
Increased Exercise Tolerance in G6PD African Variant Mice Driven by Metabolic Adaptations and Erythrophagocytosis

 , ,
, ,  , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. G6PDd Mouse Model
2.2. Treadmill Exercise and Constant Speed Tests
2.3. Data Modeling for the Determination of CS
2.4. Hemodynamics
2.5. Mass Spectrometry-Based Proteomics
2.6. Mass Spectrometry-Based Metabolomics and Lipidomics
2.7. Mass Spectrometry-Based Metal Analysis and Inductively Coupled Plasma Mass Spectrometry Instrumentation
2.8. Macrophage Isolation and Erythrophagocytosis
2.9. Scanning Electron Microscopy
2.10. Statistics and Modeling
3. Results
3.1. G6PDA Mice Maintain Higher CS
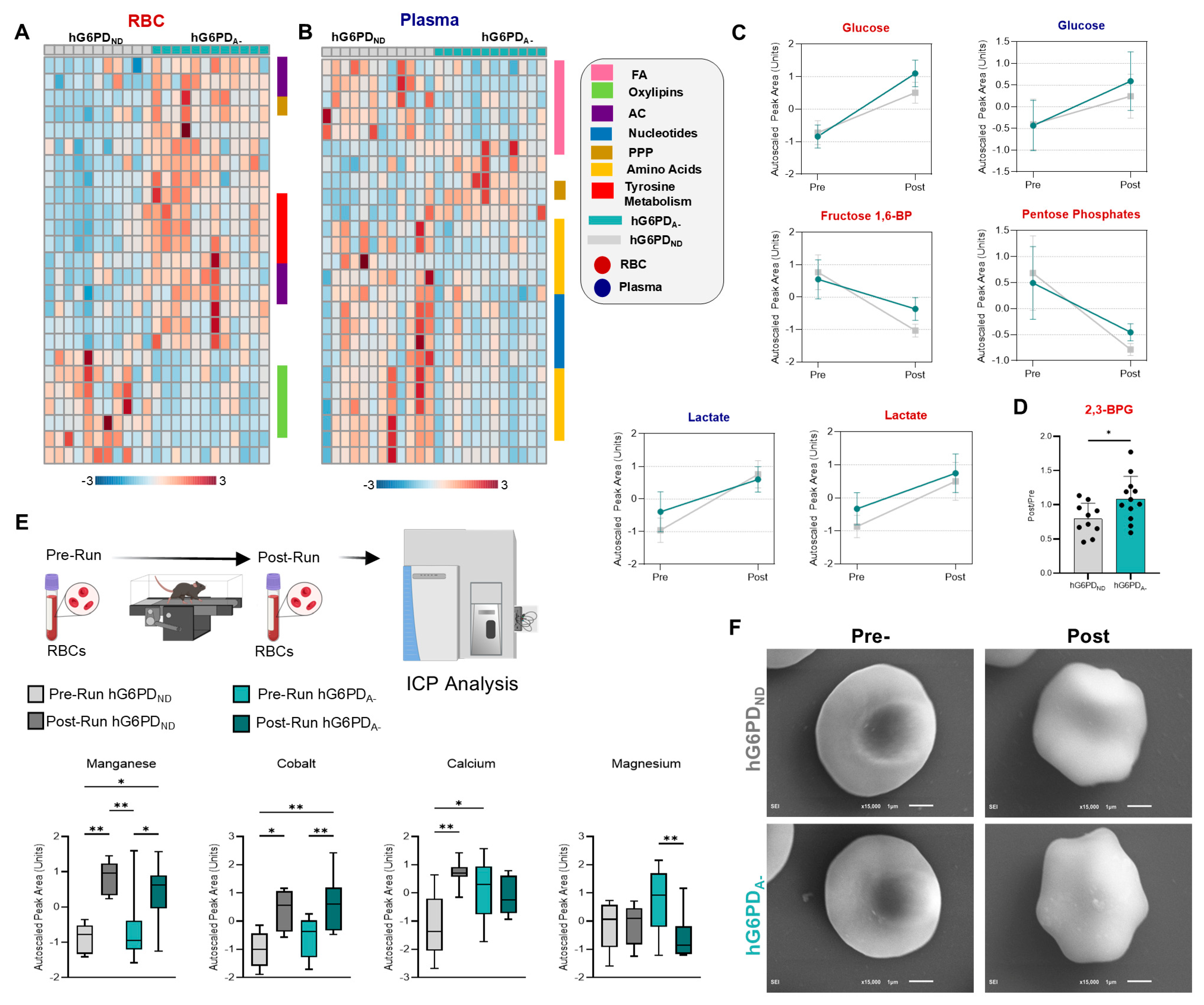
3.2. Pre- and Post-Exercise Blood Analysis
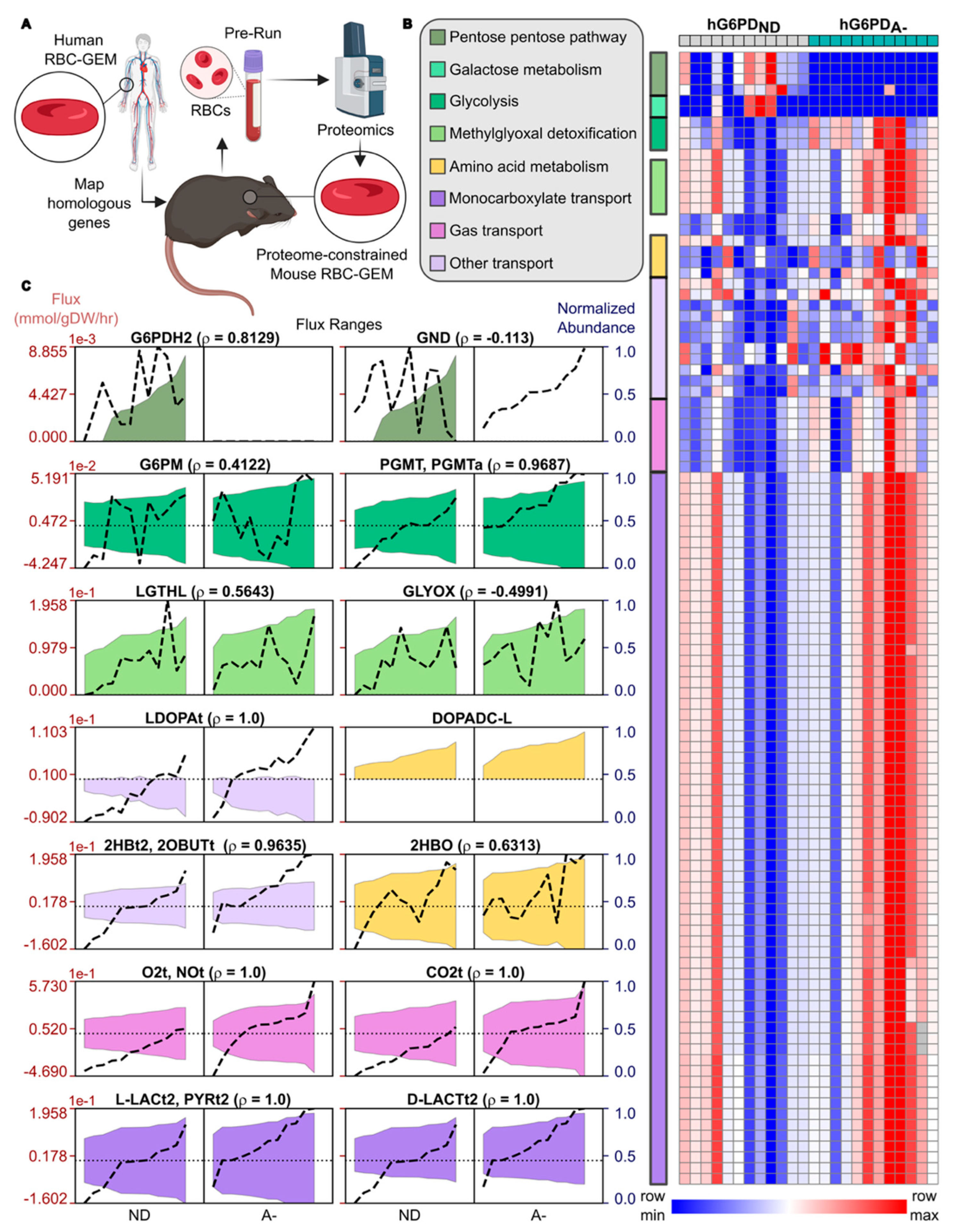
3.3. Proteome-Constrained Modeling Highlights hG6PDA− RBCs Reprogram Metabolism to Enhance Oxygen Delivery
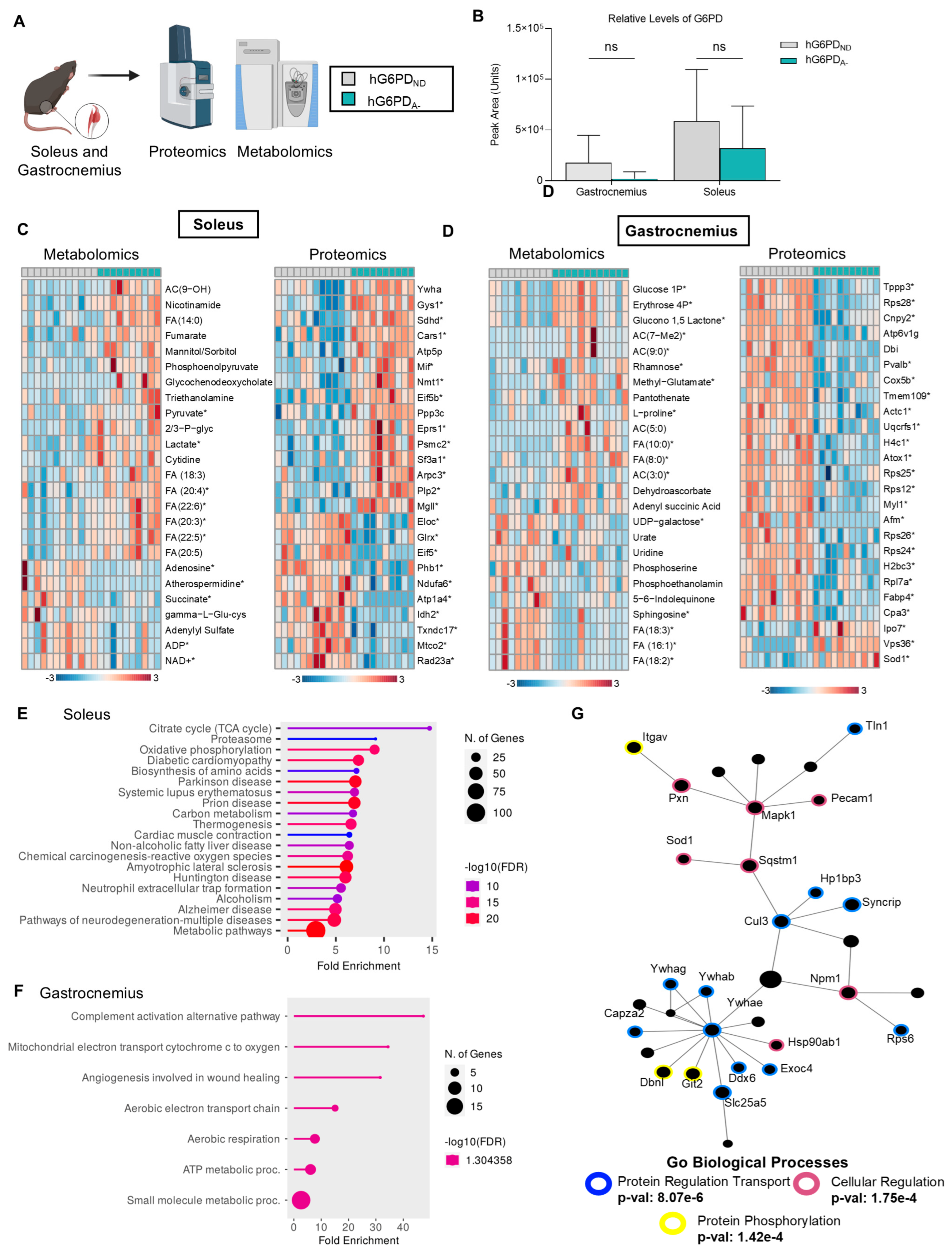
3.4. Skeletal Muscle Omics Profiles Reveal Higher Levels of Oxidation and Cellular Regulation
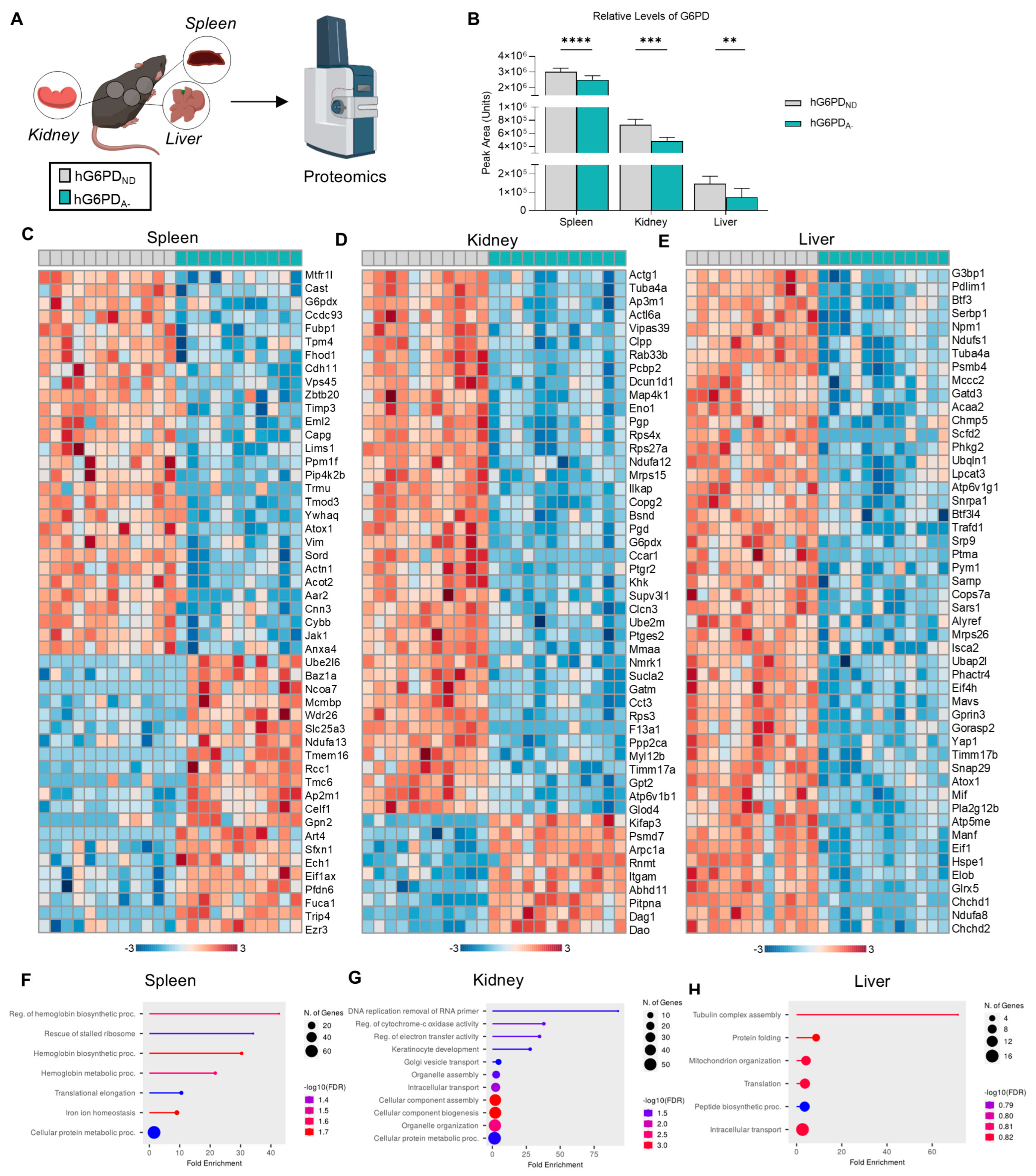
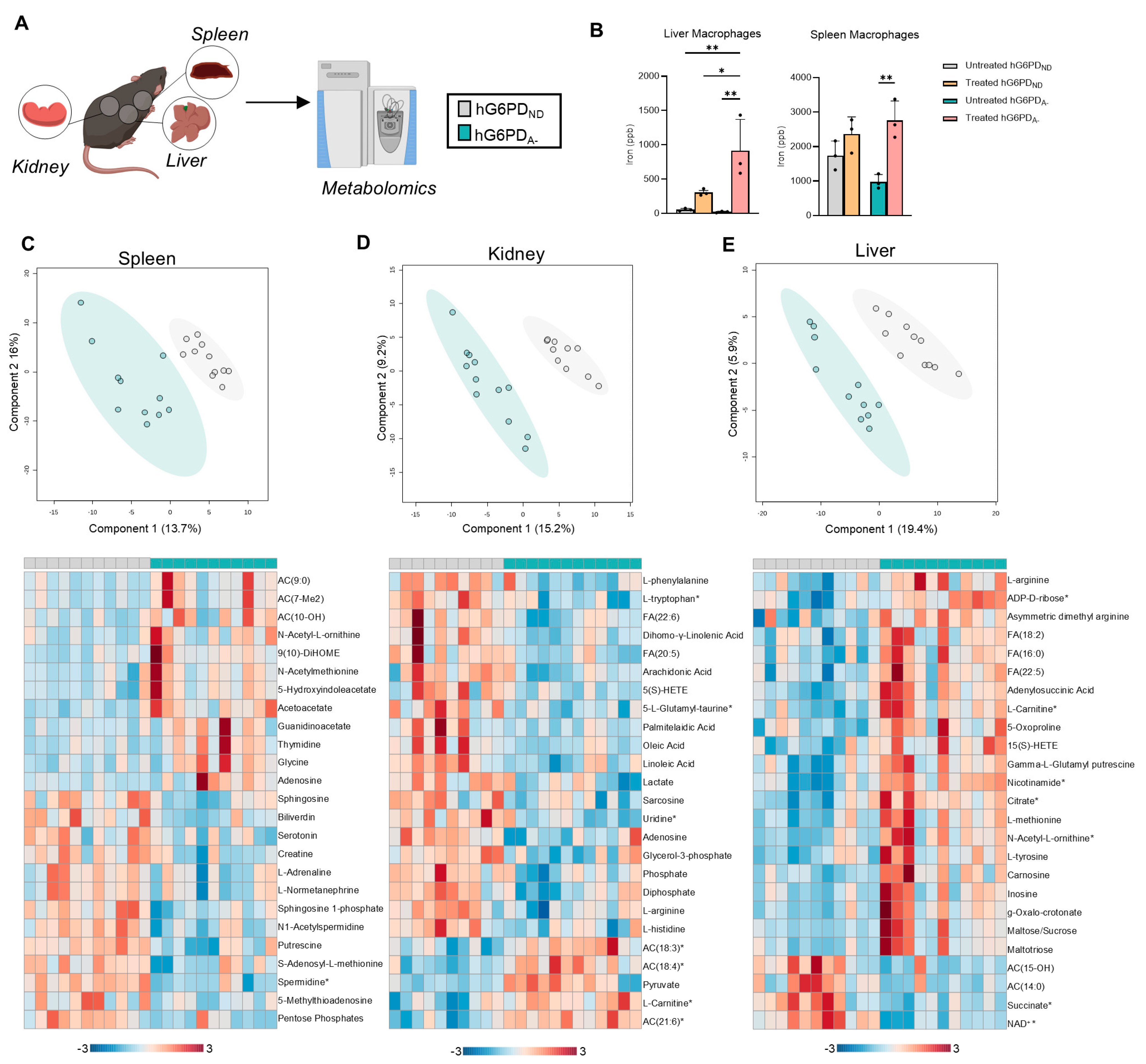
3.5. Impact of G6PD Status and Exercise on Spleen, Kidney, and Liver
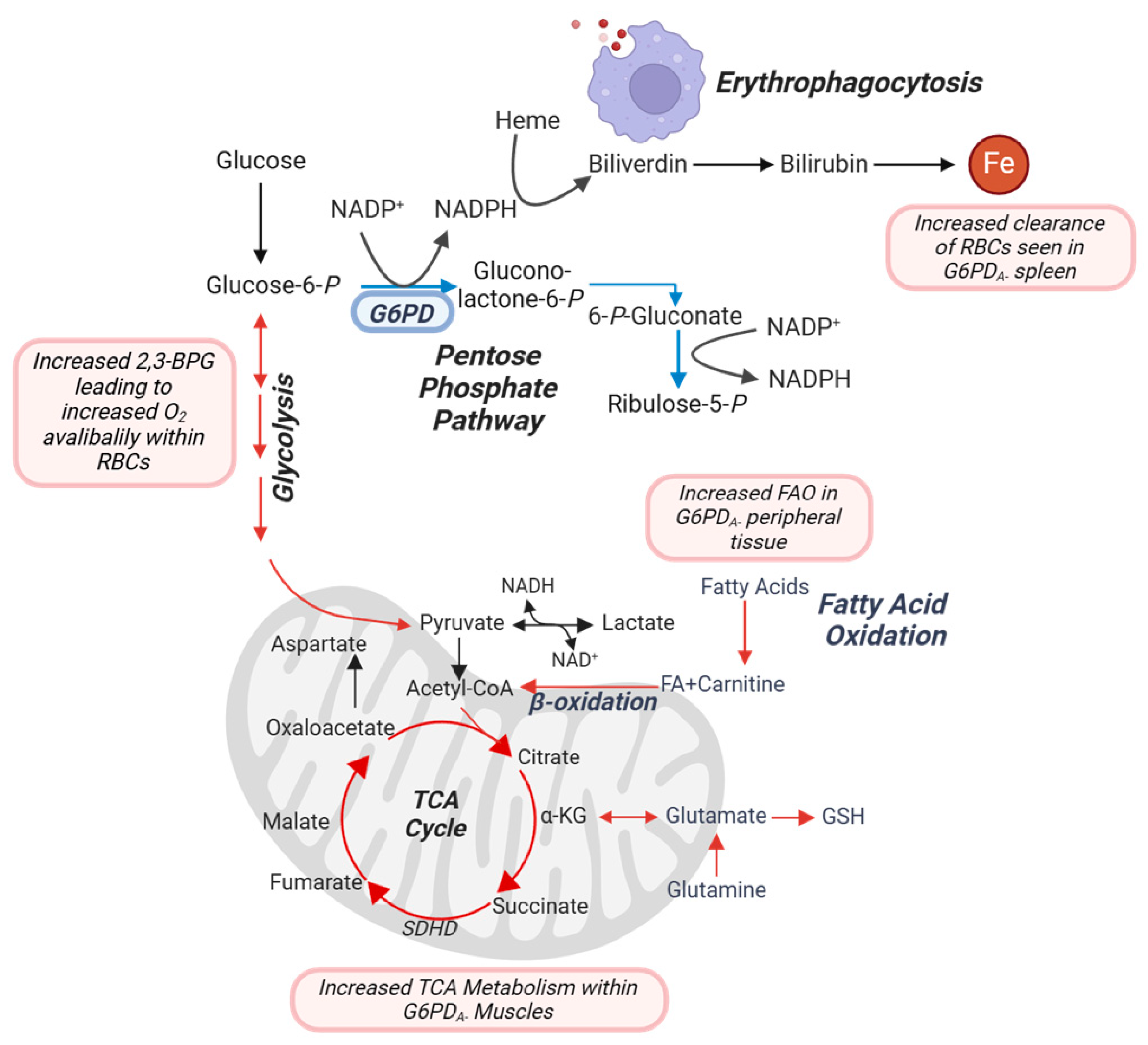
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luzzatto, L.; Ally, M.; Notaro, R. Glucose-6-phosphate dehydrogenase deficiency. Blood 2020, 136, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Joshi, S.R.; Kitagawa, A.; Jacob, C.; Hashimoto, R.; Dhagia, V.; Ramesh, A.; Zheng, C.; Zhang, H.; Jordan, A.; Waddell, I.; et al. Hypoxic activation of glucose-6-phosphate dehydrogenase controls the expression of genes involved in the pathogenesis of pulmonary hypertension through the regulation of DNA methylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L773–L786. [Google Scholar] [CrossRef]
- Miller, C.G.; Holmgren, A.; Arner, E.S.J.; Schmidt, E.E. NADPH-dependent and -independent disulfide reductase systems. Free Radic. Biol. Med. 2018, 127, 248–261. [Google Scholar] [CrossRef]
- Richardson, S.R.; O’Malley, G.F. Glucose-6-Phosphate Dehydrogenase Deficiency; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- D’Alessandro, A.; Anastasiadi, A.T.; Tzounakas, V.L.; Nemkov, T.; Reisz, J.A.; Kriebardis, A.G.; Zimring, J.C.; Spitalnik, S.L.; Busch, M.P. Red Blood Cell Metabolism In Vivo and In Vitro. Metabolites 2023, 13, 793. [Google Scholar] [CrossRef]
- Karafin, M.S.; Francis, R.O. Impact of G6PD status on red cell storage and transfusion outcomes. Blood Transfus. 2019, 17, 289–295. [Google Scholar] [CrossRef]
