The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling


Abstract
1. Introduction
2. Pulmonary Airway and Vascular Remodeling
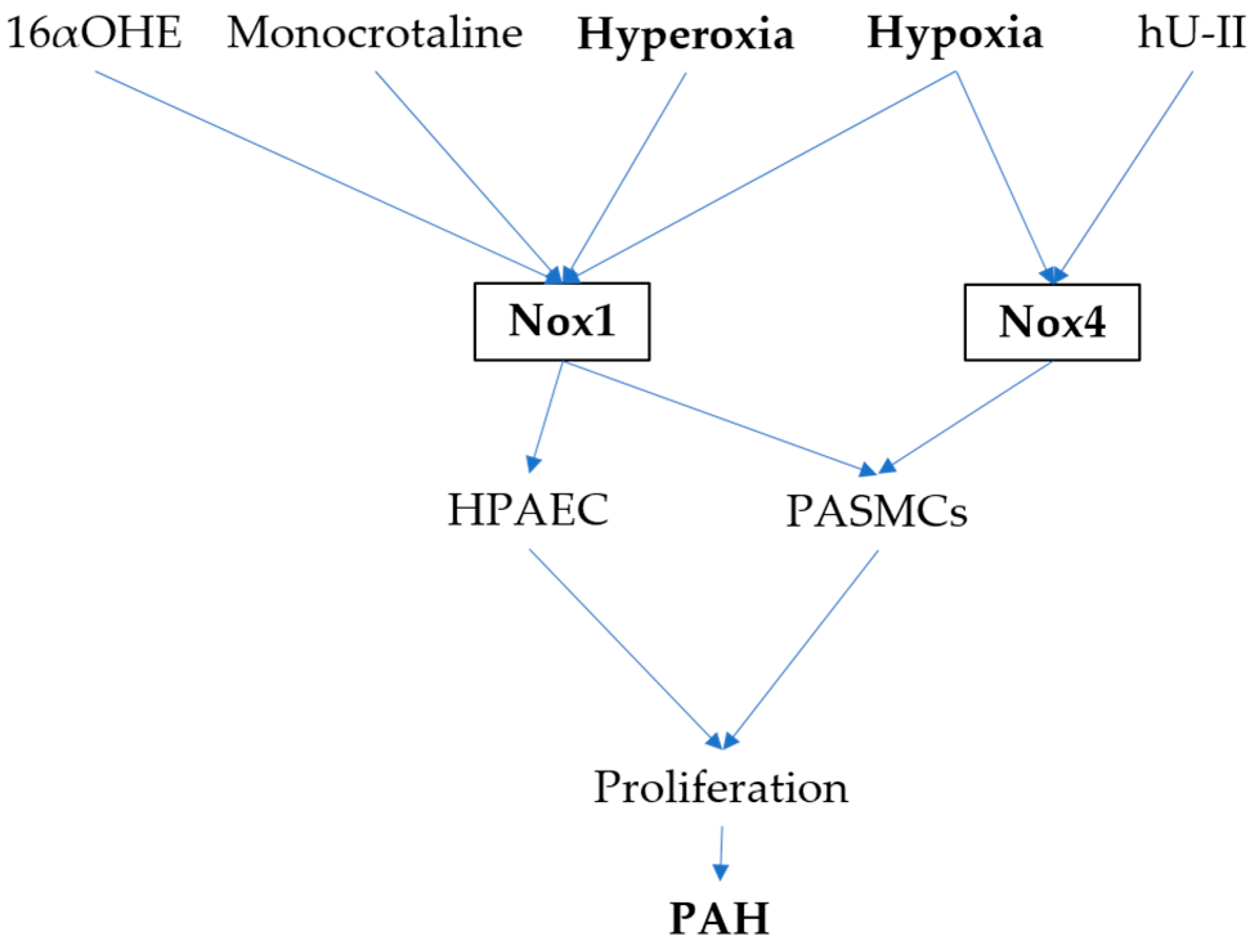
3. NADPH Oxidases and PAH
4. NADPH Oxidases and COPD
5. NADPH Oxidases and Asthma
6. NADPH Oxidases in Neonatal BPD
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khan, M.A.; Assiri, A.M.; Broering, D.C. Complement mediators: Key regulators of airway tissue remodeling in asthma. J. Transl. Med. 2015, 13, 272. [Google Scholar] [CrossRef] [PubMed]
- Al-Muhsen, S.; Johnson, J.R.; Hamid, Q. Remodeling in asthma. J. Allergy Clin. Immunol. 2011, 128, 451–462; quiz 463–454. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Laurie, S.S. Vascular remodeling in pulmonary hypertension. J. Mol. Med. (Berl.) 2013, 91, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J.R.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Domej, W.; Oettl, K.; Renner, W. Oxidative stress and free radicals in COPD–implications and relevance for treatment. Int. J. Chronic Obstruct. Pulm. Dis. 2014, 9, 1207–1224. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, S.; Nolin, J.; McMillan, D.; Wouters, E.; Janssen-Heininger, Y.; Reynaert, N. Thiol redox chemistry: Role of protein cysteine oxidation and altered redox homeostasis in allergic inflammation and asthma. J. Cell. Biochem. 2015, 116, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Leto, T.L. The Nox family of NAD(P)H oxidases: Host defense and beyond. J. Biol. Chem. 2004, 279, 51715–51718. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Terada, L.S. Specificity in reactive oxidant signaling: Think globally, act locally. J. Cell Biol. 2006, 174, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.D.; Hancock, J.T.; Morice, A.H. NADPH oxidase: A universal oxygen sensor? Free Radic. Biol. Med. 2000, 29, 416–424. [Google Scholar] [CrossRef]
- Royce, S.G.; Li, X.; Tortorella, S.; Goodings, L.; Chow, B.S.; Giraud, A.S.; Tang, M.L.; Samuel, C.S. Mechanistic insights into the contribution of epithelial damage to airway remodeling. Novel therapeutic targets for asthma. Am. J. Respir. Cell Mol. Biol. 2014, 50, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Holloway, J.; Wilson, S.; Bucchieri, F.; Puddicombe, S.; Davies, D.E. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc. Am. Thorac. Soc. 2004, 1, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.E.; Polosa, R.; Puddicombe, S.M.; Richter, A.; Holgate, S.T. The epidermal growth factor receptor and its ligand family: Their potential role in repair and remodelling in asthma. Allergy 1999, 54, 771–783. [Google Scholar] [PubMed]
- Demoly, P.; Simony-Lafontaine, J.; Chanez, P.; Pujol, J.L.; Lequeux, N.; Michel, F.B.; Bousquet, J. Cell proliferation in the bronchial mucosa of asthmatics and chronic bronchitics. Am. J. Respir. Crit. Care Med. 1994, 150, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Barsky, S.H.; Roth, M.D.; Kleerup, E.C.; Simmons, M.; Tashkin, D.P. Histopathologic and molecular alterations in bronchial epithelium in habitual smokers of marijuana, cocaine and/or tobacco. J. Natl. Cancer Inst. 1998, 90, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Vignola, A.M.; Chanez, P.; Chiappara, G.; Merendino, A.; Pace, E.; Rizzo, A.; la Rocca, A.M.; Bellia, V.; Bonsignore, G.; Bousquet, J. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1997, 156, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Lee, D.; Sung, Y.K. Prospect of vasoactive intestinal peptide therapy for COPD/PAH and asthma: A review. Respir. Res. 2011, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Shimoda, L.A. Lung Circulation. Compr. Physiol. 2016, 6, 897–943. [Google Scholar] [PubMed]
- Song, S.; Yamamura, A.; Yamamura, H.; Ayon, R.J.; Smith, K.A.; Tang, H.; Makino, A.; Yuan, J.X. Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2014, 307, C373–C383. [Google Scholar] [CrossRef] [PubMed]
