
A High-Fat Diet Induces Oxidative Stress in OPA1+/− Mouse Cortices: A Critical Double Challenge

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diet Composition
2.2. Metabolic Parameters: Intraperitoneal Glucose Tolerance Test (ipGTT)
2.3. Metabolic Parameters: Plasma Insulin Levels
2.4. Metabolic Parameters: HOMA-IR
2.5. Immunoblot Analysis
2.6. Measurement of Aconitase Activities
2.7. Protein Level Score
2.8. Statistical Analysis
3. Results
3.1. Effect of Nutritional Challenge on Mouse Body Weight
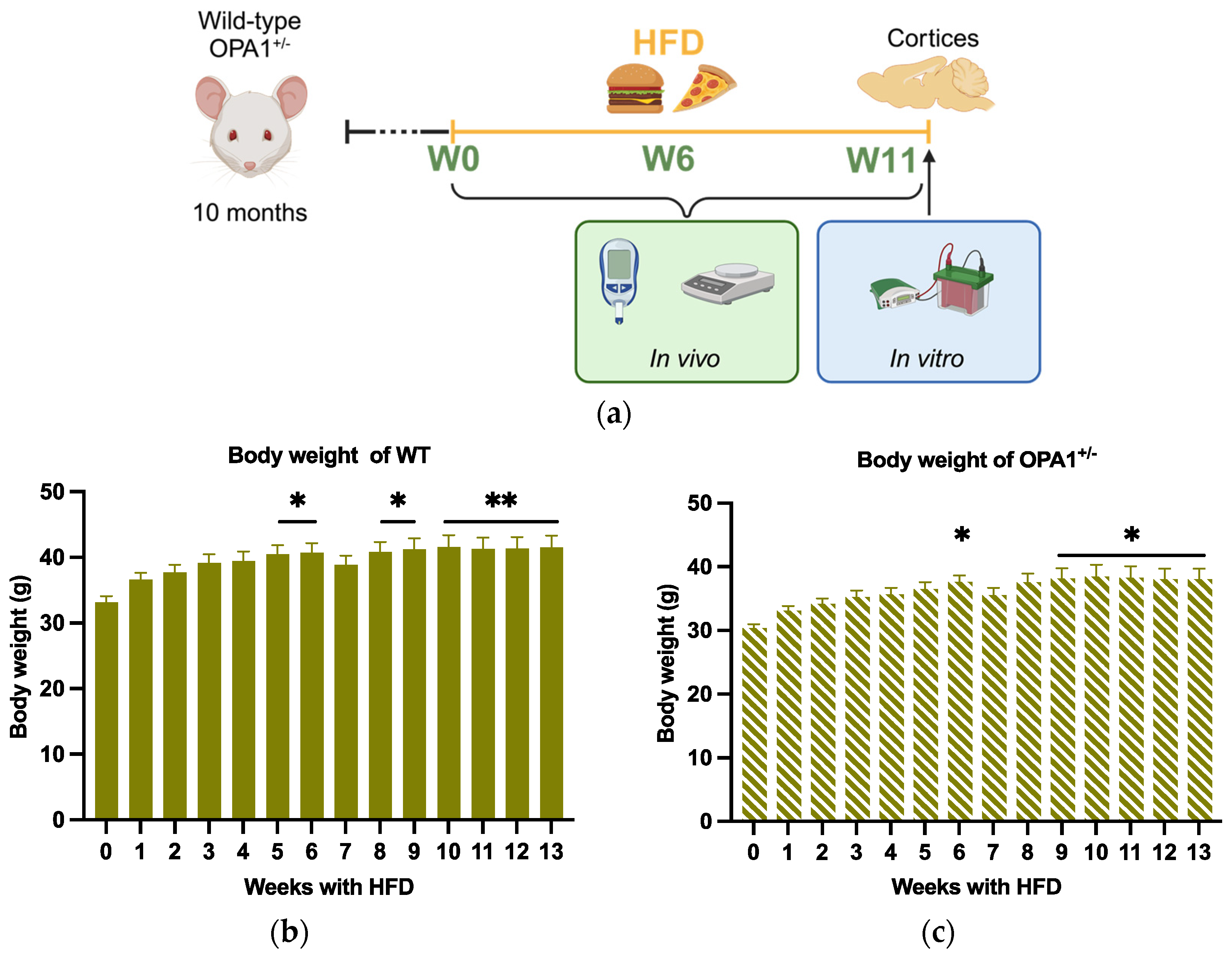
3.2. Assessing the High-Fat Diet’s Efficacy in Both WT and OPA1+/− Mice
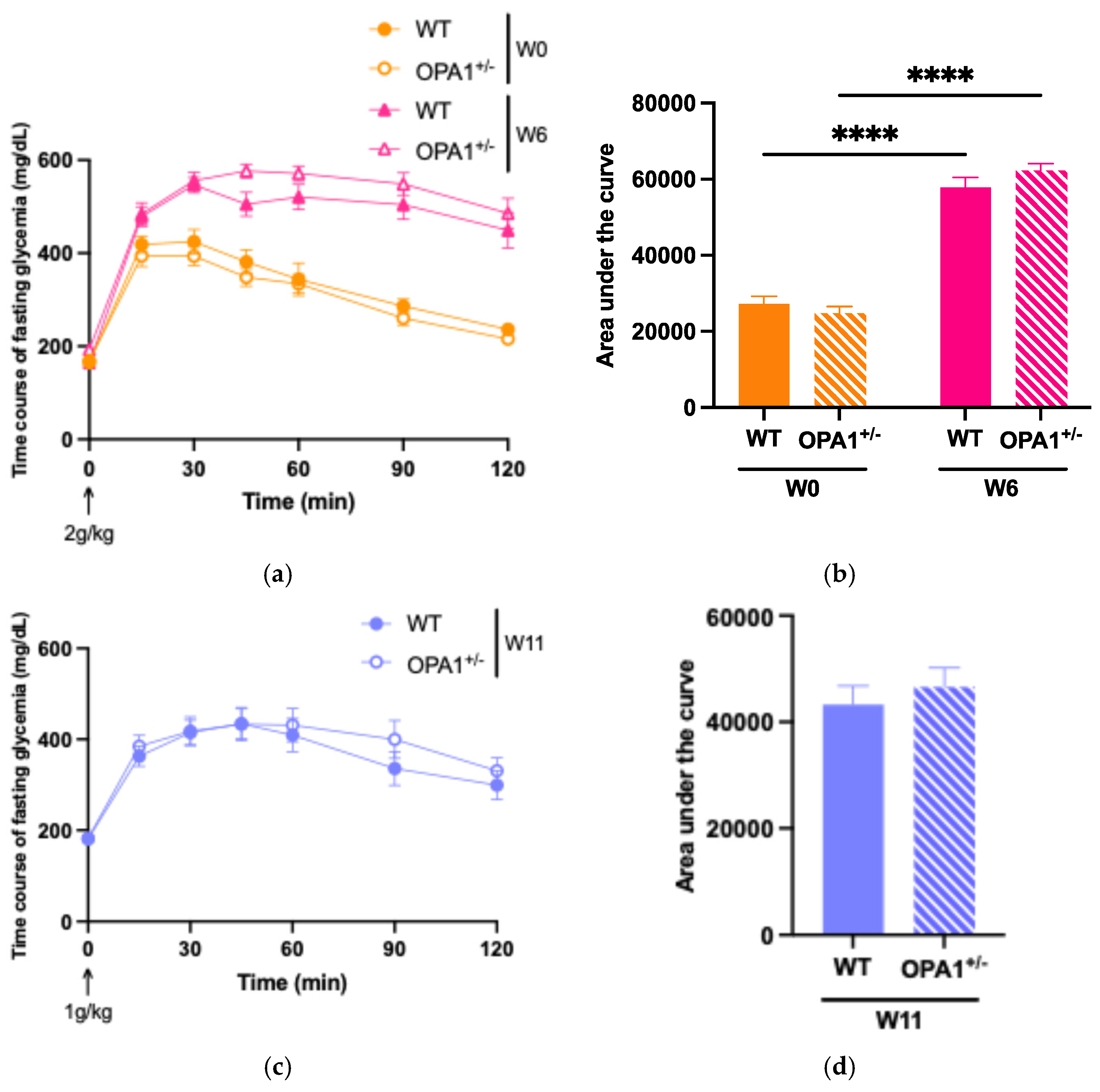
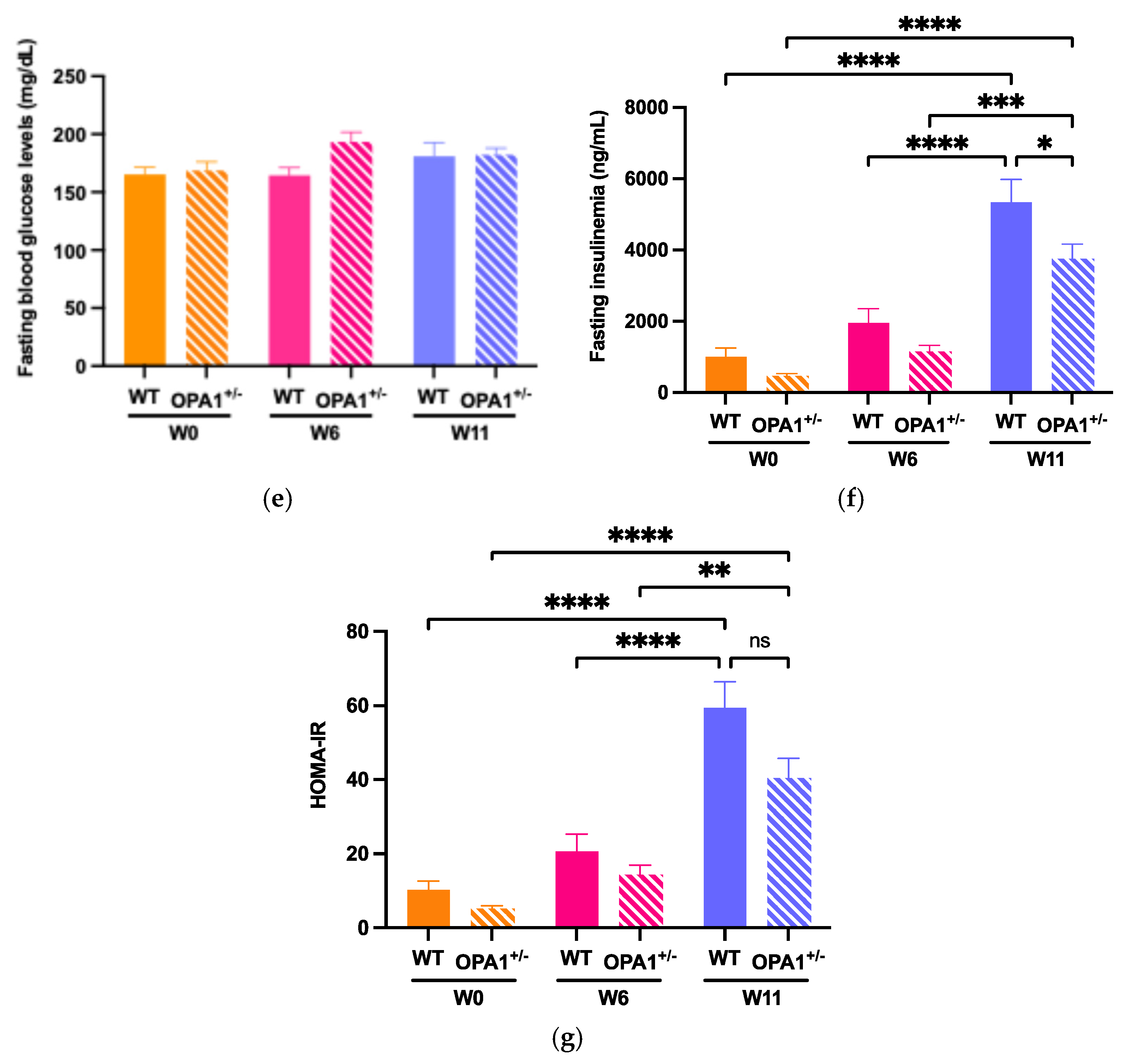
3.3. Mitochondria and Reactive Oxygen Species in Both WT and OPA1 Mice Fed a HFD

3.4. Antioxidant Defenses in Both WT and OPA1+/− Mice Fed a HFD
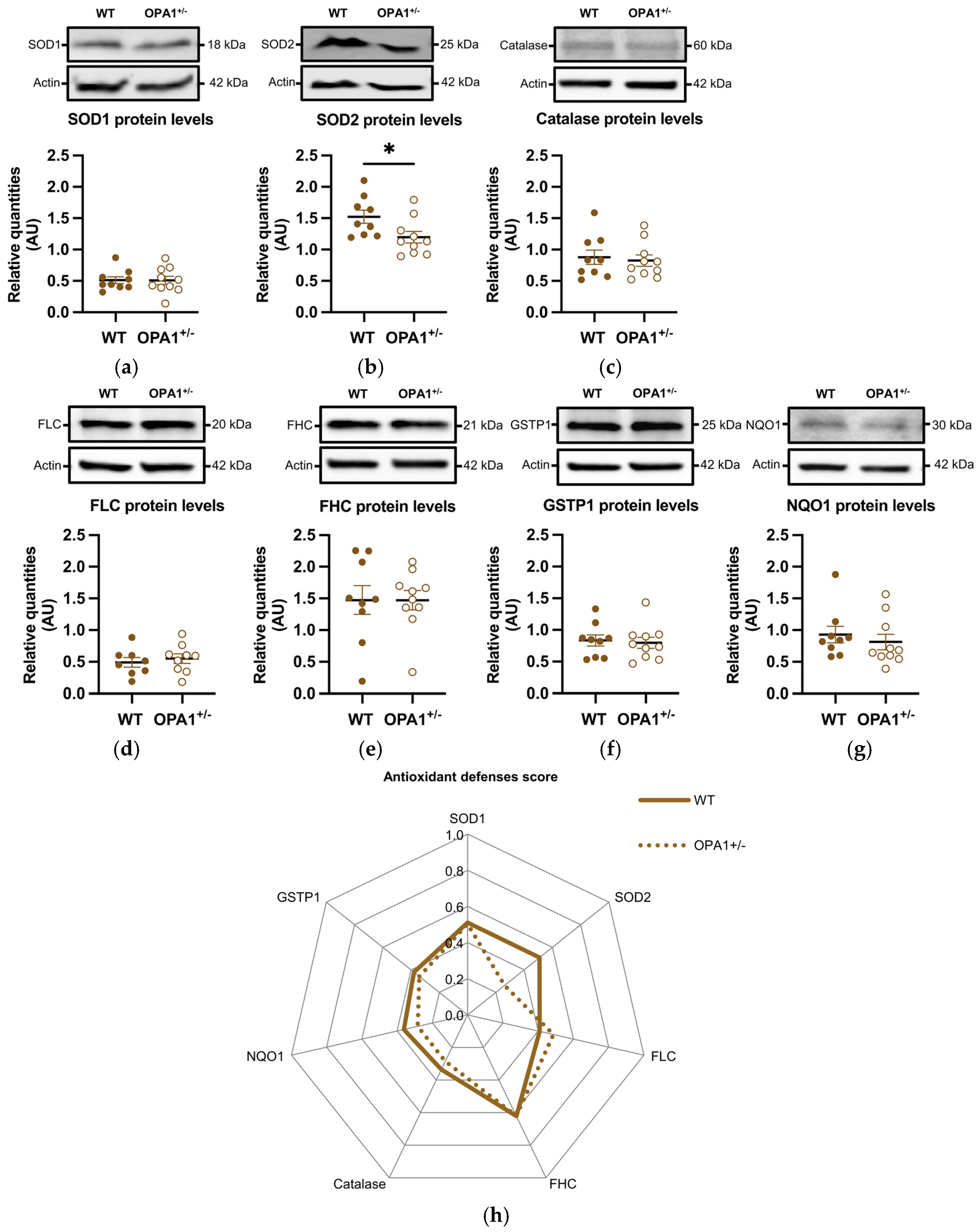
4. Discussion
4.1. A HFD Induces a Normal Response in OPA1+/− Mice
4.2. A HFD Induces a Pro-Oxidant State in OPA1+/− Mice
4.3. HFD Redox Imbalance, Neuronal Plasticity, and Cognition
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DOA | Dominant Optic Atrophy |
| AUC | Area Under the Curve |
| TBST | 0.2% Tween-20 in 1X Tris-Buffered Saline |
| FHC | Ferritin Heavy Chain |
| FLC | Ferritin Light Chain |
| GSTP1 | Glutathione S-Transferase P |
| HFD | High-Fat Diet |
| HOMA-IR | Homeostasis Model Assessment-Estimated Insulin Resistance |
| HSP60 | Heat Shock Protein 60 |
| ipGTT | intraperitoneal Glucose Tolerance Test |
| LHON | Leber Hereditary Optic Neuropathy |
| MRC1 | Mitochondrial Respiratory Complex 1 |
| NQO1 | NAD(P)H Dehydrogenase [Quinone] 1 |
| OPA1 | OPtic Atrophy 1 |
| ROS | Reactive Oxygen Species |
| SOD1 | Superoxide Dismutase 1 |
| SOD2 | Superoxide Dismutase 2 |
| WT | Wild Type |
References
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Pakiet, A.; Jakubiak, A.; Mierzejewska, P.; Zwara, A.; Liakh, I.; Sledzinski, T.; Mika, A. The Effect of a High-Fat Diet on the Fatty Acid Composition in the Hearts of Mice. Nutrients 2020, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Zemdegs, J.; Quesseveur, G.; Jarriault, D.; Pénicaud, L.; Fioramonti, X.; Guiard, B.P. High-Fat Diet-Induced Metabolic Disorders Impairs 5-HT Function and Anxiety-like Behavior in Mice. Br. J. Pharmacol. 2016, 173, 2095–2110. [Google Scholar] [CrossRef] [PubMed]
