim, B.S.; Jo, S.A.; Jo, I.; Jue, D.M.; et al. Nox 2 stimulates muscle differentiation via NF-kappaB/iNOS pathway. Free Radic. Biol. Med. 2005, 38, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Kojim, S.; Ikeda, M.; Shibukawa, A.; Kamikawa, Y. Modification of 5-hydroxytryptophan-evoked 5-hydroxytryptamine formation of guinea pig colonic mucosa by reactive oxygen species. Jpn. J. Pharmacol. 2002, 88, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Anrather, J.; Huang, J.; Speth, R.C.; Pickel, V.M.; Iadecola, C. NADPH oxidase contributes to angiotensin II signaling in the nucleus tractus solitarius. J. Neurosci. 2004, 24, 5516–5524. [Google Scholar] [CrossRef] [PubMed]
- Erdös, B.; Broxson, C.S.; King, M.A.; Scarpace, P.J.; Tümer, N. Acute pressor effect of central angiotensin II is mediated by NAD(P)H-oxidase-dependent production of superoxide in the hypothalamic cardiovascular regulatory nuclei. J. Hypertens. 2006, 24, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Petersen, D.R. An overview of the chemistry and biology of reactive aldehydes. Free Radic. Biol. Med. 2013, 59, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005, 338, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.A.; Caldwell, S.E.; Mills, K.A. Mechanisms of free radical oxidation of unsaturated lipids. Lipids 1995, 30, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008, 477, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N. Role of oxidative stress in adaptive responses in special reference to atherogenesis. J. Clin. Biochem. Nutr. 2008, 43, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Landar, A.; Watanabe, N.; Dickinson, D.A.; Noguchi, N.; Darley-Usmar, V.M. Cell signalling by oxidized lipids and the role of reactive oxygen species in the endothelium. Biochem. Soc. Trans. 2005, 33, 1385–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Serviddio, G.; Calcagnini, S.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Linking lipid peroxidation and neuropsychiatric disorders: Focus on 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 2017, 111, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winczura, A.; Zdżalik, D.; Tudek, B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic. Res. 2012, 46, 442–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, C.K.; Segall, H.J.; Haddon, W.F. Formation of cyclic adducts of deoxyguanosine with the aldehydes trans-4-hydroxy-2-hexenal and trans-4-hydroxy-2-nonenal in vitro. Cancer Res. 1986, 46, 5682–5686. [Google Scholar] [PubMed]
- Chung, F.L.; Young, R.; Hecht, S.S. Formation of cyclic 1,N2-propanodeoxyguanosine adducts in DNA upon reaction with acrolein or crotonaldehyde. Cancer Res. 1984, 44, 990–995. [Google Scholar] [PubMed]
- Cohn, J.A.; Tsai, L.; Friguet, B.; Szweda, L.I. Chemical characterization of a protein-4-hydroxy-2-nonenal cross-link: Immunochemical detection in mitochondria exposed to oxidative stress. Arch. Biochem. Biophys. 1996, 328, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Seto, H.; Okuda, T.; Takesue, T.; Ikemura, T. Reaction of malonaldehyde with nucleic acid. I. Formation of fluorescent pyrimido[1,2-a]purin-10-one nucleosides. Bull. Chem. Soc. Jpn. 1983, 56, 1799–1802. [Google Scholar] [CrossRef]
- Esterbauer, H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 1993, 57, 779S–785S. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 149–159. [Google Scholar] [CrossRef]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteom. 2013, 92, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Rindgen, D.; Nakajima, M.; Wehrli, S.; Xu, K.; Blair, I.A. Covalent modifications to 2′-deoxyguanosine by 4-oxo-2-nonenal, a novel product of lipid peroxidation. Chem. Res. Toxicol. 1999, 12, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rindgen, D.; Bible, R.H., Jr.; Hajdu, E.; Blair, I.A. Characterization of 2′-deoxyadenosine adducts derived from 4-oxo-2-nonenal, a novel product of lipid peroxidation. Chem. Res. Toxicol. 2000, 13, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Pollack, M.; Oe, T.; Lee, S.H.; Silva Elipe, M.V.; Arison, B.H.; Blair, I.A. Characterization of 2′-deoxycytidine adducts derived from 4-oxo-2-nonenal, a novel lipid peroxidation product. Chem. Res. Toxicol. 2003, 16, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.V.; Lee, S.H.; Pollack, M.; Blair, I.A. Endogenous lipid hydroperoxide-mediated DNA-adduct formation in min mice. J. Biol. Chem. 2006, 281, 10127–10133. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, J.; Zhu, H.; Xiong, Y.L. Mass spectrometric evidence of malonaldehyde and 4-hydroxynonenal adductions to radical-scavenging soy peptides. J. Agric. Food Chem. 2012, 60, 9727–9736. [Google Scholar] [CrossRef] [PubMed]
- Mukai, F.H.; Goldstein, B.D. Mutagenicity of malonaldehyde, a decomposition product of peroxidized polyunsaturated fatty acids. Science 1976, 191, 868–869. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K.; Marnett, L.J. Unequivocal demonstration that malondialdehyde is a mutagen. Carcinogenesis 1983, 4, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J.; Hurd, H.K.; Hollstein, M.C.; Levin, D.E.; Esterbauer, H.; Ames, B.N. Naturally occurring carbonyl compounds are mutagens in Salmonella tester strain TA104. Mutat. Res. 1985, 148, 25–34. [Google Scholar] [CrossRef]
- Vasanthi, P.; Nalini, G.; Rajasekhar, G. Status of oxidative stress in rheumatoid arthritis. Int. J. Rheum. Dis. 2009, 12, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Singh, A.; Chandra, V.; Negi, M.P.; Tripathy, B.C.; Prakash, J.; Gupta, V. A comparative analysis of serological parameters and oxidative stress in osteoarthritis and rheumatoid arthritis. Rheumatol. Int. 2012, 32, 2377–2382. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Wanchu, A.; Bhatnagar, A. Interaction between oxidative stress and chemokines: Possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology 2011, 216, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.C.; Chang, Y.S.; Cheng, C.W.; Chi, W.M.; Tsai, K.L.; Chen, W.J.; Kung, T.S.; Tai, C.C.; Lin, Y.F.; Lin, H.T.; et al. Isotypes of autoantibodies against differentially expressed novel malondialdehyde-modified peptide adducts in serum of Taiwanese women with rheumatoid arthritis. J. Proteom. 2018, 170, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, J.T.; Wållberg-Jonsson, S.; Ahmed, E.; Rantapää-Dahlqvist, S.; Lefvert, A.K. Increased levels of autoantibodies against copper-oxidized low density lipoprotein, malondialdehyde-modified low density lipoprotein and cardiolipin in patients with rheumatoid arthritis. Rheumatology 2002, 41, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wållberg-Jonsson, S.; Cvetkovic, J.T.; Sundqvist, K.G.; Lefvert, A.K.; Rantapää-Dahlqvist, S. Activation of the immune system and inflammatory activity in relation to markers of atherothrombotic disease and atherosclerosis in rheumatoid arthritis. J. Rheumatol. 2002, 29, 875–882. [Google Scholar] [PubMed]
- Chung, F.L.; Chen, H.J.; Nath, R.G. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis 1996, 17, 2105–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, F.L.; Nath, R.G.; Nagao, M.; Nishikawa, A.; Zhou, G.D.; Randerath, K. Endogenous formation and significance of 1,N2-propanodeoxyguanosine adducts. Mutat. Res. 1999, 424, 71–81. [Google Scholar] [CrossRef]
- Wang, M.Y.; Chung, F.L.; Hecht, S.S. Identification of crotonaldehyde as a hepatic microsomal metabolite formed by alpha-hydroxylation of the carcinogen N-nitrosopyrrolidine. Chem. Res. Toxicol. 1988, 1, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of paraldol-deoxyguanosine adducts in DNA reacted with crotonaldehyde. Chem. Res. Toxicol. 2000, 13, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. In Dry Cleaning, Some Chlorinated Solvents and Other Industrial Chemicals; International Agency for Research on Cancer: Lyon, France, 1995; Volume 63, pp. 373–391. [Google Scholar]
- Chung, F.L.; Tanaka, T.; Hecht, S.S. Induction of liver tumors in F344 rats by crotonaldehyde. Cancer Res. 1986, 46, 1285–1289. [Google Scholar] [PubMed]