- Kriebardis, A.G.; Antonelou, M.H.; Stamoulis, K.E.; Economou-Petersen, E.; Margaritis, L.H.; Papassideri, I.S. Progressive oxidation of cytoskeletal proteins and accumulation of denatured hemoglobin in stored red cells. J. Cell. Mol. Med. 2007, 11, 148–155. [Google Scholar] [CrossRef]
- Klei, T.R.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.S.; Stone, E.F.; Francis, R.O.; Karafin, M.S. The global role of G6PD in infection and immunity. Front. Immunol. 2024, 15, 1393213. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Shaya, G.E.; Al-Mallah, M.H.; Hung, R.K.; Nasir, K.; Blumenthal, R.S.; Ehrman, J.K.; Keteyian, S.J.; Brawner, C.A.; Qureshi, W.T.; Blaha, M.J. High Exercise Capacity Attenuates the Risk of Early Mortality After a First Myocardial Infarction: The Henry Ford Exercise Testing (FIT) Project. Mayo Clin. Proc. 2016, 91, 129–139. [Google Scholar] [CrossRef]
- Leeper, N.J.; Myers, J.; Zhou, M.; Nead, K.T.; Syed, A.; Kojima, Y.; Caceres, R.D.; Cooke, J.P. Exercise capacity is the strongest predictor of mortality in patients with peripheral arterial disease. J. Vasc. Surg. 2013, 57, 728–733. [Google Scholar] [CrossRef]
- Bermudez-Munoz, J.M.; Celaya, A.M.; Hijazo-Pechero, S.; Wang, J.; Serrano, M.; Varela-Nieto, I. G6PD overexpression protects from oxidative stress and age-related hearing loss. Aging Cell 2020, 19, e13275. [Google Scholar] [CrossRef] [PubMed]
- Nobrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Vina, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef] [PubMed]
- Correas, A.G.; Olaso-Gonzalez, G.; Roca, M.; Blanco-Gandia, M.C.; Nascimento, C.; Lahoz, A.; Rodriguez-Arias, M.; Minarro, J.; Gomez-Cabrera, M.C.; Vina, J. Glucose 6 phosphate dehydrogenase overexpression rescues the loss of cognition in the double transgenic APP/PS1 mouse model of Alzheimer’s disease. Redox Biol. 2024, 75, 103242. [Google Scholar] [CrossRef] [PubMed]
- Nemkov, T.; Skinner, S.C.; Nader, E.; Stefanoni, D.; Robert, M.; Cendali, F.; Stauffer, E.; Cibiel, A.; Boisson, C.; Connes, P.; et al. Acute Cycling Exercise Induces Changes in Red Blood Cell Deformability and Membrane Lipid Remodeling. Int. J. Mol. Sci. 2021, 22, 896. [Google Scholar] [CrossRef]
- San-Millan, I.; Stefanoni, D.; Martinez, J.L.; Hansen, K.C.; D’Alessandro, A.; Nemkov, T. Metabolomics of Endurance Capacity in World Tour Professional Cyclists. Front. Physiol. 2020, 11, 578. [Google Scholar] [CrossRef]
- Nemkov, T.; Cendali, F.; Stefanoni, D.; Martinez, J.L.; Hansen, K.C.; San-Millan, I.; D’Alessandro, A. Metabolic Signatures of Performance in Elite World Tour Professional Male Cyclists. Sports Med. 2023, 53, 1651–1665. [Google Scholar] [CrossRef]