- Papamatheakis, D.G.; Blood, A.B.; Kim, J.H.; Wilson, S.M. Antenatal hypoxia and pulmonary vascular function and remodeling. Curr. Vasc. Pharmacol. 2013, 11, 616–640. [Google Scholar] [CrossRef] [PubMed]
- Eickelberg, O.; Morty, R.E. Transforming growth factor beta/bone morphogenic protein signaling in pulmonary arterial hypertension: Remodeling revisited. Trends Cardiovasc. Med. 2007, 17, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, P.M.; Mouthon, L.; Barbera, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, growth factors and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 2009, 54 (Suppl. S1), S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Arcot, S.S.; Fagerland, J.A.; Lipke, D.W.; Gillespie, M.N.; Olson, J.W. Basic fibroblast growth factor alterations during development of monocrotaline-induced pulmonary hypertension in rats. Growth Factors 1995, 12, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.T.; Breen, E.C.; Fu, Z.; Mathieu-Costello, O.; West, J.B. Alveolar hypoxia increases gene expression of extracellular matrix proteins and platelet-derived growth factor-B in lung parenchyma. Am. J. Respir. Crit. Care Med. 1998, 158, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Katayose, D.; Ohe, M.; Yamauchi, K.; Ogata, M.; Shirato, K.; Fujita, H.; Shibahara, S.; Takishima, T. Increased expression of PDGF A- and B-chain genes in rat lungs with hypoxic pulmonary hypertension. Am. J. Physiol. 1993, 264 Pt 1, L100–L106. [Google Scholar] [PubMed]
- Perkett, E.A.; Badesch, D.B.; Roessler, M.K.; Stenmark, K.R.; Meyrick, B. Insulin-like growth factor I and pulmonary hypertension induced by continuous air embolization in sheep. Am. J. Respir. Cell Mol. Biol. 1992, 6, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; MacNee, W. Regulation of redox glutathione levels and gene transcription in lung inflammation: Therapeutic approaches. Free Radic. Biol. Med. 2000, 28, 1405–1420. [Google Scholar] [CrossRef]
- Rahman, I.; Mulier, B.; Gilmour, P.S.; Watchorn, T.; Donaldson, K.; Jeffery, P.K.; MacNee, W. Oxidant-mediated lung epithelial cell tolerance: The role of intracellular glutathione and nuclear factor-kB. Biochem. Pharmacol. 2001, 62, 787–794. [Google Scholar] [CrossRef]
- Lee, J.H.; Kagan, E. Role of nicotinamide adenine dinucleotide phosphate oxidase in mediating vesicant-induced interleukin-6 secretion in human airway epithelial cell. Am. J. Respir. Cell Mol. Biol. 2014, 50, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Valente, A.J. Nuclear factor B activation by NADPH oxidase. Mech. Ageing Dev. 2004, 125, 199–810. [Google Scholar] [CrossRef] [PubMed]
- Gabelloni, M.L.; Sabbione, F.; Jancic, C.; Fuxman Bass, J.; Keitelman, I.; Iula, L.; Oleastro, M.; Geffner, J.R.; Trevani, A.S. NADPH oxidase derive reactive oxygen species are involved in human neutrophil IL-1β secretion but not in inflammasome activation. Eur. J. Immunol. 2013, 43, 3324–3325. [Google Scholar] [CrossRef] [PubMed]
- Levenerence, J.T.; Medhora, M.; Konduri, G.G.; Sampath, V. Lipopolysaccharide-induced cytokine expression in alveolar epithelial cells: Role of PKCζ-mediated p47phox phosphorylation. Chem. Biol. Interact. 2011, 189, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2008, 118, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Metrailler-Ruchonnet, I.; Schappi, M.; Donati, Y.; Matthay, M.A.; Krause, K.H.; Barazzone Argiroffo, C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Berkelhamer, S.K.; Kim, G.A.; Radder, J.E.; Wedgwood, S.; Czech, L.; Steinhorn, R.H.; Schumacker, P.T. Developmental differences in hyperoxia-induced oxidative stress and cellular responses in the murine lung. Free Radic. Biol. Med. 2013, 61, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.E.; Aschner, J.L.; Milatovic, D.; Schmidt, J.W.; Aschner, M.; Kaplowitz, M.R.; Zhang, Y.; Fike, C.D. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L596–L607. [Google Scholar] [CrossRef] [PubMed]