- Al Haj Ahmad, R.M.; Ababneh, N.A.; Al-Domi, H.A. Brain Insulin Resistance as a Mechanistic Mediator Links Peripheral Metabolic Disorders with Declining Cognition. Diabetes Metab. Syndr. Clin. Res. Rev. 2022, 16, 102468. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Hasselwander, S.; Li, H.; Xia, N. Effects of Different Diets Used in Diet-Induced Obesity Models on Insulin Resistance and Vascular Dysfunction in C57BL/6 Mice. Sci. Rep. 2019, 9, 19556. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, R.A.; Sheedfar, F.; Morrison, M.C.; Hommelberg, P.P.; Kor, D.; Kloosterhuis, N.J.; Gruben, N.; Youssef, S.A.; de Bruin, A.; Hofker, M.H.; et al. High-Fat Diet Induced Obesity Primes Inflammation in Adipose Tissue Prior to Liver in C57BL/6j Mice. Aging 2015, 7, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bai, Y.; Song, F. High-Fat Diet and Neuroinflammation: The Role of Mitochondria. Pharmacol. Res. 2025, 212, 107615. [Google Scholar] [CrossRef] [PubMed]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic Mitochondrial Abnormalities Occur Downstream of Inflammation in Diet-Induced Obesity. Mol. Cell. Endocrinol. 2018, 460, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High Fat Induces Acute and Chronic Inflammation in the Hypothalamus: Effect of High-Fat Diet, Palmitate and TNF-α on Appetite-Regulating NPY Neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Kozlov, A.V.; Camara, A.K.S. Mitochondria in Health and Diseases. Cells 2020, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Battersby, B.J. Mitochondrial Diseases: The Contribution of Organelle Stress Responses to Pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P. Nutrition and Training Influences on the Regulation of Mitochondrial Adenosine Diphosphate Sensitivity and Bioenergetics. Sports Med. 2017, 47, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial Dynamics in Health and Disease: Mechanisms and Potential Targets. Sig. Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The Role of Mitochondria in Metabolism and Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Millet, A.M.C.; Coustham, C.; Champigny, C.; Botella, M.; Demeilliers, C.; Devin, A.; Galinier, A.; Belenguer, P.; Bordeneuve-Guibé, J.; Davezac, N. OPA1 Deficiency Impairs Oxidative Metabolism in Cycling Cells, Underlining a Translational Approach for Degenerative Diseases. Dis. Model. Mech. 2023, 16, dmm050266. [Google Scholar] [CrossRef] [PubMed]
- Zanfardino, P.; Amati, A.; Perrone, M.; Petruzzella, V. The Balance of MFN2 and OPA1 in Mitochondrial Dynamics, Cellular Homeostasis, and Disease. Biomolecules 2025, 15, 433. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Cabrera, R.; Scorrano, L. Determinants and Outcomes of Mitochondrial Dynamics. Mol. Cell 2023, 83, 857–876. [Google Scholar] [CrossRef] [PubMed]
- Chao de la Barca, J.M.; Prunier-Mirebeau, D.; Amati-Bonneau, P.; Ferré, M.; Sarzi, E.; Bris, C.; Leruez, S.; Chevrollier, A.; Desquiret-Dumas, V.; Gueguen, N.; et al. OPA1-Related Disorders: Diversity of Clinical Expression, Modes of Inheritance and Pathophysiology. Neurobiol. Dis. 2016, 90, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Atamena, D.; Gurram, V.; Petsophonsakul, P.; Khosrobakhsh, F.; Arrázola, M.S.; Botella, M.; Wissinger, B.; Szelechowski, M.; Belenguer, P. Genetic Background Modulates Phenotypic Expressivity in OPA1 Mutated Mice, Relevance to DOA Pathogenesis. Front. Mol. Neurosci. 2023, 16, 1241222. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Yu-Wai-Man, P.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; De Coo, I.F.; Klopstock, T.; Chinnery, P.F. Gene–Environment Interactions in Leber Hereditary Optic Neuropathy. Brain 2009, 132, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Zappaterra, M.D.; Hasegawa, A.; Wright, A.P.; Newman-Smith, E.D.; Buttle, K.F.; McDonald, K.; Mannella, C.A.; van der Bliek, A.M. The C. Elegans Opa1 Homologue EAT-3 Is Essential for Resistance to Free Radicals. PLoS Genet. 2008, 4, e1000022. [Google Scholar] [CrossRef] [PubMed]