- Qiang, Y.; Liu, J.; Dao, M.; Suresh, S.; Du, E. Mechanical fatigue of human red blood cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19828–19834. [Google Scholar] [CrossRef]
- Mairbaurl, H. Red blood cells in sports: Effects of exercise and training on oxygen supply by red blood cells. Front. Physiol. 2013, 4, 332. [Google Scholar] [CrossRef]
- Nader, E.; Skinner, S.; Romana, M.; Fort, R.; Lemonne, N.; Guillot, N.; Gauthier, A.; Antoine-Jonville, S.; Renoux, C.; Hardy-Dessources, M.-D.; et al. Blood Rheology: Key Parameters, Impact on Blood Flow, Role in Sickle Cell Disease and Effects of Exercise. Front. Physiol. 2019, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- Diederich, L.; Suvorava, T.; Sansone, R.; Keller, T.C.S.t.; Barbarino, F.; Sutton, T.R.; Kramer, C.M.; Luckstadt, W.; Isakson, B.E.; Gohlke, H.; et al. On the Effects of Reactive Oxygen Species and Nitric Oxide on Red Blood Cell Deformability. Front. Physiol. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wiltshire, H.D.; Baker, J.S.; Wang, Q. Effects of High Intensity Exercise on Oxidative Stress and Antioxidant Status in Untrained Humans: A Systematic Review. Biology 2021, 10, 1272. [Google Scholar] [CrossRef]
- Mason, S.A.; Trewin, A.J.; Parker, L.; Wadley, G.D. Antioxidant supplements and endurance exercise: Current evidence and mechanistic insights. Redox Biol. 2020, 35, 101471. [Google Scholar] [CrossRef]
- Howes, R.E.; Piel, F.B.; Patil, A.P.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Hogg, M.M.; Battle, K.E.; Padilla, C.D.; Baird, J.K.; et al. G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: A geostatistical model-based map. PLoS Med. 2012, 9, e1001339. [Google Scholar] [CrossRef]
- Lee, J.; Poitras, B.T. Prevalence of glucose-6-phosphate dehydrogenase deficiency, U.S. Armed Forces, May 2004-September 2018. MSMR 2019, 26, 14–17. [Google Scholar]
- Luzzatto, L. G6PD deficiency: A polymorphism balanced by heterozygote advantage against malaria. Lancet Haematol. 2015, 2, e400–e401. [Google Scholar] [CrossRef]
- Luzzatto, L.; Bancone, G.; Dugue, P.A.; Jiang, W.; Minucci, A.; Nannelli, C.; Pfeffer, D.; Prchal, J.; Sirdah, M.; Sodeinde, O.; et al. New WHO classification of genetic variants causing G6PD deficiency. Bull. World Health Organ. 2024, 102, 615–617. [Google Scholar] [CrossRef]
- Hecker, P.A.; Leopold, J.A.; Gupte, S.A.; Recchia, F.A.; Stanley, W.C. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H491–H500. [Google Scholar] [CrossRef]