- Veit, F.; Pak, O.; Egemnazarov, B.; Roth, M.; Kosanovic, D.; Seimetz, M.; Sommer, N.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; et al. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid. Redox Signal. 2013, 19, 2213–2231. [Google Scholar] [CrossRef] [PubMed]
- Kameshima, S.; Kazama, K.; Okada, M.; Yamawaki, H. Eukaryotic elongation factor 2 kinase mediates monocrotaline-induced pulmonary arterial hypertension via reactive oxygen species-dependent vascular remodeling. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1298–H1305. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16alpha-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Ghouleh, I.A.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; de Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial Nox1 Oxidase Assembly in Human Pulmonary Arterial Hypertension; Driver of Gremlin1-Medziated Proliferation. Clin. Sci. (Lond.) 2017, 131, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Ikami, K.; Matsuno, K.; Yamashita, T.; Shiba, D.; Ibi, M.; Matsumoto, M.; Katsuyama, M.; Cui, W.; Zhang, J.; et al. Deficiency of NOX1/nicotinamide adenine dinucleotide phosphate, reduced form oxidase leads to pulmonary vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.A.; Steinhorn, R.H.; Wedgwood, S.; Mata-Greenwood, E.; Roark, E.A.; Russell, J.A.; Black, S.M. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: A role for NADPH oxidase. Circ. Res. 2003, 92, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Fike, C.D.; Slaughter, J.C.; Kaplowitz, M.R.; Zhang, Y.; Aschner, J.L. Reactive oxygen species from NADPH oxidase contribute to altered pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L881–L888. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Lakshminrusimha, S.; Farrow, K.N.; Czech, L.; Gugino, S.F.; Soares, F.; Russell, J.A.; Steinhorn, R.H. Apocynin improves oxygenation and increases eNOS in persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L616–L626. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, T.; Peng, J.J.; Zhang, J.J.; Liu, W.Q.; Luo, X.J.; Ma, Q.L.; Gong, Z.C.; Peng, J. Non-muscle myosin light chain promotes endothelial progenitor cells senescence and dysfunction in pulmonary hypertensive rats through up-regulation of NADPH oxidase. Eur. J. Pharmacol. 2016, 775, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Luo, X.J.; Yang, Z.B.; Zhang, J.J.; Li, T.B.; Zhang, X.J.; Ma, Q.L.; Zhang, G.G.; Hu, C.P.; Peng, J. Inhibition of NOX/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 2014, 63, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, T.; BelAiba, R.S.; Bonello, S.; Pfeilschifter, J.; Hess, J.; Gorlach, A. Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Huecksteadt, T.P.; Norman, K.; Sanders, K.; Murphy, T.M.; Chitano, P.; Wilson, K.; Hoidal, J.R.; Kennedy, T.P. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation and hypertrophy in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1543–L1555. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef] [PubMed]
- Ge, A.; Ma, Y.; Liu, Y.N.; Li, Y.S.; Gu, H.; Zhang, J.X.; Wang, Q.X.; Zeng, X.N.; Huang, M. Diosmetin prevents TGF-beta1-induced epithelial-mesenchymal transition via ROS/MAPK signaling pathways. Life Sci. 2016, 153, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Hou, X.; Jourd’heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGFβ1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ. Res. 2010, 107, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C. Vascular smooth muscle growth: Autocrine growth mechanisms. Physiol. Rev. 2001, 81, 999–1030. [Google Scholar] [PubMed]