- Ichihashi, K.; Osawa, T.; Toyokuni, S.; Uchida, K. Endogenous formation of protein adducts with carcinogenic aldehydes: Implications for oxidative stress. J. Biol. Chem. 2001, 276, 23903–23913. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, A.; Nakamura, M.; Osawa, T.; Uchida, K. Thiolation of protein-bound carcinogenic aldehyde. An electrophilic acrolein-lysine adduct that covalently binds to thiols. J. Biol. Chem. 2002, 277, 27919–27926. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Kanematsu, M.; Morimitsu, Y.; Osawa, T.; Noguchi, N.; Niki, E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. Biol. Chem. 1998, 273, 16058–16066. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Cohen, G.; Riahi, Y.; Sunda, V.; Deplano, S.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic. Biol. Med. 2013, 65, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Cohen, G.; Shamni, O.; Sasson, S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E879–E886. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal-A Bioactive Lipid Peroxidation Product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Bachmann, K.A.; Bailer, A.J.; Bolger, P.M.; Borak, J.; Cai, L.; Cedergreen, N.; Cherian, M.G.; Chiueh, C.C.; Clarkson, T.W.; et al. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007, 222, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Pizzimenti, S.; Barrera, G.; Dianzani, M.U.; Brüsselbach, S. Inhibition of D1, D2, and A-cyclin expression in HL-60 cells by the lipid peroxydation product 4-hydroxynonenal. Free Radic. Biol. Med. 1999, 26, 1578–1586. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Kong, A.N. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2012, 2, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sano, M.; Shinmura, K.; Tamaki, K.; Katsumata, Y.; Matsuhashi, T.; Morizane, S.; Ito, H.; Hishiki, T.; Endo, J.; et al. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 2010, 49, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Siow, R.C.; Ishii, T.; Mann, G.E. Modulation of antioxidant gene expression by 4-hydroxynonenal: Atheroprotective role of the Nrf2/ARE transcription pathway. Redox Rep. 2007, 12, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Agbaga, M.P.; Anderson, R.E. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic. Biol. Med. 2007, 42, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Cascio, M.G.; Di Marzo, V. The endocannabinoid system: A general view and latest additions. Br. J. Pharmacol. 2004, 141, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Fride, E.; Di Marzo, V. Endocannabinoids. Eur. J. Pharmacol. 1998, 359, 1–18. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Sierra, S.; Luquin, N.; Navarro-Otano, J. The endocannabinoid system in cardiovascular function: Novel insights and clinical implications. Clin. Auton Res. 2018, 28, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Costa, M.A.; Almada, M.; Correia-da-Silva, G.; Teixeira, N.A. Endogenous cannabinoids revisited: A biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013, 102–103, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, B.M.; Correia-da-Silva, G.; Taylor, A.H.; Lam, P.M.; Marczylo, T.H.; Bell, S.C.; Konje, J.C.; Teixeira, N.A. The endocannabinoid 2-arachidonoylglycerol (2-AG) and metabolizing enzymes during rat fetoplacental development: A role in uterine remodelling. Int. J. Biochem. Cell Biol. 2010, 42, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Correia-da-Silva, G.; Taylor, A.H.; Lam, P.M.; Marczylo, T.H.; Konje, J.C.; Bell, S.C.; Teixeira, N.A. N-acylethanolamine levels and expression of their metabolizing enzymes during pregnancy. Endocrinology 2010, 151, 3965–3974. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Gonsiorek, W.; Lunn, C.; Fan, X.; Narula, S.; Lundell, D.; Hipkin, R.W. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: Antagonism by anandamide. Mol. Pharmacol. 2000, 57, 1045–1050. [Google Scholar] [PubMed]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30 (Suppl. 1), S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Steffens, S. The emerging role of the endocannabinoid system in cardiovascular disease. Semin. Immunopathol. 2009, 31, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.; Hinden, L.; Drori, A.; Udi, S.; Azar, S.; Baraghithy, S. The therapeutic potential of targeting the peripheral endocannabinoid/CB1 receptor system. Eur. J. Intern. Med. 2018, 49, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.; Mackie, K. Distribution of the Endocannabinoid System in the Central Nervous System. Handb. Exp. Pharmacol. 2015, 231, 59–93. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease—Successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoid receptor ligands: Clinical and neuropharmacological considerations, relevant to future drug discovery and development. Expert Opin. Investig. Drugs 2000, 9, 1553–1571. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.D.; Carey, K.D.; Stork, P.J.; Iyengar, R. Modulation of rap activity by direct interaction of Galpha(o) with Rap1 GTPase-activating protein. J. Biol. Chem. 1999, 274, 21507–21510. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef] [PubMed]
- Maneuf, Y.P.; Brotchie, J.M. Paradoxical action of the cannabinoid WIN 55, 212–212 in stimulated and basal cyclic AMP accumulation in rat globus pallidus slices. Br. J. Pharmacol. 1997, 120, 1397–1398. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. Cannabinoid receptor signaling. Handb. Exp. Pharmacol. 2005, 168, 53–79. [Google Scholar] [CrossRef]
- Turu, G.; Hunyady, L. Signal transduction of the CB1 cannabinoid receptor. J. Mol. Endocrinol. 2010, 44, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and pharmacology of endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; De Petrocellis, L. Endocannabinoids as regulators of transient receptor potential (TRP) channels: A further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr. Med. Chem. 2010, 17, 1430–1449. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Haugh, O.; Penman, J.; Irving, A.J.; Campbell, V.A. The emerging role of the cannabinoid receptor family in peripheral and neuro-immune interactions. Curr. Drug Targets 2016, 17, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Luo, Z.; Ma, S.; Wong, W.T.; Ma, L.; Zhong, J.; He, H.; Zhao, Z.; Cao, T.; Yan, Z.; et al. Activation of TRPV1 by dietary capsaicin improves endothelium-dependent vasorelaxation and prevents hypertension. Cell Metab. 2010, 12, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Poblete, I.M.; Orliac, M.L.; Briones, R.; Adler-Graschinsky, E.; Huidobro-Toro, J.P. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J. Physiol. 2005, 568, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randhawa, P.K.; Jaggi, A.S. TRPV1 channels in cardiovascular system: A double edged sword? Int. J. Cardiol. 2017, 228, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Bisogno, T.; De Petrocellis, L. Anandamide: Some like it hot. Trends Pharmacol. Sci. 2001, 22, 346–349. [Google Scholar] [CrossRef]
- Starowicz, K.; Makuch, W.; Osikowicz, M.; Piscitelli, F.; Petrosino, S.; Di Marzo, V.; Przewlocka, B. Spinal anandamide produces analgesia in neuropathic rats: Possible CB1- and TRPV1-mediated mechanisms. Neuropharmacology 2012, 62, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Santoro, A.; Pirozzi, C.; d’Emmanuele di Villa Bianca, R.; Mitidieri, E.; Paciello, O.; Pagano, T.B.; Orefice, N.S.; et al. N-Palmitoylethanolamide protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress. Pharmacol. Res. 2013, 76, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Lim, J.H.; Chung, S.; Youn, D.Y.; Chung, H.W.; Kim, H.W.; Lee, J.-H.; Chang, Y.S.; Park, C.W.; et al. Peroxisome proliferator-activated receptor-alpha activator fenofibrate prevents high-fat diet-induced renal lipotoxicity in spontaneously hypertensive rats. Hypertens. Res. 2009, 32, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Gelosa, P.; Banfi, C.; Gianella, A.; Brioschi, M.; Pignieri, A.; Nobili, E.; Castiglioni, L.; Cimino, M.; Tremoli, E.; Sironi, L. Peroxisome proliferator-activated receptor α agonism prevents renal damage and the oxidative stress and inflammatory processes affecting the brains of stroke-prone rats. J. Pharmacol. Exp. Ther. 2010, 335, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Wolff, D.A.; Huskin, A.L.; Helenowski, I.B.; Rademaker, A.W. Fenofibrate therapy ameliorates fasting and postprandial lipoproteinemia, oxidative stress, and the inflammatory response in subjects with hypertriglyceridemia and the metabolic syndrome. Diabetes Care 2007, 30, 1945–1951. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Alexander, S.P.H.; Kendall, D.A.; Bennett, A.J. Cannabinoids and PPARalpha signalling. Biochem. Soc. Trans. 2006, 34, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Bouaboula, M.; Hilairet, S.; Marchand, J.; Fajas, L.; Le Fur, G.; Casellas, P. Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 2005, 517, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Chen, X.; Zhang, J.; Chen, C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br. J. Pharmacol. 2011, 163, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Kowalski, T.J. GPR119 agonists for the potential treatment of type 2 diabetes and related metabolic disorders. Vitam. Horm. 2010, 84, 415–448. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno-Shosaku, T.; Maejima, T.; Kano, M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 2001, 29, 729–738. [Google Scholar] [CrossRef]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Endocannabinoids and synaptic function in the CNS. Neuroscientist 2007, 13, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Oddi, S.; Fezza, F.; Pasquariello, N.; De Simone, C.; Rapino, C.; Dainese, E.; Finazzi-Agrò, A.; Maccarrone, M. Evidence for the intracellular accumulation of anandamide in adiposomes. Cell. Mol. Life Sci. 2008, 65, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ueda, N. Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat. 2009, 89, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Seillier, A.; Advani, T.; Cassano, T.; Hensler, J.G.; Giuffrida, A. Inhibition of fatty-acid amide hydrolase and CB1 receptor antagonism differentially affect behavioural responses in normal and PCP-treated rats. Int. J. Neuropsychopharmacol. 2010, 13, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Colangeli, R.; Lavecchia, A.M.; Romano, A.; Altieri, F.; Cifani, C.; Cassano, T.; Gaetani, S. Role of the basolateral amygdala in mediating the effects of the fatty acid amide hydrolase inhibitor URB597 on HPA axis response to stress. Eur. Neuropsychopharmacol. 2014, 24, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Tsuboi, K.; Uyama, T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog. Lipid Res. 2010, 49, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Tsuboi, K.; Uyama, T.; Ohnishi, T. Biosynthesis and degradation of the endocannabinoid 2-arachidonoylglycerol. Biofactors 2011, 37, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, andcytochromes P450: Cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, H.B.; Walker, J.M. The expanding field of cannabimimetic and related lipid mediators. Br. J. Pharmacol. 2009, 144, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V.; Schmid, P.C.; Krebsbach, R.J.; Hillard, C.J.; Huang, C.; Chen, N.; Dong, Z.; Schmid, H.H. Cannabinoid-receptor-independent cell signalling by N-acylethanolamines. Biochem. J. 2001, 360, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Azari, E.K.; Ramachandran, D.; Weibel, S.; Arnold, M.; Romano, A.; Gaetani, S.; Langhans, W.; Mansouri, A. Vagal afferents are not necessary for the satiety effect of the gut lipid messenger oleoylethanolamide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R167–R178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provensi, G.; Coccurello, R.; Umehara, H.; Munari, L.; Giacovazzo, G.; Galeotti, N.; Nosi, D.; Gaetani, S.; Romano, A.; Moles, A.; et al. Satiety factor oleoylethanolamide recruits the brain histaminergic system to inhibit food intake. Proc. Natl. Acad. Sci. USA 2014, 111, 11527–11532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Cassano, T.; Tempesta, B.; Cianci, S.; Dipasquale, P.; Coccurello, R.; Cuomo, V.; Gaetani, S. The satiety signal oleoylethanolamide stimulates oxytocin neurosecretion from rat hypothalamic neurons. Peptides 2013, 49, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Karimian Azari, E.; Tempesta, B.; Mansouri, A.; Micioni Di Bonaventura, M.V.; Ramachandran, D.; Lutz, T.A.; Bedse, G.; Langhans, W.; Gaetani, S. High dietary fat intake influences the activation of specific hindbrain and hypothalamic nuclei by the satiety factor oleoylethanolamide. Physiol. Behav. 2014, 136, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Coccurello, R.; Giacovazzo, G.; Bedse, G.; Moles, A.; Gaetani, S. Oleoylethanolamide: A novel potential pharmacological alternative to cannabinoid antagonists for the control of appetite. Biomed. Res. Int. 2014, 2014, 203425. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, S.; Kaye, W.H.; Cuomo, V.; Piomelli, D. Role of endocannabinoids and their analogues in obesity and eating disorders. Eat. Weight Disord. 2008, 13, e42–e48. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Fanelli, F.; Ceru, M.P.; Moreno, S. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARα) and its lipid ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Romano, A.; Lavecchia, A.M.; Cassano, T.; Gaetani, S. The role of endocannabinoid signaling in the molecular mechanisms of neurodegeneration in Alzheimer’s disease. J. Alzheimer Dis. 2015, 43, 1115–1136. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L., Jr.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxid. Med. Cell. Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 3662–3665. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.C.; Sauer, J.M.; Knierman, M.D.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Console-Bram, L.; Mundy, C.; Popoff, S.N.; Kapur, A.; Abood, M.E. The Endocannabinoids Anandamide and Virodhamine Modulate the Activity of the Candidate Cannabinoid Receptor GPR55. J. Neuroimmune Pharmacol. 2012, 7, 856–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, M.; Hasegawa, H.; Inoue, A.; Muraoka, M.; Miyazaki, T.; Oka, K.; Yasukawa, M. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun. 2006, 347, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Lipina, C.; Hundal, H.S. Modulation of cellular redox homeostasis by the endocannabinoid system. Open Biol. 2016, 6, 150276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, B.; Mukhopadhyay, P.; Kechrid, M.; Patel, V.; Tanchian, G.; Wink, D.A.; Gertsch, J.; Pacher, P. β-Caryophyllene ameliorates cisplatin-induced nephrotoxicity in a cannabinoid 2 receptor-dependent manner. Free Radic. Biol. Med. 2012, 52, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Pan, H.; Rajesh, M.; Batkai, S.; Patel, V.; Harvey-White, J.; Mukhopadhyay, B.; Hasko, G.; Gao, B.; Mackie, K.; et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br. J. Pharmacol. 2010, 160, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Pan, H.; Patel, V.; Mukhopadhyay, B.; Bátkai, S.; Gao, B.; Haskó, G.; Pacher, P. Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic. Biol. Med. 2010, 48, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Baggelaar, M.; Erdelyi, K.; Cao, Z.; Cinar, R.; Fezza, F.; Ignatowska-Janlowska, B.; Wilkerson, J.; van Gils, N.; Hansen, T.; et al. The novel, orally available and peripherally restricted selective cannabinoid CB2 receptor agonist LEI-101 prevents cisplatin-induced nephrotoxicity. Br. J. Pharmacol. 2016, 173, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify the risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Barutta, F.; Kunos, G.; Pacher, P. Role of the endocannabinoid system in diabetes and diabetic complications. Br. J. Pharmacol. 2016, 173, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Hasko, G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2008, 153, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, C.; Ligresti, A.; Di Marzo, V. Peripheral effects of the endocannabinoid system in energy homeostasis: Adipose tissue, liver and skeletal muscle. Rev. Endocr. Metab. Disord. 2011, 12, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P. Role of free radicals in liver diseases. Hepatol. Int. 2009, 3, 526–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Matsumoto, S.; Saito, K.; Bátkai, S.; Patel, V.; Tanchian, G.; Gao, R.Y.; Cravatt, B.F.; et al. Fatty acid amide hydrolase is a key regulator of endocannabinoid-induced myocardial tissue injury. Free Radic. Biol. Med. 2011, 50, 179–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Haskó, G.; Liaudet, L.; Mackie, K.; Pacher, P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br. J. Pharmacol. 2010, 160, 688–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bátkai, S.; Osei-Hyiaman, D.; Pan, H.; El-Assal, O.; Rajesh, M.; Mukhopadhyay, P.; Hong, F.; Harvey-White, J.; Jafri, A.; Haskó, G.; et al. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J. 2007, 21, 1788–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, M.; Pan, H.; Mukhopadhyay, P.; Batkai, S.; Osei-Hyiaman, D.; Hasko, G.; Liaudet, L.; Gao, B.; Pacher, P. Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response, and apoptosis. J. Leukoc. Biol. 2007, 82, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Lenglet, S.; Braunersreuther, V.; Burger, F.; Pelli, G.; Bertolotto, M.; Mach, F.; Steffens, S. CB2 cannabinoid receptor activation is cardioprotective in a mouse model of ischemia/reperfusion. J. Mol. Cell. Cardiol. 2009, 46, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Adler, M.W.; Abood, M.E.; Ganea, D.; Jallo, J.; Tuma, R.F. CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc. Res. 2009, 78, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Murikinati, S.; Juttler, E.; Keinert, T.; Ridder, D.A.; Muhammad, S.; Waibler, Z.; Ledent, C.; Zimmer, A.; Kalinke, U.; Schwaninger, M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2009. [Google Scholar] [CrossRef] [PubMed]