- Arai, Y. G6PD Deficiency: A Possible Cardiovascular Risk Factor in Older People. J. Atheroscler. Thromb. 2021, 28, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Javed, Z.; Haisum Maqsood, M.; Yahya, T.; Amin, Z.; Acquah, I.; Valero-Elizondo, J.; Andrieni, J.; Dubey, P.; Jackson, R.K.; Daffin, M.A.; et al. Race, Racism, and Cardiovascular Health: Applying a Social Determinants of Health Framework to Racial/Ethnic Disparities in Cardiovascular Disease. Circ. Cardiovasc. Qual. Outcomes 2022, 15, e007917. [Google Scholar] [CrossRef]
- Beutler, E. Glucose-6-phosphate dehydrogenase deficiency: A historical perspective. Blood 2008, 111, 16–24. [Google Scholar] [CrossRef]
- Bancone, G.; Chu, C.S. G6PD Variants and Haemolytic Sensitivity to Primaquine and Other Drugs. Front. Pharmacol. 2021, 12, 638885. [Google Scholar] [CrossRef]
- Al-Dubai, H.; Al-Mashdali, A.; Hailan, Y. Acute hemolysis and methemoglobinemia secondary to fava beans ingestion in a patient with G6PD deficiency: A case report of a rare co-occurrence. Medicine 2021, 100, e27904. [Google Scholar] [CrossRef]
- Gupte, S.A.; Levine, R.J.; Gupte, R.S.; Young, M.E.; Lionetti, V.; Labinskyy, V.; Floyd, B.C.; Ojaimi, C.; Bellomo, M.; Wolin, M.S.; et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell. Cardiol. 2006, 41, 340–349. [Google Scholar] [CrossRef]
- Georgakouli, K.; Fatouros, I.G.; Draganidis, D.; Papanikolaou, K.; Tsimeas, P.; Deli, C.K.; Jamurtas, A.Z. Exercise in Glucose-6-Phosphate Dehydrogenase Deficiency: Harmful or Harmless? A Narrative Review. Oxid. Med. Cell. Longev. 2019, 2019, 8060193. [Google Scholar] [CrossRef]
- Theodorou, A.A.; Nikolaidis, M.G.; Paschalis, V.; Sakellariou, G.K.; Fatouros, I.G.; Koutedakis, Y.; Jamurtas, A.Z. Comparison between glucose-6-phosphate dehydrogenase-deficient and normal individuals after eccentric exercise. Med. Sci. Sports Exerc. 2010, 42, 1113–1121. [Google Scholar] [CrossRef]
- Cendali, F.I.; Lisk, C.; Dzieciatkowska, M.; LaCroix, I.S.; Reisz, J.A.; Harral, J.; Stephenson, D.; Hay, A.M.; Wartchow, E.; Darehshouri, A.; et al. Increased exercise tolerance in humanized G6PD deficient mice. Blood Adv. 2024, 9, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Dziewulska-Cronk, K.; Reisz, J.A.; Nemkov, T.; Hay, A.M.; Issaian, A.; Cendali, F.; Dzieciatkowska, M.; Gamboni, F.; Lamb, D.; Palha, M.; et al. Mechanisms of Primaquine Induced Hemolysis in a Novel Humanized Murine Model of Mediterranean G6PD Deficiency. Blood 2023, 142, 1070. [Google Scholar] [CrossRef]
- Dziewulska-Cronk, K.H.; Reisz, J.A.; Hay, A.M.; Nemkov, T.; Cendali, F.I.; Issaian, A.; Lamb, D.R.; Palha, M.S.; Legenzov, E.A.; Kao, J.P.Y.; et al. Primaquine-5,6-Orthoquinone Is Directly Hemolytic to Older G6PD Deficient RBCs in a Humanized Mouse Model. J. Pharmacol. Exp. Ther. 2024, 391, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.C.; Jones, A.M. Critical power: A paradigm-shift for benchmarking exercise testing and prescription. Exp. Physiol. 2023, 108, 539–540. [Google Scholar] [CrossRef]