- Morrell, N.W.; Yang, X.; Upton, P.D.; Jourdan, K.B.; Morgan, N.; Sheares, K.K.; Trembath, R.C. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation 2001, 104, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Perkett, E.A.; Lyons, R.M.; Moses, H.L.; Brigham, K.L.; Meyrick, B. Transforming growth factor-beta activity in sheep lung lymph during the development of pulmonary hypertension. J. Clin. Investig. 1990, 86, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Sheares, K.K.; Jeffery, T.K.; Long, L.; Yang, X.; Morrell, N.W. Differential effects of TGF-beta1 and BMP-4 on the hypoxic induction of cyclooxygenase-2 in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L919–L927. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ambalavanan, N.; Liu, H.; Sun, Y.; Jhala, N.; Bradley, W.E.; Dell’Italia, L.J.; Michalek, S.; Wu, H.; Steele, C.; et al. TLR4 regulates pulmonary vascular homeostasis and remodeling via redox signaling. Front. Biosci. (Landmark Ed.) 2016, 21, 397–409. [Google Scholar] [PubMed]
- Lu, X.; Murphy, T.C.; Nanes, M.S.; Hart, C.M. PPARλ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L559–L566. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Marshall, N.A.; Barker, R.N.; Vickers, M.A. Immunomodulation against leukemias and lymphomas: A realistic future treatment? Crit. Rev. Oncol. Hematol. 2008, 65, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Fu, P.; Mohan, V.; Epshtein, Y.; Jacobson, J.R.; Gomez-Cambronero, J.; Wary, K.K.; Bindokas, V.; Dudek, S.M.; Salgia, R.; et al. Role of c-Met/phosphatidylinositol 3-kinase (PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation and motility of lung endothelial cells. J. Biol. Chem. 2014, 289, 13476–13491. [Google Scholar] [CrossRef] [PubMed]
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2 and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L337–L351. [Google Scholar] [PubMed]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Hunt, J.; Barnes, P.J.; Alving, K.; Antczak, A.; Baraldi, E.; Becher, G.; van Beurden, W.J.; Corradi, M.; Dekhuijzen, R.; et al. Exhaled breath condensate: Methodological recommendations and unresolved questions. Eur. Respir. J. 2005, 26, 523–548. [Google Scholar] [CrossRef] [PubMed]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154 Pt 1, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Kasielski, M.; Antczak, A.; Pietras, T.; Bialasiewicz, P. Increased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: No significant effect of cigarette smoking. Respir. Med. 1999, 93, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., 2nd. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.L.; Novelli, F.; Costa, F.; Malagrino, L.; Melosini, L.; Bacci, E.; Cianchetti, S.; Dente, F.L.; Di Franco, A.; Vagaggini, B.; et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediat. Inflamm. 2011, 2011, 891752. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, B.; Ma, A.; He, J.; Liu, X.; Chen, J. The Expression of NOX4 in Smooth Muscles of Small Airway Correlates with the Disease Severity of COPD. BioMed Res. Int. 2016, 2016, 2891810. [Google Scholar] [CrossRef] [PubMed]
- Van Eeden, S.F.; Sin, D.D. Oxidative stress in chronic obstructive pulmonary disease: A lung and systemic process. Can. Respir. J. 2013, 20, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Hollins, F.; Sutcliffe, A.; Gomez, E.; Berair, R.; Russell, R.; Szyndralewiez, C.; Saunders, R.; Brightling, C. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir. Res. 2016, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-beta regulates Nox4, MnSOD and catalase expression and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L295–L304. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, A.; Hollins, F.; Gomez, E.; Saunders, R.; Doe, C.; Cooke, M.; Challiss, R.A.; Brightling, C.E. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Kennedy, T.P.; Sturrock, A.B.; Huecksteadt, T.P.; Quinn, M.T.; Murphy, T.M.; Chitano, P.; Hoidal, J.R. NADPH oxidase promotes NF-κB activation and proliferation in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L782–L795. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Li, J.; Hodge, J.A.; Liu, P.T.; Vanden Hoek, T.L.; Becker, L.B.; Pestell, R.G.; Rosner, M.R.; Hershenson, M.B. Characterization of a Rac1 signaling pathway to cyclin D(1) expression in airway smooth muscle cells. J. Biol. Chem. 1999, 274, 22065–22071. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.; Lin, J.C.; Lahjouji, K.; Roy, J.P.; Senecal, J.; Morin, A.; Couture, R. Cigarette smoke-induced kinin B1 receptor promotes NADPH oxidase activity in cultured human alveolar epithelial cells. Peptides 2011, 32, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Ganesan, S.; Comstock, A.T.; Meldrum, C.A.; Mahidhara, R.; Goldsmith, A.M.; Curtis, J.L.; Martinez, F.J.; Hershenson, M.B.; Sajjan, U. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Betsuyaku, T.; Suzuki, M.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid. Redox Signal. 2008, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Bidan, C.M.; Veldsink, A.C.; Meurs, H.; Gosens, R. Airway and Extracellular Matrix Mechanics in COPD. Front. Physiol. 2015, 6, 346. [Google Scholar] [CrossRef] [PubMed]
- Black, P.N.; Ching, P.S.; Beaumont, B.; Ranasinghe, S.; Taylor, G.; Merrilees, M.J. Changes in elastic fibres in the small airways and alveoli in COPD. Eur. Respir. J. 2008, 31, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, O.; Nihlberg, K.; Dahlback, M.; Bjermer, L.; Eriksson, L.T.; Erjefalt, J.S.; Lofdahl, C.G.; Westergren-Thorsson, G. Altered fibroblast proteoglycan production in COPD. Respir. Res. 2010, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.D.; Kennett, E.C.; Whitelock, J.M.; Davies, M.J. Oxidative damage to extracellular matrix and its role in human pathologies. Free Radic. Biol. Med. 2008, 44, 1973–2001. [Google Scholar] [CrossRef] [PubMed]
- Barth, D.; Kyrieleis, O.; Frank, S.; Renner, C.; Moroder, L. The role of cystine knots in collagen folding and stability, part II. Conformational properties of (Pro-Hyp-Gly)n model trimers with N- and C-terminal collagen type III cystine knots. Chemistry 2003, 9, 3703–3714. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.S.; Chew, T.; Flaherty, K.R.; Frye, S.; Gibson, K.F.; Kaminski, N.; Klemsz, M.J.; Lange, W.; Noth, I.; Rothhaar, K. Oral immunotherapy with type V collagen in idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Voelkel, N.F. Molecular pathogenesis of emphysema. J. Clin. Investig. 2008, 118, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.C.; Krause, K.H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2015, 235, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Noguera, A.; Busquets, X.; Sauleda, J.; Villaverde, J.M.; MacNee, W.; Agusti, A.G. Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 158 Pt 1, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agusti, A.G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.J.; Brown, E.J. CR3 (Mac-1, alpha M beta 2, CD11b/CD18) and Fc gamma RIII cooperate in generation of a neutrophil respiratory burst: Requirement for Fc gamma RIII and tyrosine phosphorylation. J. Cell Biol. 1994, 125, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Lowell, C.A.; Fumagalli, L.; Berton, G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J. Cell Biol. 1996, 133, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Liles, W.C.; Ledbetter, J.A.; Waltersdorph, A.W.; Klebanoff, S.J. Cross-linking of CD18 primes human neutrophils for activation of the respiratory burst in response to specific stimuli: Implications for adhesion-dependent physiological responses in neutrophils. J. Leukoc. Biol. 1995, 58, 690–697. [Google Scholar] [PubMed]