- Steffens, S.; Veillard, N.R.; Arnaud, C.; Pelli, G.; Burger, F.; Staub, C.; Karsak, M.; Zimmer, A.; Frossard, J.L.; Mach, F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature 2005, 434, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Batkai, S.; Hasko, G.; Liaudet, L.; Huffman, J.W.; Csiszar, A.; Ungvari, Z.; Mackie, K.; Chatterjee, S.; et al. CB2-receptor stimulation attenuates TNF-alpha-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte–endothelial adhesion. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2210–H2218. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Mechoulam, R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog. Lipid Res. 2011, 50, 193–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, A.T.; Lee, J.H.; Borazjani, A.; Mangum, L.C.; Hou, X.; Ross, M.K. Oxyradical stress increases the biosynthesis of 2-arachidonoylglycerol: Involvement of NADPH oxidase. Am. J. Physiol. Cell Physiol. 2016, 311, C960–C974. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, F.F.; Steinmetz, M.; Zimmer, S.; Becker, A.; Lütjohann, D.; Buchalla, R.; Zimmer, A.; Nickenig, G. Atheroprotection via cannabinoid receptor-2 is mediated by circulating and vascular cells in vivo. J. Mol. Cell. Cardiol. 2011, 51, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Tiyerili, V.; Zimmer, S.; Jung, S.; Wassmann, K.; Naehle, C.P.; Lütjohann, D.; Zimmer, A.; Nickenig, G.; Wassmann, S. CB1 receptor inhibition leads to decreased vascular AT1 receptor expression, inhibition of oxidative stress and improved endothelial function. Basic Res. Cardiol. 2010, 105, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Zolese, G.; Bacchetti, T.; Masciangelo, S.; Ragni, L.; Ambrosi, S.; Ambrosini, A.; Marini, M.; Ferretti, G. Effect of acylethanolamides on lipid peroxidation and paraoxonase activity. Biofactors 2008, 33, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Gulaya, N.M.; Kuzmenko, A.I.; Margitich, V.M.; Govseeva, N.M.; Melnichuk, S.D.; Goridko, T.M.; Zhukov, A.D. Long-chain N-acylethanolamines inhibit lipid peroxidation in rat liver mitochondria under acute hypoxic hypoxia. Chem. Phys. Lipids 1998, 97, 49–54. [Google Scholar] [CrossRef]
- Poli, G.; Albano, E.; Dianzani, M.U. The role of lipid peroxidation in liver damage. Chem. Phys. Lipids 1987, 45, 117–142. [Google Scholar] [CrossRef]
- Emerit, J.; Chaudiere, J. Free Radicals and Lipid Peroxidation in Cell Biology; Handbook of Free Radicals and Antioxidants in Biomedicine; CRC Press: Boca Raton, FL, USA, 1989; pp. 177–185. [Google Scholar]
- Gulaya, N.M.; Melnik, A.A.; Balkov, D.I.; Volkov, G.L.; Vysotskiy, M.V.; Vaskovsky, V.E. The effect of long-chain N-acylethanolamines on some membrane-associated functions of neuroblastoma C1300 N18 cells. Biochim. Biophys. Acta 1993, 1152, 280–288. [Google Scholar] [CrossRef]
- Parinandi, N.L.; Schmid, H.H. Effects of long-chain N-acylethanolamines on lipid peroxidation in cardiac mitochondria. FEBS Lett. 1988, 237, 49–52. [Google Scholar] [CrossRef]
- Zolese, G.; Bacchetti, T.; Ambrosini, A.; Wozniak, M.; Bertoli, E.; Ferretti, G. Increased plasma concentrations of palmitoylethanolamide, an endogenous fatty acid amide, affect oxidative damage of human low-density lipoproteins: An in vitro study. Atherosclerosis 2005, 182, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, M.; Łuczaj, W.; Gęgotek, A.; Toczek, M.; Bielawska, K.; Skrzydlewska, E. Crosstalk between liver antioxidant and the endocannabinoid systems after chronic administration of the FAAH inhibitor, URB597, to hypertensive rats. Toxicol. Appl. Pharmacol. 2016, 301, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Maccarrone, M. FAAH and anandamide: Is 2-AG really the odd one out? Trends Pharmacol. Sci. 2008, 29, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.P.; Aloysius, M.M.; Shah, N.J.; Brown, R.S., Jr. Review article: The endocannabinoid system in liver disease, a potential therapeutic target. Aliment. Pharmacol. Ther. 2014, 39, 790–801. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Wang, X.; Kanel, G.C.; Atkinson, R.D.; McCuskey, R.S. Prevention of hepatic fibrosis in a murine model of metabolic syndrome with nonalcoholic steatohepatitis. Am. J. Pathol. 2008, 173, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Mallat, A.; Teixeira-Clerc, F.; Lotersztajn, S. Cannabinoid signaling and liver therapeutics. J. Hepatol. 2013, 59, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Simonian, N.A.; Coyle, J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Alzheimer’s disease and oxidative stress: Implications for novel therapeutic approaches. Prog. Neurobiol. 1999, 57, 301–323. [Google Scholar] [CrossRef]
- Bedse, G.; Romano, A.; Cianci, S.; Lavecchia, A.M.; Lorenzo, P.; Elphick, M.R.; Laferla, F.M.; Vendemiale, G.; Grillo, C.; Altieri, F.; et al. Altered expression of the CB1 cannabinoid receptor in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimer Dis. 2014, 40, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Gatta, E.; Lefebvre, T.; Gaetani, S.; dos Santos, M.; Marrocco, J.; Mir, A.M.; Cassano, T.; Maccari, S.; Nicoletti, F.; Mairesse, J. Evidence for an imbalance between tau O-GlcNAcylation and phosphorylation in the hippocampus of a mouse model of Alzheimer’s disease. Pharmacol. Res. 2016, 105, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Milton, N.G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: Implications for treatment. Drugs Aging 2004, 21, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Ano, Y.; Sakudo, A.; Kimata, T.; Uraki, R.; Sugiura, K.; Onodera, T. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci. Lett. 2010, 469, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Huang, B.; Zhang, X.; Zhu, Y.; Chen, X. Astaxanthin protects against MPP+-induced oxidative stress in PC12 cells via the HO-1/NOX2 axis. BMC Neurosci. 2012, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Ma, L.; Wu, M.; Zhang, L.; Zhang, X.; Zhai, Q.; Jiang, T.; Wang, Q.; Xiong, L. Anandamide protects HT22 cells exposed to hydrogen peroxide by inhibiting CB1 receptor-mediated type 2 NADPH oxidase. Oxid. Med. Cell. Longev. 2014, 2014, 893516. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Bok, E.; Huh, S.H.; Park, J.Y.; Yoon, S.H.; Kim, S.R.; Kim, Y.S.; Maeng, S.; Park, S.H.; Jin, B.K. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 2011, 187, 6508–6517. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Jayant, S.; Sharma, B.M.; Bansal, R.; Sharma, B. Pharmacological benefits of selective modulation of cannabinoid receptor type 2 (CB2) in experimental Alzheimer’s disease. Pharmacol. Biochem. Behav. 2016, 140, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Koppel, J.; Vingtdeux, V.; Marambaud, P.; d’Abramo, C.; Jimenez, H.; Stauber, M.; Friedman, R.; Davies, P. CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer’s disease. Mol. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Paloczi, J.; Varga, Z.V.; Hasko, G.; Pacher, P. Neuroprotection in Oxidative Stress-Related Neurodegenerative Diseases: Role of Endocannabinoid System Modulation. Antioxid. Redox Signal. 2018, 29, 75–108. [Google Scholar] [CrossRef] [PubMed]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflamm. 2015, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Kessiova, M.; Alexandrova, A.; Georgieva, A.; Kirkova, M.; Todorov, S. In vitro effects of CB1 receptor ligands on lipid peroxidation and antioxidant defense systems in the rat brain. Pharmacol. Rep. 2006, 58, 870–875. [Google Scholar] [PubMed]
- Sun, H.J.; Lu, Y.; Wang, H.W.; Zhang, H.; Wang, S.R.; Xu, W.Y.; Fu, H.L.; Yao, X.Y.; Yang, F.; Yuan, H.B. Activation of Endocannabinoid Receptor 2 as a Mechanism of Propofol Pretreatment-Induced Cardioprotection against Ischemia-Reperfusion Injury in Rats. Oxid. Med. Cell. Longev. 2017, 2017, 2186383. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.H.; Ballesteros, I.; de Miguel, F.; Coyle, C.H.; Tyagi, S.; Yoshimura, N.; Chancellor, M.B.; Tyagi, P. Functional and immunohistochemical characterization of CB1 and CB2 receptors in rat bladder. Urology 2008, 72, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Wang, P.; Bjorling, D.E. Activation of cannabinoid receptor 2 inhibits experimental cystitis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R846–R853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanson, D.J.; Marnett, L.J. Cannabinoids endocannabinoids, and cancer. Cancer Metastasis Rev. 2011, 30, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014, 5, 2475–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapham, D.E.; Runnels, L.W.; Strübing, C. The TRP ion channel family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Harteneck, C.; Plant, T.D.; Schultz, G. From worm to man: Three subfamilies of TRP channels. Trends Neurosci. 2000, 23, 159–166. [Google Scholar] [CrossRef]