- Copp, S.W.; Hirai, D.M.; Musch, T.I.; Poole, D.C. Critical speed in the rat: Implications for hindlimb muscle blood flow distribution and fibre recruitment. J. Physiol. 2010, 588, 5077–5087. [Google Scholar] [CrossRef]
- Cendali, F.I.; Nemkov, T.; Lisk, C.; Lacroix, I.S.; Nouraie, S.M.; Zhang, Y.; Gordeuk, V.R.; Buehler, P.W.; Irwin, D.; D’Alessandro, A. Metabolic correlates to critical speed in murine models of sickle cell disease. Front. Physiol. 2023, 14, 1151268. [Google Scholar] [CrossRef] [PubMed]
- Nemkov, T.; Reisz, J.A.; Gehrke, S.; Hansen, K.C.; D’Alessandro, A. High-Throughput Metabolomics: Isocratic and Gradient Mass Spectrometry-Based Methods. Methods Mol. Biol. 2019, 1978, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Nemkov, T.; Qadri, S.M.; Sheffield, W.P.; D’Alessandro, A. Inductively-Coupled Plasma Mass Spectrometry-Novel Insights From an Old Technology Into Stressed Red Blood Cell Physiology. Front. Physiol. 2022, 13, 828087. [Google Scholar] [CrossRef]
- Lisk, C.; Cendali, F.; Setua, S.; Thangaraju, K.; Pak, D.I.; Swindle, D.; Dzieciatkowska, M.; Gamboni, F.; Hassell, K.; Nuss, R.; et al. Metabolic and Proteomic Divergence Is Present in Circulating Monocytes and Tissue-Resident Macrophages from Berkeley Sickle Cell Anemia and beta-Thalassemia Mice. J. Proteome Res. 2023, 22, 2925–2935. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; D’Amici, G.M.; Vaglio, S.; Zolla, L. Time-course investigation of SAGM-stored leukocyte-filtered red bood cell concentrates: From metabolism to proteomics. Haematologica 2012, 97, 107–115. [Google Scholar] [CrossRef]
- Haiman, Z.B.; Key, A.; D’Alessandro, A.; Palsson, B.O. RBC-GEM: A genome-scale metabolic model for systems biology of the human red blood cell. PLoS Comput. Biol. 2025, 21, e1012109. [Google Scholar] [CrossRef]
- Baldarelli, R.M.; Smith, C.L.; Ringwald, M.; Richardson, J.E.; Bult, C.J. Mouse Genome Informatics: An integrated knowledgebase system for the laboratory mouse. Genetics 2024, 227, iyae031. [Google Scholar] [CrossRef]
- Bryk, A.H.; Wiśniewski, J.R. Quantitative Analysis of Human Red Blood Cell Proteome. J. Proteome Res. 2017, 16, 2752–2761. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Rakus, D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J. Proteomics 2014, 109, 322–331. [Google Scholar] [CrossRef]