- Zhang, X.; Shan, P.; Qureshi, S.; Homer, R.; Medzhitov, R.; Noble, P.W.; Lee, P.J. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J. Immunol. 2005, 175, 4834–4838. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.T.; Zhang, X.; Aberg, E.; Bousette, N.; Giaid, A.; Shan, P.; Medzhitov, R.M.; Lee, P.J. Inducible activation of TLR4 confers resistance to hyperoxia-induced pulmonary apoptosis. J. Immunol. 2006, 176, 4950–4958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Edirisinghe, I.; Yang, S.R.; Rajendrasozhan, S.; Kode, A.; Caito, S.; Adenuga, D.; Rahman, I. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am. J. Pathol. 2008, 172, 1222–1237. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.M. The role of the tracheobronchial circulation in aerosol clearance. J. Aerosol Med. 1995, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Csete, M.E.; Chediak, A.D.; Abraham, W.M.; Wanner, A. Airway blood flow modifies allergic airway smooth muscle contraction. Am. Rev. Respir. Dis. 1991, 144, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Flood-Page, P.; Menzies-Gow, A.; Phipps, S.; Ying, S.; Wangoo, A.; Ludwig, M.S.; Barnes, N.; Robinson, D.; Kay, A.B. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Investig. 2003, 112, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Le Cras, T.D.; Acciani, T.H.; Mushaben, E.M.; Kramer, E.L.; Pastura, P.A.; Hardie, W.D.; Korfhagen, T.R.; Sivaprasad, U.; Ericksen, M.; Gibson, A.M.; et al. Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L414–L421. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.S.; Yuan, S.; Innes, A.L.; Kerr, S.; Woodruff, P.G.; Hou, L.; Muller, S.J.; Fahy, J.V. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production and collagen gel elasticity in asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 14170–14175. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A.A.; Hazen, S.L.; Comhair, S.A.; Erzurum, S.C. Oxidative and nitrosative events in asthma. Free Radic. Biol. Med. 2003, 35, 213–225. [Google Scholar] [CrossRef]
- Wedes, S.H.; Khatri, S.B.; Zhang, R.; Wu, W.; Comhair, S.A.; Wenzel, S.; Teague, W.G.; Israel, E.; Erzurum, S.C.; Hazen, S.L. Noninvasive markers of airway inflammation in asthma. Clin. Transl. Sci. 2009, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Owayed, A.; Dhaunsi, G.S.; Al-Mukhaizeem, F. Nitric oxide-mediated activation of NADPH oxidase by salbutamol during acute asthma in children. Cell Biochem. Funct. 2008, 26, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003, 17, 1502–1504. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, C.; Machen, T.E.; Illek, B.; Fischer, H. NADPH oxidase-dependent acid production in airway epithelial cells. J. Biol. Chem. 2004, 279, 36454–36461. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Gonzales, L.K.; Kolla, V.; Schwarzer, C.; Miot, F.; Illek, B.; Ballard, P.L. Developmental regulation of DUOX1 expression and function in human fetal lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1506–L1514. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Kim, E.Y.; Ide, K.; Pelletier, M.R.; Roswit, W.T.; Morton, J.D.; Battaile, J.T.; Patel, A.C.; Patterson, G.A.; Castro, M.; et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J. Clin. Investig. 2006, 116, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Habibovic, A.; Hristova, M.; Heppner, D.E.; Danyal, K.; Ather, J.L.; Janssen-Heininger, Y.M.; Irvin, C.G.; Poynter, M.E.; Lundblad, L.K.; Dixon, A.E.; et al. DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia and airway remodeling during allergic asthma. JCI Insight 2016, 1, e88811. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.X.; Nadel, J.A. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.J.; Senior, R.M. Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 2003, 28, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Gaston, B.; Hunt, J. Acid stress in the pathology of asthma. J. Allergy Clin. Immunol. 2004, 113, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.Y.; Hollins, F.; Haste, L.; Woodman, L.; Hirst, R.A.; Bolton, S.; Gomez, E.; Sutcliffe, A.; Desai, D.; Chachi, L.; et al. NADPH Oxidase-4 Overexpression Is Associated With Epithelial Ciliary Dysfunction in Neutrophilic Asthma. Chest 2016, 149, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Bachoual, R.; Desmard, M.; Golda, S.; Guichard, C.; Lanone, S.; Aubier, M.; Ogier-Denis, E.; Boczkowski, J. Diesel exhaust particles induce matrix metalloprotease-1 in human lung epithelial cells via a NADP(H) oxidase/NOX4 redox-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L170–L181. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, Y.D.; Moon, U.Y.; Kim, J.H.; Jeon, J.H.; Lee, J.G.; Bae, Y.S.; Yoon, J.H. The role of Nox4 in oxidative stress-induced MUC5AC overexpression in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.A.; Taranova, A.G.; Lee, N.A.; Lee, J.J. Eosinophils: Singularly destructive effector cells or purveyors of immunoregulation? J. Allergy Clin. Immunol. 2007, 119, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Eosinophilic and neutrophilic inflammation in asthma: Insights from clinical studies. Proc. Am. Thorac. Soc. 2009, 6, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, E.R.; Henderson, W.R., Jr. Role of T cells in a gp91phox knockout murine model of acute allergic asthma. Allergy Asthma Clin. Immunol. 2013, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, E.R.; Henderson, W.R., Jr. Defining the molecular role of gp91phox in the immune manifestation of acute allergic asthma using a preclinical murine model. Clin. Mol. Allergy 2012, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Abdala-Valencia, H.; Earwood, J.; Bansal, S.; Jansen, M.; Babcock, G.; Garvy, B.; Wills-Karp, M.; Cook-Mills, J.M. Nonhematopoietic NADPH oxidase regulation of lung eosinophilia and airway hyperresponsiveness in experimentally induced asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1111–L1125. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Bacsi, A.; Choudhury, B.K.; Dharajiya, N.; Alam, R.; Hazra, T.K.; Mitra, S.; Goldblum, R.M.; Sur, S. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J. Clin. Investig. 2005, 115, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Kim, G.A.; Taylor, J.M.; Gugino, S.F.; Farrow, K.N.; Schumacker, P.T.; Berkelhamer, S.K. Mouse lung development and NOX1 induction during hyperoxia are developmentally regulated and mitochondrial ROS dependent. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L369–L377. [Google Scholar] [CrossRef] [PubMed]
- Rajashekhar, G.; Kamocka, M.; Marin, A.; Suckow, M.A.; Wolter, W.R.; Badve, S.; Sanjeevaiah, A.R.; Pumiglia, K.; Rosen, E.; Clauss, M. Pro-inflammatory angiogenesis is mediated by p38 MAP kinase. J. Cell. Physiol. 2011, 226, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Menden, H.; Tate, E.; Hogg, N.; Sampath, V. LPS-mediated endothelial activation in pulmonary endothelial cells: Role of Nox2-dependent IKK-beta phosphorylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L445–L455. [Google Scholar] [CrossRef] [PubMed]
- Menden, H.; Welak, S.; Cossette, S.; Ramchandran, R.; Sampath, V. Lipopolysaccharide (LPS)-mediated angiopoietin-2-dependent autocrine angiogenesis is regulated by NADPH oxidase 2 (Nox2) in human pulmonary microvascular endothelial cells. J. Biol. Chem. 2015, 290, 5449–5461. [Google Scholar] [CrossRef] [PubMed]