- Wu, L.J.; Sweet, T.B.; Clapham, D.E. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol. Rev. 2010, 62, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.N.; Clapham, D.E. TRP channels and mice deficient in TRP channels. Pflugers Arch. 2005, 451, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, D.J.; Chesler, A.T.; Jackson, A.C.; Sigal, Y.M.; Yamanaka, H.; Grant, R.; O’Donnell, D.; Nicoll, R.A.; Shah, N.M.; Julius, D.; et al. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J. Neurosci. 2011, 31, 5067–5077. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Jordt, S.E.; Tominaga, M.; Julius, D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA 2000, 97, 8134–8139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, T.; Moriyama, T.; Kobata, K.; Morita, A.; Murayama, N.; Hashizume, S.; Fushiki, T.; Yazawa, S.; Watanabe, T.; Tominaga, M. TRPV1 activation and induction of nociceptive response by a non-pungent capsaicin-like compound, capsiate. Neuropharmacology 2003, 44, 958–967. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Harris, R.A. Deletion of vanilloid receptor (TRPV1) in mice alters behavioral effects of ethanol. Neuropharmacology 2009, 56, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellingson, J.M.; Silbaugh, B.C.; Brasser, S.M. Reduced oral ethanol avoidance in mice lacking transient receptor potential channel vanilloid receptor 1. Behav. Genet. 2009, 39, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ahern, G.P. Activation of TRPV1 by the satiety factor oleoylethanolamide. J. Biol. Chem. 2003, 278, 30429–30434. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; De Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.J.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.J.; Huang, S.M.; De Petrocellis, L.; Bisogno, T.; Ewing, S.A.; Miller, J.D.; Zipkin, R.E.; Daddario, N.; Appendino, G.; Di Marzo, V.; et al. N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J. Biol. Chem. 2003, 278, 13633–13639. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numazaki, M.; Tominaga, T.; Takeuchi, K.; Murayama, N.; Toyooka, H.; Tominaga, M. Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc. Natl. Acad. Sci. USA 2003, 100, 8002–8006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, T.; Gordon-Shaag, A.; Munari, M.; Gordon, S.E. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J. Gen. Physiol. 2004, 123, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lishko, P.V.; Procko, E.; Jin, X.; Phelps, C.B.; Gaudet, R. The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 2007, 54, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Hofmann, T.; Montell, C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol. Cell 2007, 25, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, L.S.; Ahern, G.P. Induction of vanilloid receptor channel activity by protein kinase C. Nature 2000, 408, 985–990. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Harrison, S.; Bisogno, T.; Tognetto, M.; Brandi, I.; Smith, G.D.; Creminon, C.; Davis, J.B.; Geppetti, P.; Di Marzo, V. The vanilloid receptor (VR1)-mediated effects of anandamide are potently enhanced by the cAMP-dependent protein kinase. J. Neurochem. 2001, 77, 1660–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, R.J.; Yeats, J.C.; Bevan, S.; Boddeke, H.W. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 1996, 431, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Birder, L.A.; Nakamura, Y.; Kiss, S.; Nealen, M.L.; Barrick, S.; Kanai, A.J.; Wang, E.; Ruiz, G.; De Groat, W.C.; Apodaca, G.; et al. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat. Neurosci. 2002, 5, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Rong, W.; Hillsley, K.; Davis, J.B.; Hicks, G.; Winchester, W.J.; Grundy, D. Jejunal afferent nerve sensitivity in wild-type and TRPV1 knockout mice. J. Physiol. 2004, 560, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treesukosol, Y.; Lyall, V.; Heck, G.L.; DeSimone, J.A.; Spector, A.C. A psychophysical and electrophysiological analysis of salt taste in Trpv1 null mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1799–R1809. [Google Scholar] [CrossRef] [PubMed]
- Cagiano, R.; Cassano, T.; Coluccia, A.; Gaetani, S.; Giustino, A.; Steardo, L.; Tattoli, M.; Trabace, L.; Cuomo, V. Genetic factors involved in the effects of developmental low-level alcohol induced behavioral alterations in rats. Neuropsychopharmacology 2002, 26, 191–203. [Google Scholar] [CrossRef]
- Geppetti, P.; Materazzi, S.; Nicoletti, P. The transient receptor potential vanilloid 1: Role in airway inflammation and disease. Eur. J. Pharmacol. 2006, 533, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sharma, B.; Singh, P.; Sharma, B.M. Modulation of transient receptor potential vanilloid subtype 1 (TRPV1) and norepinephrine transporters (NET) protect against oxidative stress, cellular injury, and vascular dementia. Curr. Neurovasc. Res. 2014, 11, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sharma, B. Pharmacological benefits of agomelatine and vanillin in experimental model of Huntington’s disease. Pharmacol. Biochem. Behav. 2014, 122, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Balasubramanian, A.; Marrelli, S.P. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R149–R156. [Google Scholar] [CrossRef] [PubMed]
- Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Wang, Y.; Flores, C.M.; Qin, N. Activation properties of heterologously expressed mammalian TRPV2: Evidence for species dependence. J. Biol. Chem. 2007, 282, 15894–15902. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Szöllősi, A.G.; Oláh, A.; Tóth, I.B.; Papp, F.; Czifra, G.; Panyi, G.; Bíró, T. Transient receptor potential vanilloid-2 mediates the effects of transient heat shock on endocytosis of human monocyte-derived dendritic cells. FEBS Lett. 2013, 587, 1440–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Hanson, P.I.; Schlesinger, P. Luminal chloride-dependent activation of endosome calcium channels: Patch clamp study of enlarged endosomes. J. Biol. Chem. 2007, 282, 27327–27333. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Puertollano, R. Role of TRP channels in the regulation of the endosomal pathway. Physiology 2011, 26, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Lévêque, M.; Penna, A.; Le Trionnaire, S.; Belleguic, C.; Desrues, B.; Brinchault, G.; Jouneau, S.; Lagadic-Gossmann, D.; Martin-Chouly, C. Phagocytosis depends on TRPV2-mediated calcium influx and requires TRPV2 in lipids rafts: Alteration in macrophages from patients with cystic fibrosis. Sci. Rep. 2018, 8, 4310. [Google Scholar] [CrossRef] [PubMed]
- Link, T.M.; Park, U.; Vonakis, B.M.; Raben, D.M.; Soloski, M.J.; Caterina, M.J. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat. Immunol. 2010, 11, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peier, A.M.; Reeve, A.J.; Andersson, D.A.; Moqrich, A.; Earley, T.J.; Hergarden, A.C.; Story, G.M.; Colley, S.; Hogenesch, J.B.; McIntyre, P.; et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 2002, 296, 2046–2049. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Gunthorpe, M.J.; Kelsell, R.E.; Hayes, P.D.; Reilly, P.; Facer, P.; Wright, J.E.; Jerman, J.C.; Walhin, J.P.; Ooi, L.; Egerton, J.; et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 2002, 418, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Moqrich, A.; Hwang, S.W.; Earley, T.J.; Petrus, M.J.; Murray, A.N.; Spencer, K.S.; Andahazy, M.; Story, G.M.; Patapoutian, A. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 2005, 307, 1468–1472. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Delling, M.; Jun, J.C.; Clapham, D.E. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat. Neurosci. 2006, 9, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Phelps, C.B.; Wang, R.R.; Choo, S.S.; Gaudet, R. Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J. Biol. Chem. 2010, 285, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Mandadi, S.; Sokabe, T.; Shibasaki, K.; Katanosaka, K.; Mizuno, A.; Moqrich, A.; Patapoutian, A.; Fukumi-Tominaga, T.; Mizumura, K.; Tominaga, M. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch. 2009, 458, 1093–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.M.; Lee, H.; Chung, M.K.; Park, U.; Yu, Y.Y.; Bradshaw, H.B.; Coulombe, P.A.; Walker, J.M.; Caterina, M.J. Overexpressed transient receptor potential vanilloid 3 ion channels in skin keratinocytes modulate pain sensitivity via prostaglandin E2. J. Neurosci. 2008, 28, 13727–13737. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Lee, H.; Mizuno, A.; Suzuki, M.; Caterina, M.J. TRPV3 and TRPV4 mediate warmth-evoked currents in primary mouse keratinocytes. J. Biol. Chem. 2004, 279, 21569–21575. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Choe, Y.; Martí-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef]
- Güler, A.D.; Lee, H.; Iida, T.; Shimizu, I.; Tominaga, M.; Caterina, M. Heat-evoked activation of the ion channel, TRPV4. J. Neurosci. 2002, 22, 6408–6414. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T.; Nilius, B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Fisslthaler, B.; Suzuki, M.; Janssens, A.; Voets, T.; Morisseau, C.; Hammock, B.D.; Fleming, I.; Busse, R.; et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005, 97, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Strotmann, R.; Semtner, M.; Kepura, F.; Plant, T.D.; Schöneberg, T. Interdomain interactions control Ca2+-dependent potentiation in the cation channel TRPV4. PLoS ONE 2010, 5, e10580. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Hamada-Nakahara, S.; Itoh, Y.; Takemura, K.; Shimada, A.; Ueda, Y.; Kitamata, M.; Matsuoka, R.; Hanawa-Suetsugu, K.; Senju, Y.; et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P2. Nat. Commun. 2014, 5, 4994. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wu, L.; O’Neil, R.G. Temperature-modulated diversity of TRPV4 channel gating: Activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J. Biol. Chem. 2003, 278, 27129–27137. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Zhang, X.; McNaughton, P.A. Activation of the TRPV4 ion channel is enhanced by phosphorylation. J. Biol. Chem. 2009, 284, 27884–27891. [Google Scholar] [CrossRef] [PubMed]