- Yao, H.; Dahal, S.; Yang, L. Novel context-specific genome-scale modelling explores the potential of triacylglycerol production by Chlamydomonas reinhardtii. Microb. Cell Fact. 2023, 22, 13. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Ebrahim, A.; Beber, M.E.; Mandal, S.; König, M.; Redestig, H.; Diener, C.; Scientist, D.; John, P.S.; akaviaLab; Hemant_Yadav; et al. Opencobra/Cobrapy: 0.29.1; Zenodo: Geneva, Switzerland, 2024. [Google Scholar]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinform. 2010, 11, 489. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Reisz, J.A.; Wither, M.J.; Dzieciatkowska, M.; Nemkov, T.; Issaian, A.; Yoshida, T.; Dunham, A.J.; Hill, R.C.; Hansen, K.C.; D’Alessandro, A. Oxidative modifications of glyceraldehyde 3-phosphate dehydrogenase regulate metabolic reprogramming of stored red blood cells. Blood 2016, 128, e32–e42. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.H.; Ghatge, M.S.; Safo, M.K. Hemoglobin: Structure, Function and Allostery. Subcell. Biochem. 2020, 94, 345–382. [Google Scholar] [CrossRef]
- Safo, M.K.; Ahmed, M.H.; Ghatge, M.S.; Boyiri, T. Hemoglobin–ligand binding: Understanding Hb function and allostery on atomic level. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2011, 1814, 797–809. [Google Scholar] [CrossRef]
- Yuan, Y.; Tam, M.F.; Simplaceanu, V.; Ho, C. New look at hemoglobin allostery. Chem. Rev. 2015, 115, 1702–1724. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Earley, E.J.; Nemkov, T.; Stephenson, D.; Dzieciatkowska, M.; Hansen, K.C.; Minetti, G.; Champigneulle, B.; Stauffer, E.; Pichon, A.; et al. Genetic polymorphisms and expression of Rhesus blood group RHCE are associated with 2,3-bisphosphoglycerate in humans at high altitude. Proc. Natl. Acad. Sci. USA 2024, 121, e2315930120. [Google Scholar] [CrossRef]
- Ploszczyca, K.; Czuba, M.; Chalimoniuk, M.; Gajda, R.; Baranowski, M. Red Blood Cell 2,3-Diphosphoglycerate Decreases in Response to a 30 km Time Trial Under Hypoxia in Cyclists. Front. Physiol. 2021, 12, 670977. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Nemkov, T.; Sun, K.; Liu, H.; Song, A.; Monte, A.A.; Subudhi, A.W.; Lovering, A.T.; Dvorkin, D.; Julian, C.G.; et al. AltitudeOmics: Red Blood Cell Metabolic Adaptation to High Altitude Hypoxia. J. Proteome Res. 2016, 15, 3883–3895. [Google Scholar] [CrossRef] [PubMed]
- Forbes, L.M.; Cendali, F.I.; Nemkov, T.; Bull, T.M.; D’Alessandro, A.; Rawlinson, K.E.; Roach, R.C.; Subudhi, A.W.; Lahm, T.; Cornwell, W.K., 3rd. Red Blood Cell Metabolic Responses during Acute Hypoxic Exercise in Healthy Adults. Am. J. Respir. Cell Mol. Biol. 2025, 72, 456–459. [Google Scholar] [CrossRef]