- Shahrestani, P.; Leung, H.-T.; Le, P.K.; Pak, W.L.; Tse, S.; Ocorr, K.; Huang, T. Heterozygous Mutation of Drosophila Opa1 Causes the Development of Multiple Organ Abnormalities in an Age-Dependent and Organ-Specific Manner. PLoS ONE 2009, 4, e6867. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Le, P.K.; Tse, S.; Wallace, D.C.; Huang, T. Heterozygous Mutation of Opa1 in Drosophila Shortens Lifespan Mediated through Increased Reactive Oxygen Species Production. PLoS ONE 2009, 4, e4492. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, V.S.; Baranich, T.I.; Egorova, A.V.; Akateva, A.V.; Okulova, K.M.; Ryabova, M.S.; Skvortsova, K.A.; Dmitriev, O.V.; Mudzhiri, N.M.; Voronkov, D.N.; et al. Mitochondrial Dynamics in Brain Cells During Normal and Pathological Aging. Int. J. Mol. Sci. 2024, 25, 12855. [Google Scholar] [CrossRef] [PubMed]
- Daloyau, M.; Millet, A.M.C.; Miquel, M.-C.; Mils, V.; Wissinger, B.; Belenguer, P.; Davezac, N. Brains from Aged OPA1+/− (B6;C3-Opa1 329-355del) Mouse Strain Are in a Pro-Oxidative State. React. Oxyg. Species 2018, 6, 396–405. [Google Scholar] [CrossRef]
- Millet, A.M.C.; Bertholet, A.M.; Daloyau, M.; Reynier, P.; Galinier, A.; Devin, A.; Wissinguer, B.; Belenguer, P.; Davezac, N. Loss of Functional OPA1 Unbalances Redox State: Implications in Dominant Optic Atrophy Pathogenesis. Ann. Clin. Transl. Neurol. 2016, 3, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. OPA1 Regulates Respiratory Supercomplexes Assembly: The Role of Mitochondrial Swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Robert, P.; Nguyen, P.M.C.; Richard, A.; Grenier, C.; Chevrollier, A.; Munier, M.; Grimaud, L.; Proux, C.; Champin, T.; Lelièvre, E.; et al. Protective Role of the Mitochondrial Fusion Protein OPA1 in Hypertension. FASEB J. 2021, 35, e21678. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E. Effect of High-Fat Diets on Oxidative Stress, Cellular Inflammatory Response and Cognitive Function. Nutrients 2019, 11, 2579. [Google Scholar] [CrossRef] [PubMed]
- Viguier, C.; Bullich, S.; Botella, M.; Fasseu, L.; Alfonso, A.; Rekik, K.; Gauzin, S.; Guiard, B.P.; Davezac, N. Impact of Physical Activity on Brain Oxidative Metabolism and Intrinsic Capacities in Young Swiss Mice Fed a High Fat Diet. Neuropharmacology 2023, 241, 109730. [Google Scholar] [CrossRef] [PubMed]