- Wegierski, T.; Lewandrowski, U.; Müller, B.; Sickmann, A.; Walz, G. Tyrosine phosphorylation modulates the activity of TRPV4 in response to defined stimuli. J. Biol. Chem. 2009, 284, 2923–2933. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Shin, S.H.; Chun, J.; Hyun, S.; Kim, Y.; Kang, S.S. The modulation of TRPV4 channel activity through its Ser 824 residue phosphorylation by SGK1. Anim. Cells Syst. 2010, 14, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Delany, N.S.; Hurle, M.; Facer, P.; Alnadaf, T.; Plumpton, C.; Kinghorn, I.; See, C.G.; Costigan, M.; Anand, P.; Woolf, C.J.; et al. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol. Genom. 2001, 4, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Jong, B.E.; Frey, S.L.; Sudanagunta, S.P.; Capra, N.F.; Levine, J.D. The stretch-inactivated channel, a vanilloid receptor variant, is expressed in small-diameter sensory neurons in the rat. Neurosci. Lett. 2000, 287, 215–218. [Google Scholar] [CrossRef]
- Suzuki, M.; Mizuno, A.; Kodaira, K.; Imai, M. Impaired pressure sensation in mice lacking TRPV4. J. Biol. Chem. 2003, 278, 22664–22668. [Google Scholar] [CrossRef] [PubMed]
- Birder, L.; Kullmann, F.A.; Lee, H.; Barrick, S.; de Groat, W.; Kanai, A.; Caterina, M. Activation of urothelial transient receptor potential vanilloid 4 by 4alpha-phorbol 12,13-didecanoate contributes to altered bladder reflexes in the rat. J. Pharmacol. Exp. Ther. 2007, 323, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, T.; Vriens, J.; Segal, A.; Everaerts, W.; Roskams, T.; Talavera, K.; Owsianik, G.; Liedtke, W.; Daelemans, D.; Dewachter, I.; et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J. Clin. Investig. 2007, 117, 3453–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, D.F.; King, J.A.; Weber, D.; Addison, E.; Liedtke, W.; Townsley, M.I. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: A novel mechanism of acute lung injury. Circ. Res. 2006, 99, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, K.; Jian, M.Y.; Weber, D.S.; Alvarez, D.F.; Townsley, M.I.; Al-Mehdi, A.B.; King, J.A.; Liedtke, W.; Parker, J.C. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L923–L932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandri-Haber, N.; Dina, O.A.; Joseph, E.K.; Reichling, D.; Levine, J.D. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J. Neurosci. 2006, 26, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Vennekens, R.; Hoenderop, J.G.; Prenen, J.; Stuiver, M.; Willems, P.H.; Droogmans, G.; Nilius, B.; Bindels, R.J. Permeation and gating properties of the novel epithelial Ca2+ channel. J. Biol. Chem. 2000, 275, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Peng, J.B.; Hediger, M.A.; Clapham, D.E. CaT1 manifests the pore properties of the calcium-release-activated calcium channel. Nature 2001, 410, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Hoenderop, J.G.; Vennekens, R.; Müller, D.; Prenen, J.; Droogmans, G.; Bindels, R.J.; Nilius, B. Function and expression of the epithelial Ca2+ channel family: Comparison of mammalian ECaC1 and 2. J. Physiol. 2001, 537, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Lambers, T.T.; Weidema, A.F.; Nilius, B.; Hoenderop, J.G.; Bindels, R.J. Regulation of the mouse epithelial Ca2+ channel TRPV6 by the Ca2+-sensor calmodulin. J. Biol. Chem. 2004, 279, 28855–28861. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, B.A.; Bergs, C.; Wissenbach, U.; Flockerzi, V.; Trost, C. Competitive regulation of CaT-like-mediated Ca2+ entry by protein kinase C and calmodulin. Proc. Natl. Acad. Sci. USA 2001, 98, 3600–3605. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Vennekens, R.; Prenen, J.; Hoenderop, J.G.; Bindels, R.J.; Droogmans, G. Whole-cell and single channel monovalent cation currents through the novel rabbit epithelial Ca2+ channel ECaC. J. Physiol. 2000, 527 Pt 2, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Janssens, A.; Prenen, J.; Droogmans, G.; Nilius, B. Mg2+-dependent gating and strong inward rectification of the cation channel TRPV6. J. Gen. Physiol. 2003, 121, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cha, S.K.; Sun, T.J.; Huang, C.L. PIP2 activates TRPV5 and releases its inhibition by intracellular Mg2+. J. Gen. Physiol. 2005, 126, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Rohács, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Lukacs, V.; Rohacs, T. Hydrolysis of phosphatidylinositol 4,5-bisphosphate mediates calcium-induced inactivation of TRPV6 channels. J. Biol. Chem. 2008, 283, 14980–14987. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Benn, B.S.; Christakos, S.; Rohacs, T. Phospholipase C-mediated regulation of transient receptor potential vanilloid 6 channels: Implications in active intestinal Ca2+ transport. Mol. Pharmacol. 2009, 75, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Hoenderop, J.G.; van der Kemp, A.W.; Hartog, A.; van de Graaf, S.F.; van Os, C.H.; Willems, P.H.; Bindels, R.J. Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J. Biol. Chem. 1999, 274, 8375–8378. [Google Scholar] [CrossRef] [PubMed]
- Hoenderop, J.G.; van Leeuwen, J.P.; van der Eerden, B.C.; Kersten, F.F.; van der Kemp, A.W.; Mérillat, A.M.; Waarsing, J.H.; Rossier, B.C.; Vallon, V.; Hummler, E.; et al. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J. Clin. Investig. 2003, 112, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Van der Eerden, B.C.; Hoenderop, J.G.; de Vries, T.J.; Schoenmaker, T.; Buurman, C.J.; Uitterlinden, A.G.; Pols, H.A.; Bindels, R.J.; van Leeuwen, J.P. The epithelial Ca2+ channel TRPV5 is essential for proper osteoclastic bone resorption. Proc. Natl. Acad. Sci. USA 2005, 102, 17507–17512. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.B.; Chen, X.Z.; Berger, U.V.; Weremowicz, S.; Morton, C.C.; Vassilev, P.M.; Brown, E.M.; Hediger, M.A. Human calcium transport protein CaT1. Biochem. Biophys. Res. Commun. 2000, 278, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hirnet, D.; Olausson, J.; Fecher-Trost, C.; Bödding, M.; Nastainczyk, W.; Wissenbach, U.; Flockerzi, V.; Freichel, M. The TRPV6 gene, cDNA and protein. Cell Calcium 2003, 33, 509–518. [Google Scholar] [CrossRef]
- Peng, J.B.; Chen, X.Z.; Berger, U.V.; Vassilev, P.M.; Tsukaguchi, H.; Brown, E.M.; Hediger, M.A. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J. Biol. Chem. 1999, 274, 22739–22746. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Peng, J.B.; Tou, L.; Takanaga, H.; Adam, R.M.; Hediger, M.A.; Freeman, M.R. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab. Investig. 2002, 82, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, T.; Hoenderop, J.G.; van der Kemp, A.W.; Bindels, R.J. Localization and regulation of the epithelial Ca2+ channel TRPV6 in the kidney. J. Am. Soc. Nephrol. 2003, 14, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kovacs, C.S.; Takanaga, H.; Peng, J.B.; Landowski, C.P.; Hediger, M.A. Calcium channel TRPV6 is involved in murine maternal-fetal calcium transport. J. Bone Min. Res. 2008, 23, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, E.S.; Vong, C.T.; Quek, S.; Cheong, J.; Awal, S.; Gentry, C.; Aubdool, A.A.; Liang, L.; Bodkin, J.V.; Bevan, S.; et al. Superoxide generation and leukocyte accumulation: Key elements in the mediation of leukotriene B4-induced itch by transient receptor potential ankyrin 1 and transient receptor potential vanilloid 1. FASEB J. 2013, 27, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Sisignano, M.; Park, C.K.; Angioni, C.; Zhang, D.D.; von Hehn, C.; Cobos, E.J.; Ghasemlou, N.; Xu, Z.Z.; Kumaran, V.; Lu, R.; et al. 5,6-EET is released upon neuronal activity and induces mechanical pain hypersensitivity via TRPA1 on central afferent terminals. J. Neurosci. 2012, 32, 6364–6372. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Craib, S.J.; Stevenson, L.A.; Pertwee, R.G.; Henderson, A.; Toole, J.; Ellington, H.C. Pharmacological characterization of the anandamide cyclooxygenase metabolite: Prostaglandin E2 ethanolamide. J. Pharmacol. Exp. Ther. 2002, 301, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ives, D.; Ramesha, C.S. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 1997, 272, 21181–21186. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Yamamoto, K.; Yamamoto, S.; Tokunaga, T.; Shirakawa, E.; Shinkai, H.; Ogawa, M.; Sato, T.; Kudo, I.; Inoue, K.; et al. Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim. Biophys. Acta 1995, 1254, 127–134. [Google Scholar] [CrossRef]
- Ueda, N.; Kurahashi, Y.; Yamamoto, K.; Yamamoto, S.; Tokunaga, T. Enzymes for anandamide biosynthesis and metabolism. J. Lipid Mediat. Cell Signal. 1996, 14, 57–61. [Google Scholar] [CrossRef]
- Craib, S.J.; Ellington, H.C.; Pertwee, R.G.; Ross, R.A. A possible role of lipoxygenases in the activation of vanilloid receptors by anandamide in the guinea-pig bronchus. Br. J. Pharmacol. 2001, 134, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Smart, D.; Gunthorpe, M.J.; Jerman, J.C.; Nasir, S.; Gray, J.; Muir, A.I.; Chambers, J.K.; Randall, A.D.; Davis, J.B. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br. J. Pharmacol. 2000, 129, 227–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsson, S.O.; Wallin, T.; Fowler, C.J. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 2001, 299, 951–959. [Google Scholar] [PubMed]