- Brun, J.F.; Varlet-Marie, E.; Myzia, J.; Raynaud de Mauverger, E.; Pretorius, E. Metabolic Influences Modulating Erythrocyte Deformability and Eryptosis. Metabolites 2021, 12, 4. [Google Scholar] [CrossRef]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 2024, 9, 271. [Google Scholar] [CrossRef]
- Barua, S.; Ciannella, S.; Tijani, L.; Gomez-Pastora, J. Iron in blood cells: Function, relation to disease, and potential for magnetic separation. Biotechnol. Bioeng. 2023, 120, 1707–1724. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.S.; Orlando, J.; Meister, A. Decarboxylation of 3,4-dihydroxyphenylalanine (DOPA) by erythrocytes: A reaction promoted by methemoglobin and other ferriheme proteins. Proc. Natl. Acad. Sci. USA 1972, 69, 2505–2508. [Google Scholar] [CrossRef]
- Gómez-Gallego, F.; Garrido-Pertierra, A.; Mason, P.J.; Bautista, J.M. Unproductive folding of the human G6PD-deficient variant A. Faseb J. 1996, 10, 153–158. [Google Scholar] [CrossRef]
- Bandara, A.B.; Drake, J.C.; Brown, D.A. Complex II subunit SDHD is critical for cell growth and metabolism, which can be partially restored with a synthetic ubiquinone analog. BMC Mol. Cell Biol. 2021, 22, 35. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Lin, C.T.; Sanchez, D.M.; Laskin, J.; Spangenburg, E.E.; Neufer, P.D.; Fisher-Wellman, K.; Ellis, J.M. Skeletal muscle undergoes fiber type metabolic switch without myosin heavy chain switch in response to defective fatty acid oxidation. Mol. Metab. 2022, 59, 101456. [Google Scholar] [CrossRef]
- TeSlaa, T.; Ralser, M.; Fan, J.; Rabinowitz, J.D. The pentose phosphate pathway in health and disease. Nat. Metab. 2023, 5, 1275–1289. [Google Scholar] [CrossRef]
- Golub, M.S.; Hogrefe, C.E.; Malka, R.; Higgins, J.M. Developmental plasticity of red blood cell homeostasis. Am. J. Hematol. 2014, 89, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.S.; Wen, X.; Malloy, C.R. Isotopomer analyses with the tricarboxylic acid cycle intermediates and exchanging metabolites from the rat kidney. NMR Biomed. 2023, 36, e4994. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, B.; Berry, E.A.; Zhu, X.L.; Yang, W.C.; Yang, G.F. The assembly of succinate dehydrogenase: A key enzyme in bioenergetics. Cell. Mol. Life Sci. 2019, 76, 4023–4042. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Stone, S.N.; Reisig, K.V.; Saffel, H.L.; Miles, C.M. Management of Athletes With G6PD Deficiency: Does Missing an Enzyme Mean Missing More Games? Sports Health 2020, 12, 149–153. [Google Scholar] [CrossRef]
- Starke-Reed, P.E.; Oliver, C.N. Protein oxidation and proteolysis during aging and oxidative stress. Arch. Biochem. Biophys. 1989, 275, 559–567. [Google Scholar] [CrossRef]