- Carper, D.; Coué, M.; Laurens, C.; Langin, D.; Moro, C. Reappraisal of the Optimal Fasting Time for Insulin Tolerance Tests in Mice. Mol. Metab. 2020, 42, 101058. [Google Scholar] [CrossRef] [PubMed]
- Zemdegs, J.; Martin, H.; Pintana, H.; Bullich, S.; Manta, S.; Marqués, M.A.; Moro, C.; Layé, S.; Ducrocq, F.; Chattipakorn, N.; et al. Metformin Promotes Anxiolytic and Antidepressant-Like Responses in Insulin-Resistant Mice by Decreasing Circulating Branched-Chain Amino Acids. J. Neurosci. 2019, 39, 5935–5948. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase Is a Sensitive and Critical Target of Oxygen Poisoning in Cultured Mammalian Cells and in Rat Lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Raineri, I.; Epstein, L.B.; White, C.W. Superoxide Radical and Iron Modulate Aconitase Activity in Mammalian Cells. J.Biol. Chem. 1995, 270, 13399–13405. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gong, R.; Liu, M.; Deng, Y.; Zheng, X.; Hu, T. HOMA-IR Is Positively Correlated with Biological Age and Advanced Aging in the US Adult Population. Eur. J. Med. Res. 2023, 28, 470. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The Etiology of Oxidative Stress in Insulin Resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin Resistance in Brain Alters Dopamine Turnover and Causes Behavioral Disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for Neuronal Insulin Resistance in Neurodegenerative Diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Trudel, S.; Brouillard, F.; Bouillaud, F.; Colas, J.; Nguyen-Khoa, T.; Ollero, M.; Edelman, A.; Fritsch, J. Cystic Fibrosis Transmembrane Regulator Inhibitors CFTR(Inh)-172 and GlyH-101 Target Mitochondrial Functions, Independently of Chloride Channel Inhibition. J. Pharmacol. Exp. Ther. 2010, 333, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Syeda, T.; Sánchez-Tapia, M.; Orta, I.; Granados-Portillo, O.; Pérez-Jimenez, L.; Rodríguez-Callejas, J.-D.; Toribio, S.; Silva-Lucero, M.-C.; Rivera, A.