- Sarker, K.P.; Obara, S.; Nakata, M.; Kitajima, I.; Maruyama, I. Anandamide induces apoptosis of PC-12 cells: Involvement of superoxide and caspase-3. FEBS Lett. 2000, 472, 39–44. [Google Scholar] [CrossRef]
- Maccarrone, M.; Lorenzon, T.; Bari, M.; Melino, G.; Finazzi-Agro, A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000, 275, 31938–31945. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Wang, X.H.; Wang, H.P.; Hu, L.Q.; Zheng, X.M.; Li, S.W. Capsaicin mediates cell death in bladder cancer T24 cells through reactive oxygen species production and mitochondrial depolarization. Urology 2010, 75, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, L.; Westlund, K.N. Reactive oxygen species mediate TNFR1 increase after TRPV1 activation in mouse DRG neurons. Mol. Pain 2009, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.; Dias, J.P.; Lahjouji, K.; Bogo, M.R.; Campos, M.M.; Gaudreau, P.; Couture, R. Activation of TRPV1 by capsaicin induces functional kinin B(1) receptor in rat spinal cord microglia. J. Neuroinflamm. 2012, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimer Dis. 2010, 20 (Suppl. 2), S413–S426. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Teshima, Y.; Takahashi, N.; Thuc, L.C.; Saito, S.; Fukui, A.; Kume, O.; Fukunaga, N.; Hara, M.; Nakagawa, M.; et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J. Mol. Cell. Cardiol. 2012, 52, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.; Gratton, J.P.; Rafikov, R.; Black, S.M.; Fulton, D.J. Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Mol. Pharmacol. 2011, 80, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, E.; Naito, Y.; Handa, O.; Okada, H.; Mizushima, K.; Hirai, Y.; Nakabe, N.; Uchiyama, K.; Ishikawa, T.; Takagi, T.; et al. Oxidative stress-induced posttranslational modification of TRPV1 expressed in esophageal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G230–G238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Inoue, R.; Morii, T.; Takahashi, N.; Yamamoto, S.; Hara, Y.; Tominaga, M.; Shimizu, S.; Sato, Y.; Mori, Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006, 2, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Mori, Y. TRP Channels as Sensors and Signal Integrators of Redox Status Changes. Front. Pharmacol. 2011, 2, 58. [Google Scholar] [CrossRef] [PubMed]
- Susankova, K.; Tousova, K.; Vyklicky, L.; Teisinger, J.; Vlachova, V. Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol. Pharmacol. 2006, 70, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Clark, T.E.; McAlexander, M.A.; Nassenstein, C.; Sheardown, S.A.; Wilson, S.; Thornton, J.; Carr, M.J.; Undem, B.J. Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J. Physiol. 2008, 586, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Zhuang, Z.; Sang, H.; Wu, Z.; Meng, R.; He, E.Y.; Scott, G.I.; Maris, J.R.; Li, R.; Ren, J. α,β-Unsaturated aldehyde crotonaldehyde triggers cardiomyocyte contractile dysfunction: Role of TRPV1 and mitochondrial function. Pharmacol. Res. 2014, 82, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Goto, S.; Matsunaga, T.; Nakajima, T.; Awata, T.; Hokari, S.; Komoda, T.; Katayama, S. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism 2001, 50, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Girnun, G.D.; Domann, F.E.; Moore, S.A.; Robbins, M.E. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 2002, 16, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Diep, Q.N.; Amiri, F.; Touyz, R.M.; Cohn, J.S.; Endemann, D.; Neves, M.F.; Schiffrin, E.L. PPARalpha activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension 2002, 40, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47 in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.A.; Morgese, M.G.; Pisanu, A.; Macheda, T.; Paquette, M.A.; Seillier, A.; Cassano, T.; Carta, A.R.; Giuffrida, A. Activation of PPAR gamma receptors reduces levodopa-induced dyskinesias in 6-OHDA-lesioned rats. Neurobiol. Dis. 2015, 74, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impellizzeri, D.; Esposito, E.; Attley, J.; Cuzzocrea, S. Targeting inflammation: New therapeutic approaches in chronic kidney disease (CKD). Pharmacol. Res. 2014, 81, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Impellizzeri, D.; Mondello, P.; Velardi, E.; Aloisi, C.; Cappellani, A.; Esposito, E.; Cuzzocrea, S. Palmitoylethanolamide reduces early renal dysfunction and injury caused by experimental ischemia and reperfusion in mice. Shock 2012, 38, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Mugnaini, E.; Ceru, M.P. Immunocytochemical localization of catalase in the central nervous system of the rat. J. HistoChem. Cytochem. 1995, 43, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Nardacci, R.; Ceru, M.P. Regional and ultrastructural immunolocalization of copper-zinc superoxide dismutase in rat central nervous system. J. Histochem. Cytochem. 1997, 45, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Farioli-Vecchioli, S.; Moreno, S.; Ceru, M.P. lmmunocytochemical localization of acyl-CoA oxidase in the rat central nervous system. J. Neurocytol. 2001, 30, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Poynter, M.E.; Daynes, R.A. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998, 273, 32833–32841. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Benedetti, E.; Cristiano, L.; Sebastiani, P.; D’Amico, M.A.; D’Angelo, B.; Di Loreto, S. Expression of peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RXRs) in rat cortical neurons. Neuroscience 2005, 130, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative Stress and Neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Butterfield, D.A. Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimer Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Sepe, S.; D’Amelio, M.; Bernardi, C.; Cristiano, L.; Cimini, A.; Cecconi, F.; Ceru’, M.P.; Moreno, S. Age-dependent roles of peroxisomes in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R.S.; Chapman, K.D.; Koulen, P. The neuroprotective properties of palmitoylethanolamine against oxidative stress in a neuronal cell line. Mol. Neurodegener. 2009, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, G.M.; Esposito, E.; Vitiello, S.; Iacono, A.; Santoro, A.; D’Agostino, G.; Sasso, O.; Russo, R.; Piazza, P.V.; Calignano, A.; et al. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in C6 Cells and primary astrocytes: Involvement of peroxisome-proliferator activated receptor-a. J. Neuroendocrinol. 2011, 23, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, C.; Russo, R.; De Caro, C.; Cristiano, C.; La Rana, G.; Piegari, G.; Paciello, O.; Citraro, R.; Russo, E.; De Sarro, G.; et al. Palmitoylethanolamide protects mice against 6-OHDA-induced neurotoxicity and endoplasmic reticulum stress: In vivo and in vitro evidence. Pharmacol. Res. 2016, 113, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Manea, S.A.; Todirita, A.; Albulescu, I.C.; Raicu, M.; Sasson, S.; Simionescu, M. High-glucose-increased expression and activation of NADPH oxidase in human vascular smooth muscle cells is mediated by 4-hydroxynonenal-activated PPARα and PPARβ/δ. Cell Tissue Res. 2015, 361, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.; Abdulrazzaq, G.; Chan, S.L.F.; Penman, J.; Harvey, J.; Alexander, S.P.H. Cannabinoid Receptor-Related Orphan G Protein-Coupled Receptors. Adv. Pharmacol. 2017, 80, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Muraoka, A.; Yang, Y.K.; Samuelson, L.C.; Zimmerman, E.M.; Cook, H.; Yamada, T. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 1997, 42, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, V.B.; Puhl, H.L., 3rd; Ikeda, S.R. N-Arachidonyl glycine does not activate G protein-coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol. 2013, 83, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.B.; Joseph, W.R.; Grimsey, N.L.; Glass, M. GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N-Arachidonoyl Glycine. PeerJ 2016, 4, e1835. [Google Scholar] [CrossRef] [PubMed]