- Signoretti, C.; Matsumura, S.; Fatehi, S.; D’Silva, M.; Mathew, R.; Cendali, F.; D’Alessandro, A.; Alam, S.M.S.; Garcia, V.; Miano, J.M.; et al. G6pd(N126D) Variant Increases the Risk of Developing VEGFR (Vascular Endothelial Growth Factor Receptor) Blocker-Induced Pulmonary Vascular Disease. J. Am. Heart Assoc. 2024, 13, e035174. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Z.; Hu, J.; Stillman, I.E.; Leopold, J.A.; Handy, D.E.; Loscalzo, J.; Stanton, R.C. Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J. 2010, 24, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.H.; Zimring, J.C.; McNamara, C.A. Clinical associations and potential cellular mechanisms linking G6PD deficiency and atherosclerotic cardiovascular disease. NPJ Metab. Health Dis. 2025, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Parsanathan, R.; Jain, S.K. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is linked with cardiovascular disease. Hypertens. Res. 2020, 43, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Maurya, P.K.; Kumar, P.; Chandra, P. Age-dependent detection of erythrocytes glucose-6-phosphate dehydrogenase and its correlation with oxidative stress. Arch. Physiol. Biochem. 2016, 122, 61–66. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cendali, F.I.; Grier, A.L.; Lisk, C.; Dzieciatkowska, M.; Haiman, Z.; Reisz, J.A.; Harral, J.; Stephenson, D.; Hay, A.M.; Wartchow, E.P.; et al. Increased Exercise Tolerance in G6PD African Variant Mice Driven by Metabolic Adaptations and Erythrophagocytosis. Antioxidants 2025, 14, 927. https://doi.org/10.3390/antiox14080927
Cendali FI, Grier AL, Lisk C, Dzieciatkowska M, Haiman Z, Reisz JA, Harral J, Stephenson D, Hay AM, Wartchow EP, et al. Increased Exercise Tolerance in G6PD African Variant Mice Driven by Metabolic Adaptations and Erythrophagocytosis. Antioxidants. 2025; 14(8):927. https://doi.org/10.3390/antiox14080927
Chicago/Turabian StyleCendali, Francesca I., Abby L. Grier, Christina Lisk, Monika Dzieciatkowska, Zachary Haiman, Julie A. Reisz, Julie Harral, Daniel Stephenson, Ariel M. Hay, Eric P. Wartchow, and et al. 2025. "Increased Exercise Tolerance in G6PD African Variant Mice Driven by Metabolic Adaptations and Erythrophagocytosis" Antioxidants 14, no. 8: 927. https://doi.org/10.3390/antiox14080927
APA StyleCendali, F. I., Grier, A. L., Lisk, C., Dzieciatkowska, M., Haiman, Z., Reisz, J. A., Harral, J., Stephenson, D., Hay, A. M., Wartchow, E. P., Buehler, P. W., Hansen, K. C., Nemkov, T., Zimring, J. C., Irwin, D. C., & D’Alessandro, A. (2025). Increased Exercise Tolerance in G6PD African Variant Mice Driven by Metabolic Adaptations and Erythrophagocytosis. Antioxidants, 14(8), 927. https://doi.org/10.3390/antiox14080927