-L.; Tovar, A.R.; et al. Bioactive Foods Decrease Liver and Brain Alterations Induced by a High-Fat-Sucrose Diet through Restoration of Gut Microbiota and Antioxidant Enzymes. Nutrients 2021, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Nazar, M.A.; Sari, D.C.R.; Putra, A.; Arfian, N. Effect of High-Fat Diet on SOD2, GPx, NeuN and BDNF Expression on Frontal Lobe of Obese Rats. Malays. J. Med. Health Sci. 2021, 17, 162. [Google Scholar]
- Fadó, R.; Molins, A.; Rojas, R.; Casals, N. Feeding the Brain: Effect of Nutrients on Cognition, Synaptic Function, and AMPA Receptors. Nutrients 2022, 14, 4137. [Google Scholar] [CrossRef] [PubMed]
- Andraini, T.; Moulédous, L.; Petsophonsakul, P.; Florian, C.; Gauzin, S.; Botella-Daloyau, M.; Arrázola, M.; Nikolla, K.; Philip, A.; Leydier, A.; et al. Mitochondrial OPA1 Deficiency Is Associated to Reversible Defects in Spatial Memory Related to Adult Neurogenesis in Mice. eNeuro 2023, 10, ENEURO.0073-23.2023. [Google Scholar] [CrossRef] [PubMed]
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood–Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-J.; Li, M.-Q.; Li-Zhao; Chen, W.-G.; Wang, J.-L. Short-Term High-Fat Diet Favors the Appearances of Apoptosis and Gliosis by Activation of ERK1/2/p38MAPK Pathways in Brain. Aging 2021, 13, 23133–23148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Sig. Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Mangel, M. Messages from Mortality: The Evolution of Death Rates in the Old. Trends Ecol. Evol. 1999, 14, 438–442. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Dilution | Reference |
|---|---|---|
| anti-HSP60 | 1/3000 | Sigma-Aldrich, St. Louis, MI, USA; H-3524 |
| anti-GSTP1 | 1/2000 | Oxford Biochem. Research, Oxford, UK |
| anti-Actin | 1/10,000 | Millipore, Burlington, MA, USA; 04-1040 |
| anti-SOD1 | 1/2000 | Abcam, Cambridge, UK; ab51254 |
| anti-SOD2 | 1/2000 | Abcam, Cambridge, UK; ab68155 |
| anti-catalase | 1/800 | Abcam, Cambridge, UK; ab16731 |
| anti-aconitase | 1/500 | Abcam, Cambridge, UK; ab83528 |
| anti-FLC | 1/1000 | Abcam, Cambridge, UK; ab69090 |
| anti-FHC | 1/1000 | Abcam, Cambridge, UK; ab183781 |
| anti-NQO1 | 1/1000 | Abcam, Cambridge, UK; ab28947 |