- Penumarti, A.; Abdel-Rahman, A.A. The novel endocannabinoid receptor GPR18 is expressed in the rostral ventrolateral medulla and exerts tonic restraining influence on blood pressure. J. Pharmacol. Exp. Ther. 2014, 349, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Matouk, A.I.; Taye, A.; El-Moselhy, M.A.; Heeba, G.H.; Abdel-Rahman, A.A. The Effect of Chronic Activation of the Novel Endocannabinoid Receptor GPR18 on Myocardial Function and Blood Pressure in Conscious Rats. J. Cardiovasc. Pharmacol. 2017, 69, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Romero-Zerbo, S.Y.; Rafacho, A.; Díaz-Arteaga, A.; Suárez, J.; Quesada, I.; Imbernon, M.; Ross, R.A.; Dieguez, C.; Rodríguez de Fonseca, F.; Nogueiras, R.; et al. A role for the putative cannabinoid receptor GPR55 in the islets of Langerhans. J. Endocrinol. 2011, 211, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, L.S.; Ryberg, E.; Sims, N.A.; Ridge, S.A.; Mackie, K.; Greasley, P.J.; Ross, R.A.; Rogers, M.J. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 16511–16516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietr, M.; Kozela, E.; Levy, R.; Rimmerman, N.; Lin, Y.H.; Stella, N.; Vogel, Z.; Juknat, A. Differential changes in GPR55 during microglial cell activation. FEBS Lett. 2009, 583, 2071–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeck-Weiermair, M.; Zoratti, C.; Osibow, K.; Balenga, N.; Goessnitzer, E.; Waldhoer, M.; Malli, R.; Graier, W.F. Integrin clustering enables anandamide-induced Ca2+ signaling in endothelial cells via GPR55 by protection against CB1-receptor-triggered repression. J. Cell Sci. 2008, 121, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Kapur, A.; Zhao, P.; Sharir, H.; Bai, Y.; Caron, M.G.; Barak, L.S.; Abood, M.E. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J. Biol. Chem. 2009, 284, 29817–29827. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Balenga, N.A.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Toshida, T.; Maruyama, K.; Nakajima, K.; Yamashita, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem. 2009, 145, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, A.; Waldeck-Weiermair, M.; Naghdi, S.; Poteser, M.; Malli, R.; Graier, W.F. GPR55-dependent and -independent ion signalling in response to lysophosphatidylinositol in endothelial cells. Br. J. Pharmacol. 2010, 161, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Pérez-Gómez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB2-GPR55 receptor heteromers modulates cancer cell signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Höglund, P.J.; Gloriam, D.E.; Lagerström, M.C.; Schiöth, H.B. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003, 554, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Soga, T.; Ohishi, T.; Matsui, T.; Saito, T.; Matsumoto, M.; Takasaki, J.; Matsumoto, S.; Kamohara, M.; Hiyama, H.; Yoshida, S.; et al. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem. Biophys. Res. Commun. 2005, 326, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Inoue, H.; Kawakami, S.; Miyawaki, K.; Miyamoto, T.; Mizuta, K.; Itakura, M. Expression and distribution of Gpr119 in the pancreatic islets of mice and rats: Predominant localization in pancreatic polypeptide-secreting PP-cells. Biochem. Biophys. Res. Commun. 2006, 351, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.L.; Carroll, C.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Pedraza, M.; Mondala, H.; Gao, H.; Bagnol, D.; et al. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic Peptide release. Endocrinology 2008, 149, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, L.M.; Iakoubov, R.; Brubaker, P.L. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 2009, 58, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Kim, H.S.; Im, J.H.; Kim, J.W.; Jun, D.W.; Lim, S.C.; Lee, K.; Choi, J.M.; Kim, S.K.; Kang, K.W. GPR119: A promising target for nonalcoholic fatty liver disease. FASEB J. 2016, 30, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Cvijanovic, N.; Isaacs, N.J.; Rayner, C.K.; Feinle-Bisset, C.; Young, R.L.; Little, T.J. Duodenal fatty acid sensor and transporter expression following acute fat exposure in healthy lean humans. Clin. Nutr. 2017, 36, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.L.; Carroll, C.; Chen, R.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Xing, C.; Sebring, K.; Zhou, J.; et al. N-oleoyldopamine enhances glucose homeostasis through the activation of GPR119. Mol. Endocrinol. 2010, 24, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Rosenkilde, M.M.; Knop, F.K.; Wellner, N.; Diep, T.A.; Rehfeld, J.F.; Andersen, U.B.; Holst, J.J.; Hansen, H.S. 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J. Clin. Endocrinol. Metab. 2011, 96, E1409–E1417. [Google Scholar] [CrossRef] [PubMed]
- Balenga, N.A.; Aflaki, E.; Kargl, J.; Platzer, W.; Schröder, R.; Blättermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurchiù, V.; Lanuti, M.; De Bardi, M.; Battistini, L.; Maccarrone, M. The differential characterization of GPR55 receptor in human peripheral blood reveals a distinctive expression in monocytes and NK cells and a proinflammatory role in these innate cells. Int. Immunol. 2015, 27, 153–160. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, J.V.; Bannister, W.H.; Rotilio, G. Aspects of the structure, function, and applications of superoxide dismutase. CRC Crit. Rev. Biochem. 1987, 22, 111–180. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 1999, 27, 951–965. [Google Scholar] [CrossRef]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Palace, V.P.; Khaper, N.; Qin, Q.; Singal, P.K. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radic. Biol. Med. 1999, 26, 746–761. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- McCay, P.B. Vitamin E: Interactions with free radicals and ascorbate. Annu. Rev. Nutr. 1985, 5, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Paiva, S.A.; Russell, R.M. Beta-carotene and other carotenoids as antioxidants. J. Am. Coll. Nutr. 1999, 18, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; Salzet, M.; Cassano, T. Oxidative Stress in Aging Brain: Nutritional and Pharmacological Interventions for Neurodegenerative Disorders. Oxid. Med. Cell. Longev. 2018, 2018, 3416028. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Sandoval-Acuña, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, A.; Bristow, R.G.; Venkateswaran, V. Ingestion of selenium and other antioxidants during prostate cancer radiotherapy: A good thing? Cancer Treat. Rev. 2010, 36, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B.; Beck, F.W.; Kucuk, O.; Sarkar, F.H. Antioxidant effect of zinc in humans. Free Radic. Biol. Med. 2004, 37, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.M.; Berselli, P.; Zava, S.; Montorfano, G.; Negroni, M.; Corsetto, P.; Berra, B. Endogenous antioxidants and radical scavengers. Adv. Exp. Med. Biol. 2010, 698, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Poeggeler, B.; Menendez-Pelaez, A.; Chen, L.D.; Saarela, S. Melatonin as a free radical scavenger: Implications for aging and age-related diseases. Ann. N. Y. Acad. Sci. 1994, 719, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Traber, M.G. Vitamin E: Antioxidant activity, biokinetics, and bioavailability. Annu. Rev. Nutr. 1990, 10, 357–382. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Shannon, J.; Frei, B.; Kaye, J.A.; Quinn, J.F. Uric acid as a CNS antioxidant. J. Alzheimer Dis. 2010, 19, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Cotelle, N. Role of flavonoids in oxidative stress. Curr. Top. Med. Chem. 2001, 1, 569–590. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Moreno, J.J. Role of Ca2+-Independent Phospholipase A2 on Arachidonic Acid Release Induced by Reactive Oxygen Species. Arch. Biochem. Biophys. 2001, 392, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Bátkai, S.; Rajesh, M.; Mukhopadhyay, P.; Haskó, G.; Liaudet, L.; Cravatt, B.F.; Csiszár, A.; Ungvári, Z.; Pacher, P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H909–H918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Wang, X.; Wang, L. Presence and regulation of cannabinoid receptors in human retinal pigment epithelial cells. Mol. Vis. 2009, 15, 1243–1251. [Google Scholar] [PubMed]
- Wang, M.; Abais, J.M.; Meng, N.; Zhang, Y.; Ritter, J.K.; Li, P.L.; Tang, W.X. Upregulation of cannabinoid receptor-1 and fibrotic activation of mouse hepatic stellate cells during Schistosoma J. infection: Role of NADPH oxidase. Free Radic. Biol. Med. 2014, 71, 109–120. [Google Scholar] [CrossRef] [PubMed]
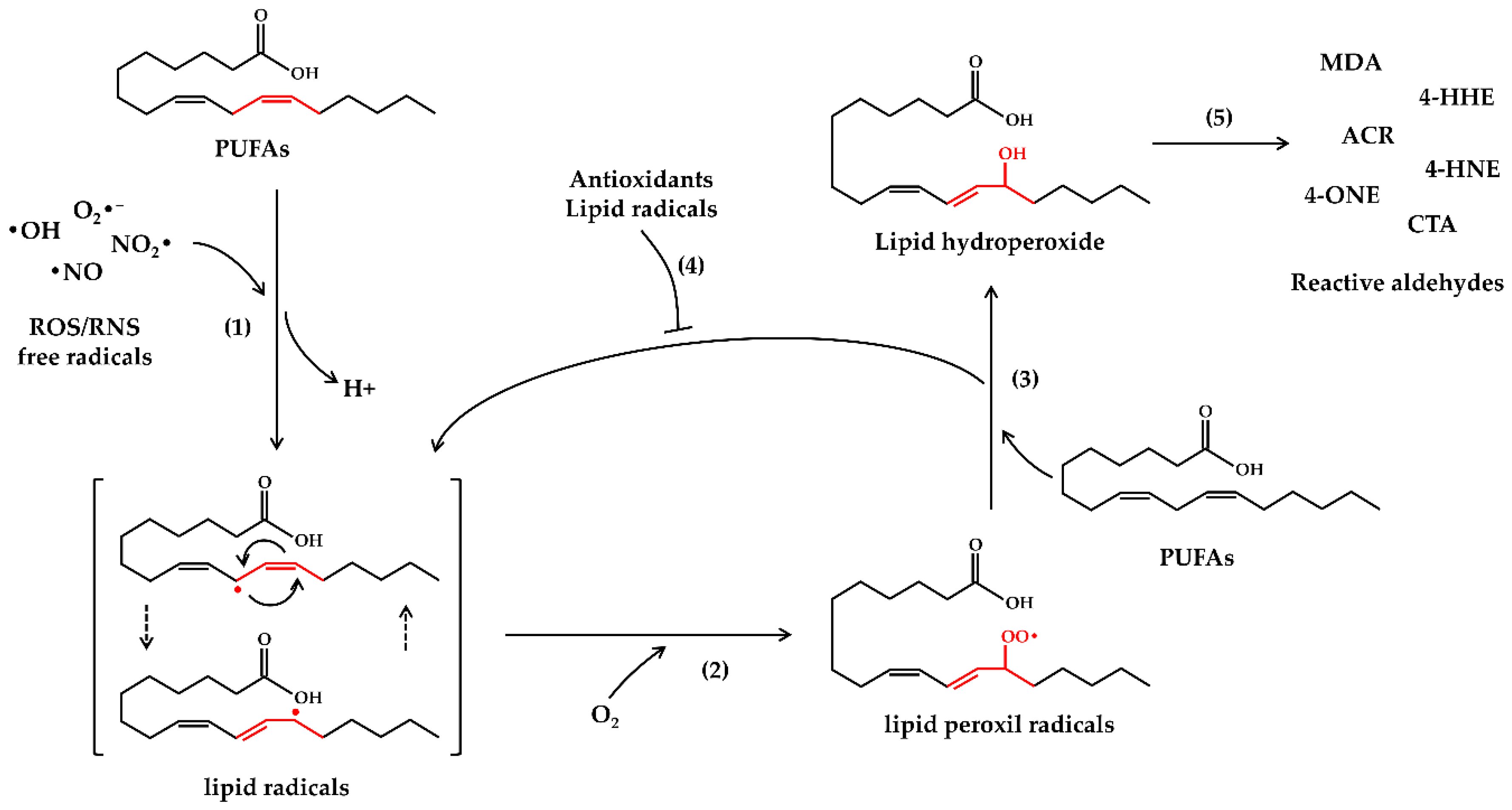
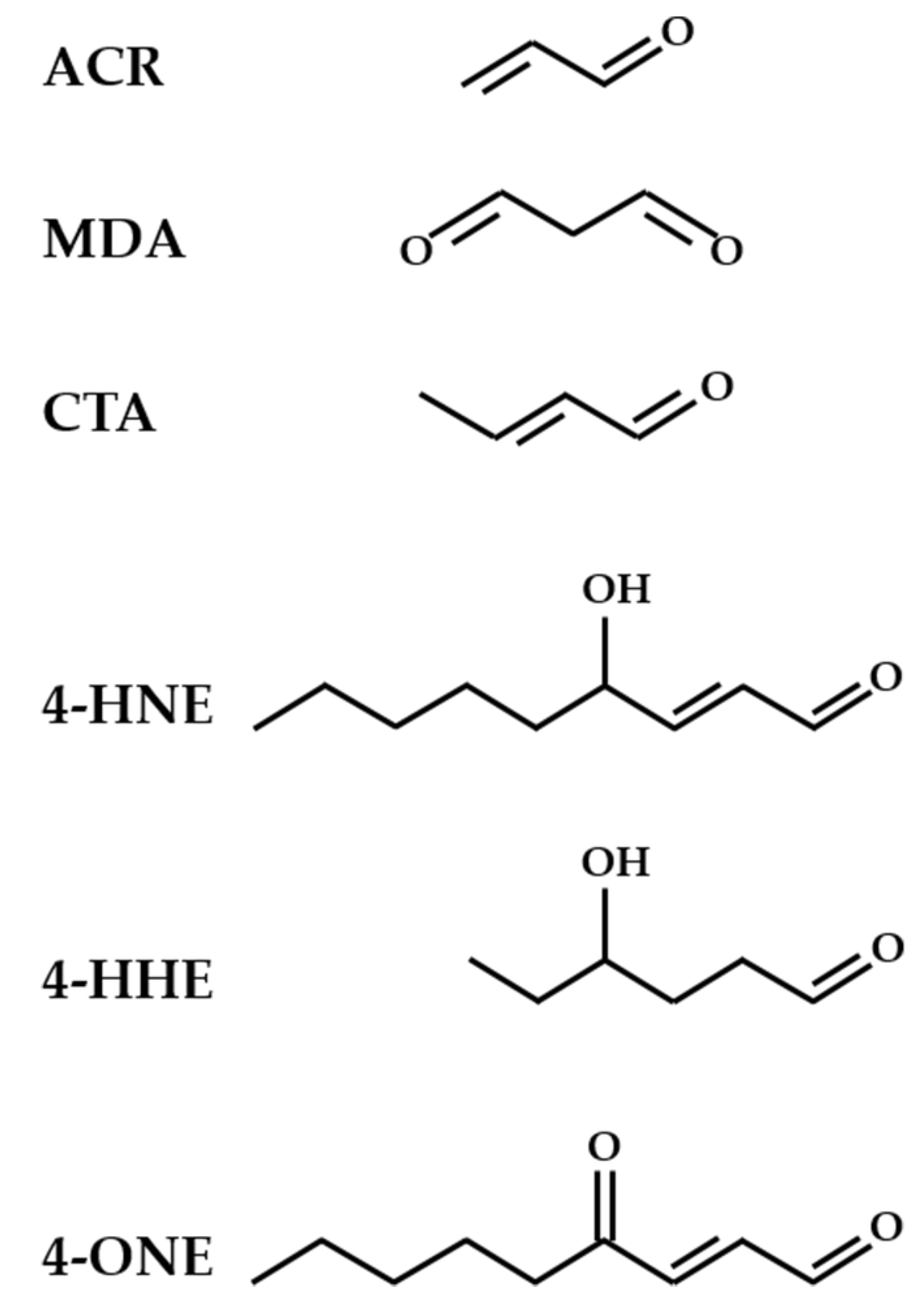
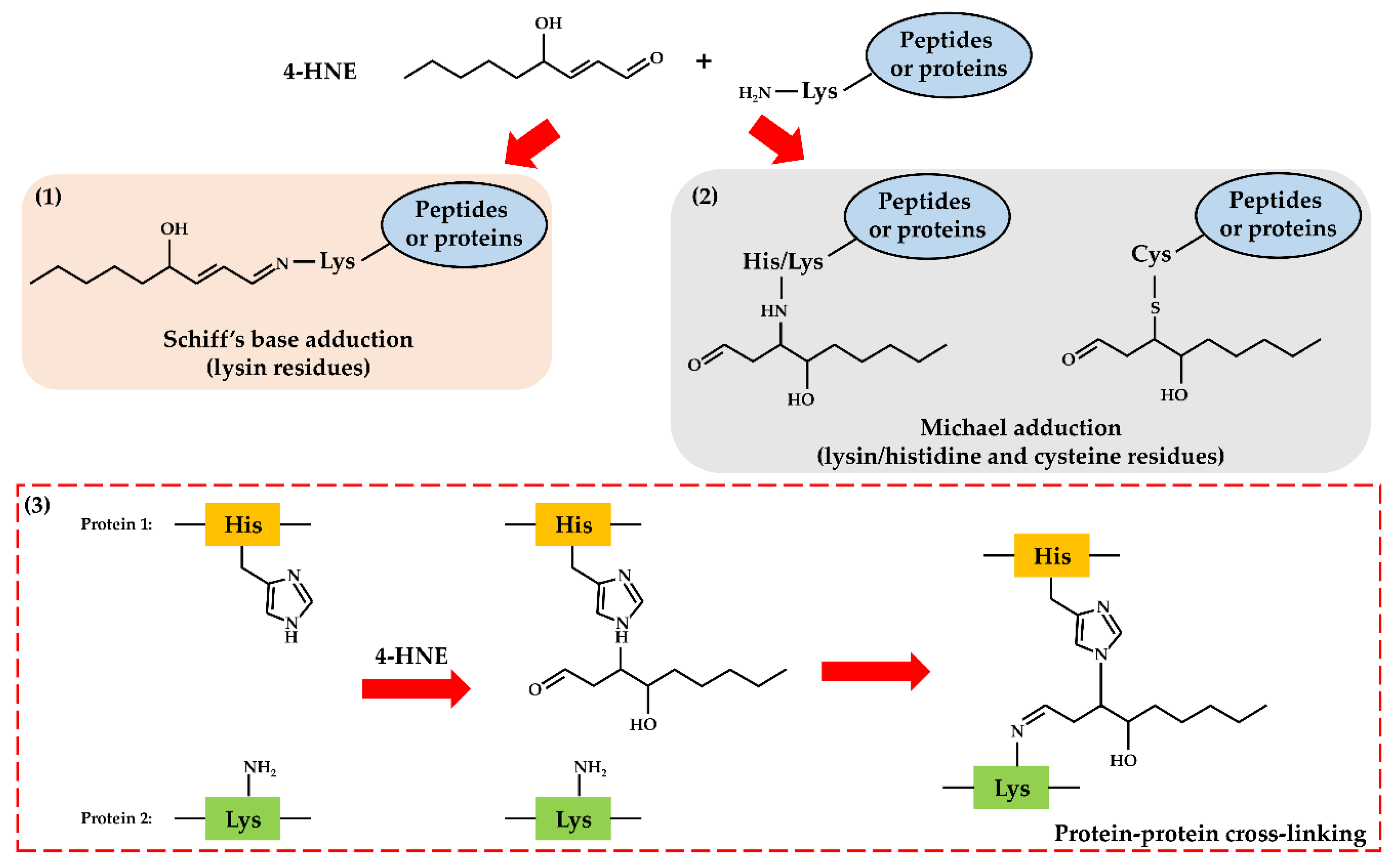
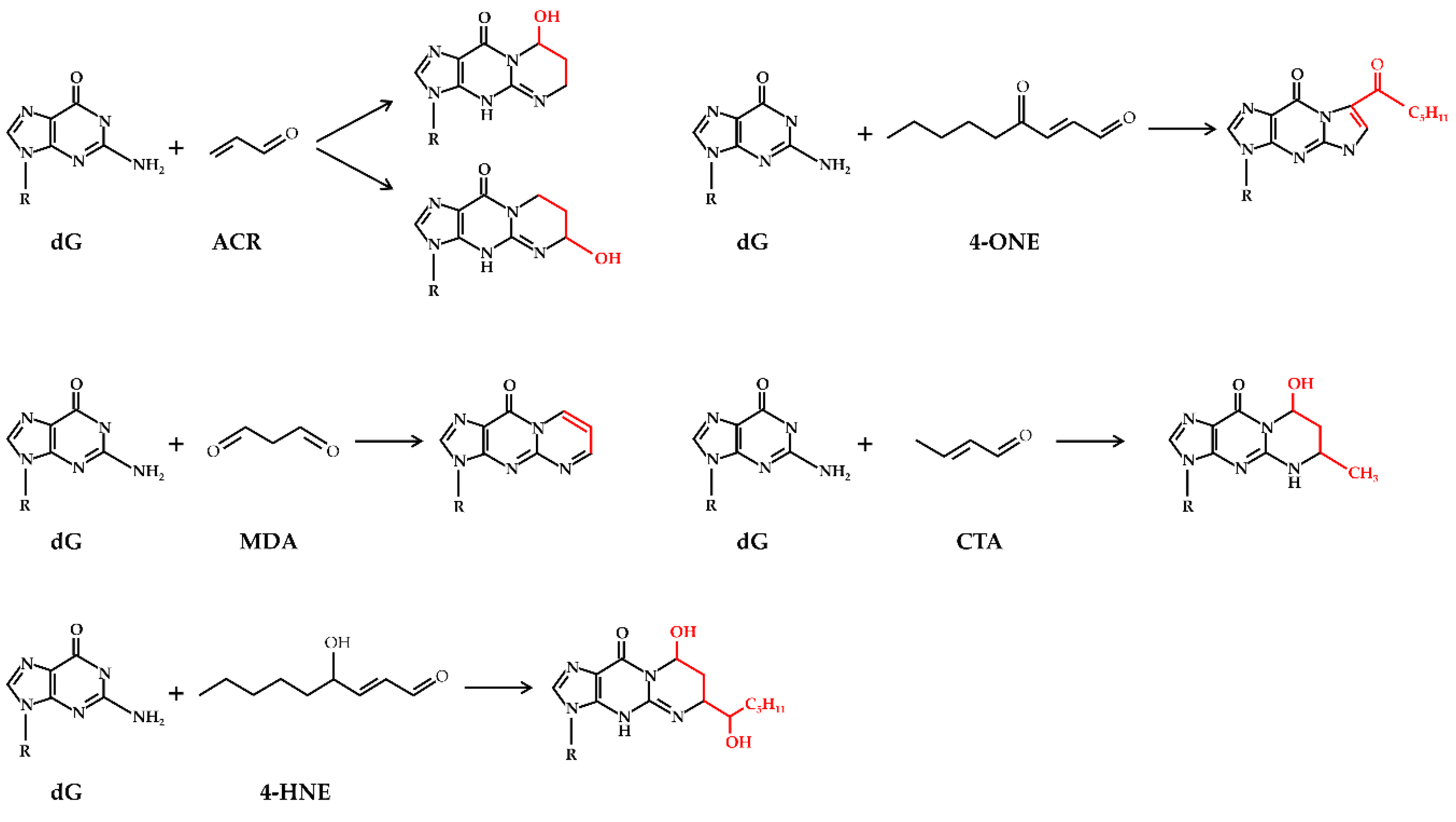
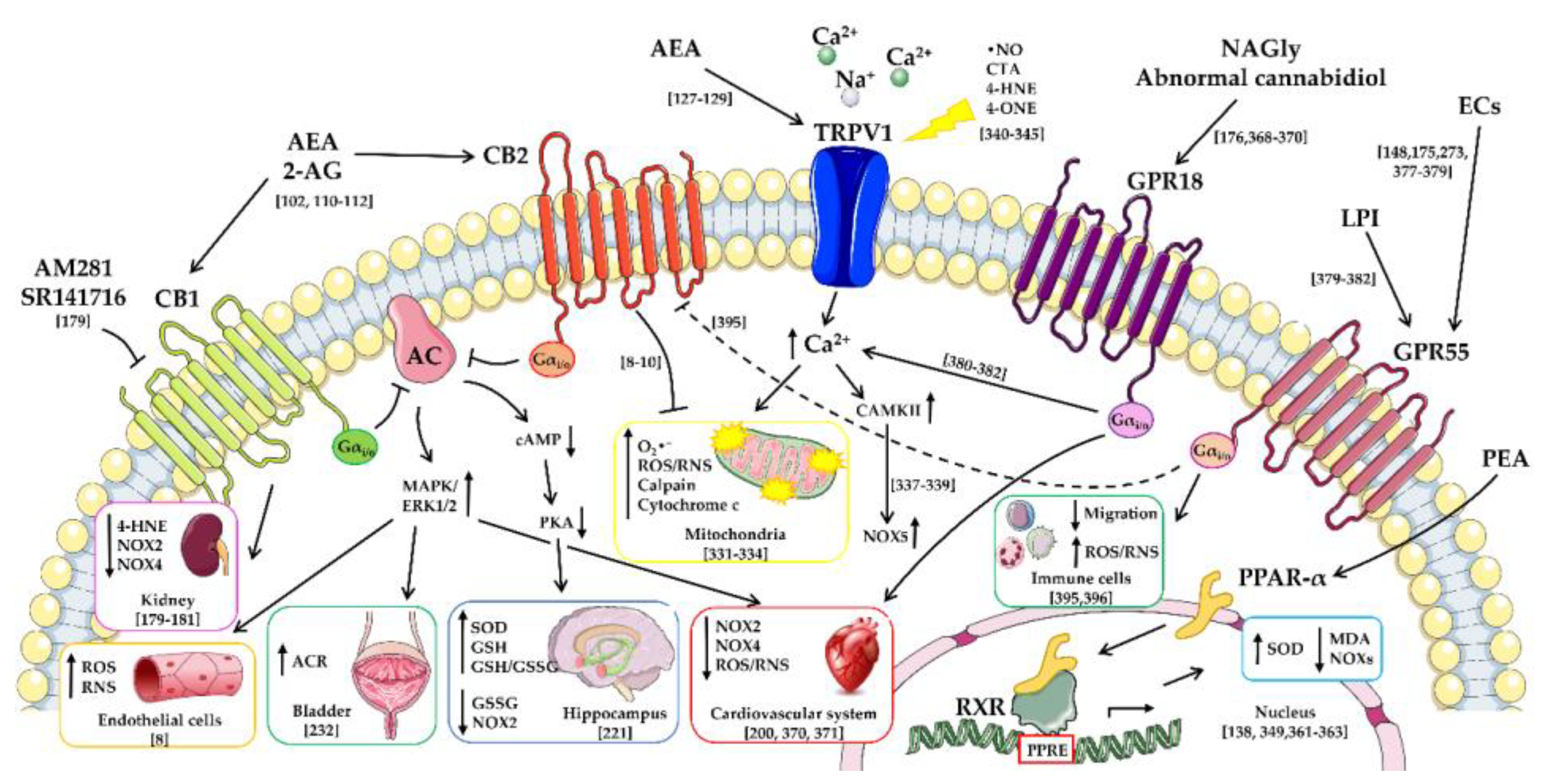
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallelli, C.A.; Calcagnini, S.; Romano, A.; Koczwara, J.B.; De Ceglia, M.; Dante, D.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Modulation of the Oxidative Stress and Lipid Peroxidation by Endocannabinoids and Their Lipid Analogues. Antioxidants 2018, 7, 93. https://doi.org/10.3390/antiox7070093
Gallelli CA, Calcagnini S, Romano A, Koczwara JB, De Ceglia M, Dante D, Villani R, Giudetti AM, Cassano T, Gaetani S. Modulation of the Oxidative Stress and Lipid Peroxidation by Endocannabinoids and Their Lipid Analogues. Antioxidants. 2018; 7(7):93. https://doi.org/10.3390/antiox7070093
Chicago/Turabian StyleGallelli, Cristina Anna, Silvio Calcagnini, Adele Romano, Justyna Barbara Koczwara, Marialuisa De Ceglia, Donatella Dante, Rosanna Villani, Anna Maria Giudetti, Tommaso Cassano, and Silvana Gaetani. 2018. "Modulation of the Oxidative Stress and Lipid Peroxidation by Endocannabinoids and Their Lipid Analogues" Antioxidants 7, no. 7: 93. https://doi.org/10.3390/antiox7070093
APA StyleGallelli, C. A., Calcagnini, S., Romano, A., Koczwara, J. B., De Ceglia, M., Dante, D., Villani, R., Giudetti, A. M., Cassano, T., & Gaetani, S. (2018). Modulation of the Oxidative Stress and Lipid Peroxidation by Endocannabinoids and Their Lipid Analogues. Antioxidants, 7(7), 93. https://doi.org/10.3390/antiox7070093