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of S1P signaling and Nox proteins. Am. J. Pathol. 2013, 183, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; LaPar, D.J.; Stone, M.L.; Zhao, Y.; Mehta, C.K.; Kron, I.L.; Laubach, V.E. NOX2 activation of natural killer T cells is blocked by the adenosine A2A receptor to inhibit lung ischemia-reperfusion injury. Am. J. Respir. Crit. Care Med. 2016, 193, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zheng, Y.M. ROS-dependent signaling mechanisms for hypoxic Ca2+ responses in pulmonary artery myocytes. Antioxid. Redox Signal. 2010, 12, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Moon, K.A.; Jo, H.Y.; Jeong, S.; Seon, S.H.; Jung, E.; Cho, Y.S.; Chun, E.; Lee, K.Y. Anti-inflammatory effects of apocynin, an inhibitor of NADPH oxidase, in airway inflammation. Immunol. Cell Biol. 2012, 90, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Moldeus, P.; Berggren, M.; Graffström, R. N-acetylcysteine protection against the toxicity of cigarette smoke and cigarette smoke condensates in various tissues and cells in vitro. Eur. J. Respir. Dis. 1985, 66 (Suppl. S139), 123–129. [Google Scholar]
- Stey, C.; Steurer, J.; Bachmann, S. The effect of oral N-acetylcysteine in chronic bronchitis: A quantitative systematic review. Eur. Respir. J. 2000, 16, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Linden, M.; Wieslander, E.; Eklund, A. Effects of oral N-acetylcysteine on cell content and macrophage function in bronchoalveolar lavage from healthy smokers. Eur. Respir. J. 1988, 1, 645–650. [Google Scholar] [PubMed]
- Drost, E.; Lannan, S.; Bridgeman, M.M.; Brown, D.; Selby, C.; Donaldson, K.; MacNee, W. Lack of effect of N-acetylcysteine on the release of oxygen radicals from neutrophils and alveolar macrophages. Eur. Respir. J. 1991, 496, 723–729. [Google Scholar]
- Jankowska, R.; Passowicz-Muszyńska, E.; Medrala, W.; Banaś, T.; Marcinkowska, A. The influence of n-acetylcysteine on chemiluminescence of granulocytes in peripheral blood of patients with chronic bronchitis. Pneumonol. Alergol. Pol. 1993, 61, 586–591. [Google Scholar] [PubMed]
- Sadowska, A.M.; Van Overveld, F.J.; Gorecka, D.; Zdral, A.; Filewska, M.; Demkow, U.A.; Luyten, C.; Saenen, E.; Zielinski, J.; De Backer, W.A. The interrelationship between markers of inflammation and oxidative stress in chronic obstructive pulmonary disease: Modulation by inhaled steroids and antioxidant. Respir. Med. 2005, 99, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. First in class, potent and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010, 53, 7715–7730. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Type of Nox | Cell Type Where Found |
|---|---|
| Nox1 | Smooth muscle, endothelium, upper airway epithelium |
| Nox2 | Inflammatory cells (macrophage and neutrophils), mesenchymal cells, smooth muscle, endothelium, upper and lower airway epithelium |
| Nox3 | Inducible in lung endothelium |
| Nox4 | Inflammatory cells (macrophage and neutrophils, mesenchymal cells, smooth muscle, endothelium, lower airway epithelial cells |
| Nox5 | Smooth muscle, endothelium |
| Duox1 | Upper airway epithelium |
| Duox2 | Upper airway epithelium |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harijith, A.; Natarajan, V.; Fu, P. The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling. Antioxidants 2017, 6, 104. https://doi.org/10.3390/antiox6040104
Harijith A, Natarajan V, Fu P. The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling. Antioxidants. 2017; 6(4):104. https://doi.org/10.3390/antiox6040104
Chicago/Turabian StyleHarijith, Anantha, Viswanathan Natarajan, and Panfeng Fu. 2017. "The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling" Antioxidants 6, no. 4: 104. https://doi.org/10.3390/antiox6040104
APA StyleHarijith, A., Natarajan, V., & Fu, P. (2017). The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling. Antioxidants, 6(4), 104. https://doi.org/10.3390/antiox6040104