| anti-OPA1 | 1/3000 | BD bioscience, San Jose, CA, USA; 612607 |
| (a) | Relative Quantities (AU) | ||||||||||||||
| Proteins | SOD1 | SOD2 | FLC | FHC | Catalase | NQO1 | GSTP1 | ||||||||
| Minimum | 0.141 | 0.895 | 0.180 | 0.192 | 0.520 | 0.390 | 0.467 | ||||||||
| Maximum | 0.871 | 2.102 | 0.939 | 2.255 | 1.588 | 1.879 | 1.433 | ||||||||
| Weight | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| (b) | #mice | Relative Quantities (AU) | Weighted Score | ||||||||||||
| SOD1 | SOD2 | FLC | FHC | Catalase | NQO1 | GSTP1 | SOD1 | SOD2 | FLC | FHC | Catalase | NQO1 | GSTP1 | ||
| WT | 728 | 0.871 | 2.102 | 0.600 | 1.422 | 0.520 | 1.133 | 0.846 | 1 | 1 | 0.553 | 0.596 | 0 | 0.499 | 0.393 |
| 755 | 0.323 | 1.858 | 0.513 | 2.071 | 0.645 | 0.583 | 0.873 | 0.250 | 0.798 | 0.438 | 0.911 | 0.117 | 0.130 | 0.420 | |
| 769 | 0.444 | 1.151 | 0.454 | 2.255 | 0.835 | 0.599 | 0.819 | 0.415 | 0.213 | 0.361 | 1 | 0.295 | 0.140 | 0.365 | |
| 771 | 0.561 | 1.683 | 0.319 | 1.486 | 0.834 | 0.840 | 0.550 | 0.576 | 0.654 | 0.184 | 0.627 | 0.294 | 0.302 | 0.086 | |
| 772 | 0.526 | 1.637 | 0.191 | 2.249 | 1.588 | 0.819 | 1.332 | 0.528 | 0.615 | 0.014 | 0.997 | 1 | 0.288 | 0.896 | |
| 777 | 0.402 | 1.410 | 0.360 | 0.800 | 1.145 | 0.906 | 1.113 | 0.358 | 0.427 | 0.237 | 0.294 | 0.585 | 0.347 | 0.668 | |
| 785 | 0.643 | 1.367 | 0.884 | 1.505 | 0.571 | 0.880 | 0.567 | 0.688 | 0.391 | 0.927 | 0.636 | 0.048 | 0.329 | 0.103 | |
| 788 | 0.443 | 1.235 | 0.600 | 1.292 | 0.654 | 0.724 | 0.869 | 0.415 | 0.282 | 0.553 | 0.533 | 0.125 | 0.224 | 0.416 | |
| 789 | 0.404 | 1.136 | - | 0.192 | 1.112 | 1.879 | 0.533 | 0.361 | 0.200 | - | 0 | 0.554 | 1 | 0.069 | |
| Average | 0.513 | 1.509 | 0.490 | 1.475 | 0.878 | 0.929 | 0.834 | 0.510 | 0.509 | 0.408 | 0.622 | 0.336 | 0.362 | 0.379 | |
| OPA1+/− | 725 | 0.427 | 1.792 | 0.519 | 1.613 | 0.522 | 0.682 | 0.576 | 0.393 | 0.744 | 0.446 | 0.689 | 0.002 | 0.196 | 0.113 |
| 726 | 0.141 | 1.058 | 0.594 | 1.961 | 0.550 | 0.644 | 0.928 | 0 | 0.136 | 0.545 | 0.857 | 0.029 | 0.171 | 0.477 | |
| 727 | 0.553 | 1.129 | 0.389 | 2.078 | 0.705 | 0.390 | 0.778 | 0.566 | 0.195 | 0.275 | 0.914 | 0.173 | 0 | 0.321 | |
| 743 | 0.364 | 1.132 | 0.344 | 1.665 | 0.623 | 0.544 | 0.711 | 0.305 | 0.197 | 0.217 | 0.714 | 0.097 | 0.103 | 0.253 | |
| 753 | 0.713 | 0.895 | 0.180 | 1.352 | 0.933 | 1.050 | 0.734 | 0.785 | 0 | 0 | 0.562 | 0.387 | 0.443 | 0.276 | |
| 754 | 0.520 | 1.303 | 0.939 | 1.482 | 1.389 | 1.354 | 1.433 | 0.519 | 0.338 | 1 | 0.625 | 0.813 | 0.648 | 1 | |
| 756 | 0.685 | 1.721 | 0.603 | 1.360 | 1.235 | 0.599 | 0.897 | 0.745 | 0.685 | 0.557 | 0.566 | 0.670 | 0.141 | 0.445 | |
| 770 | 0.864 | 1.208 | 0.763 | 1.697 | 0.809 | 1.566 | 0.530 | 0.991 | 0.260 | 0.767 | 0.729 | 0.270 | 0.790 | 0.065 | |
| 784 | 0.393 | 0.946 | 0.615 | 1.174 | 0.812 | 0.711 | 0.911 | 0.346 | 0.043 | 0.573 | 0.476 | 0.274 | 0.216 | 0.460 | |
| 787 | 0.414 | 0.921 | - | 0.339 | 0.667 | 0.585 | 0.467 | 0.375 | 0.022 | - | 0.071 | 0.138 | 0.131 | 0.00 | |
| Average | 0.507 | 1.211 | 0.549 | 1.472 | 0.824 | 0.812 | 0.797 | 0.503 | 0.262 | 0.487 | 0.620 | 0.285 | 0.284 | 0.341 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Champigny, C.; Botella, M.; Atamena, D.; Bullich, S.; Coustham, C.; Guiard, B.; Belenguer, P.; Davezac, N. A High-Fat Diet Induces Oxidative Stress in OPA1+/− Mouse Cortices: A Critical Double Challenge. Antioxidants 2025, 14, 876. https://doi.org/10.3390/antiox14070876
Champigny C, Botella M, Atamena D, Bullich S, Coustham C, Guiard B, Belenguer P, Davezac N. A High-Fat Diet Induces Oxidative Stress in OPA1+/− Mouse Cortices: A Critical Double Challenge. Antioxidants. 2025; 14(7):876. https://doi.org/10.3390/antiox14070876
Chicago/Turabian StyleChampigny, Camille, Marlène Botella, Djamaa Atamena, Sébastien Bullich, Corentin Coustham, Bruno Guiard, Pascale Belenguer, and Noélie Davezac. 2025. "A High-Fat Diet Induces Oxidative Stress in OPA1+/− Mouse Cortices: A Critical Double Challenge" Antioxidants 14, no. 7: 876. https://doi.org/10.3390/antiox14070876
APA StyleChampigny, C., Botella, M., Atamena, D., Bullich, S., Coustham, C., Guiard, B., Belenguer, P., & Davezac, N. (2025). A High-Fat Diet Induces Oxidative Stress in OPA1+/− Mouse Cortices: A Critical Double Challenge. Antioxidants, 14(7), 876. https://doi.org/10.3390/antiox14070876