
Mechanisms of Cisplatin-Induced Acute Kidney Injury: The Role of NRF2 in Mitochondrial Dysfunction and Metabolic Reprogramming

 , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cells
2.3. Animal Model Establishment and Drug Administration
2.4. Biochemical Analysis of Serum, Tissue, and Cell Samples
2.5. Measurement of Glomerular Filtration Rate (GFR) in Mice
2.6. RNA Extraction and Quantitative RT-PCR
2.7. Mouse Kidney Embedding and Sectioning
2.8. Hematoxylin and Eosin (H&E) Staining
2.9. Masson’s Trichrome Staining
2.10. Oil Red O Staining
2.11. Western Blot Analysis
2.12. Immunohistochemistry
2.13. Immunofluorescence
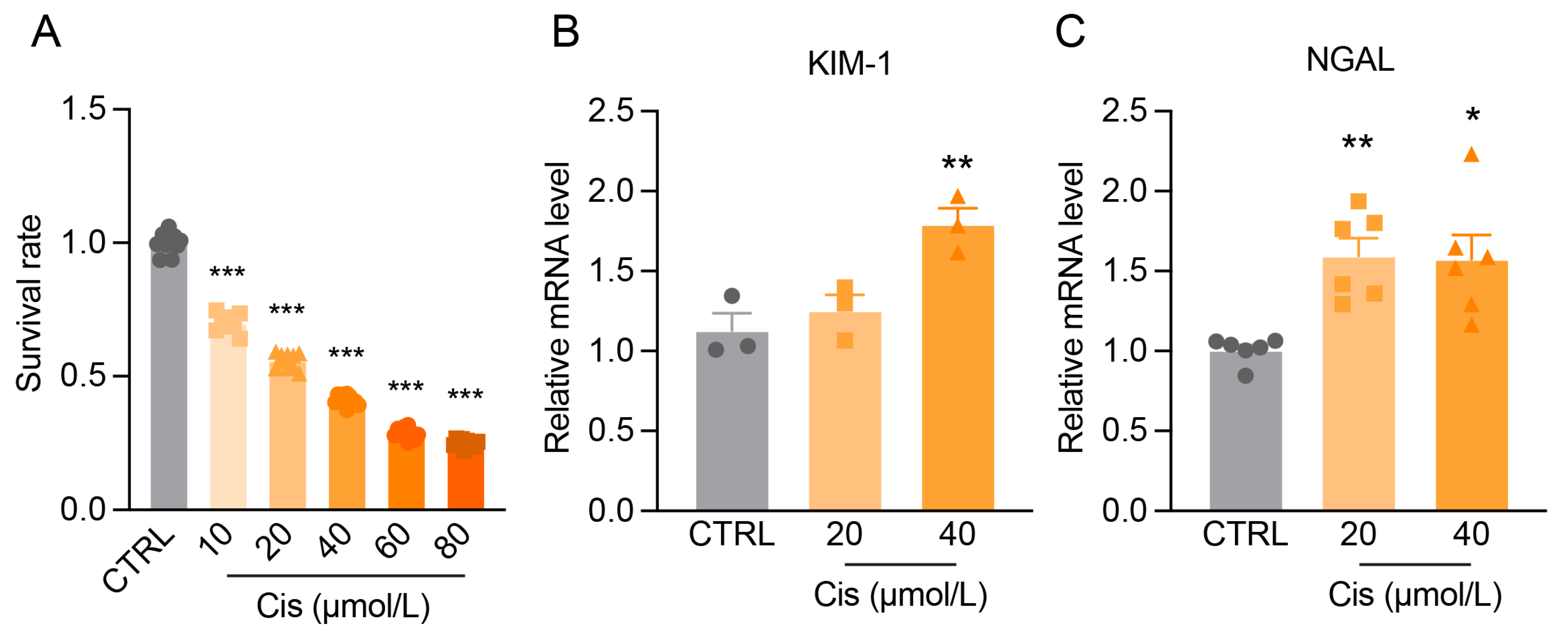
2.14. Cell Viability Assay
2.15. Mitochondrial Membrane Potential Assay
2.16. Reactive Oxygen Species (ROS) Staining
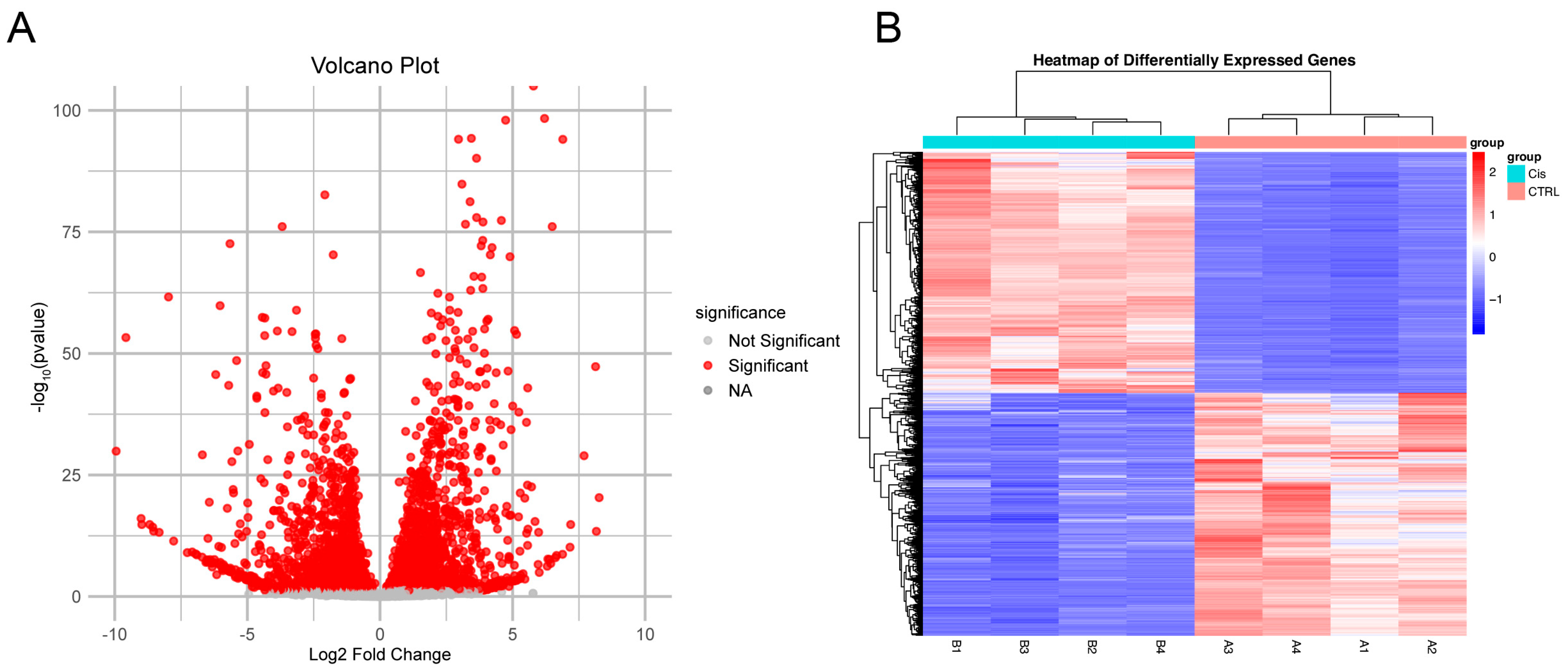
2.17. Bioinformatics Analysis of Kidney RNA Sequencing
2.18. Statistical Analysis
3. Results
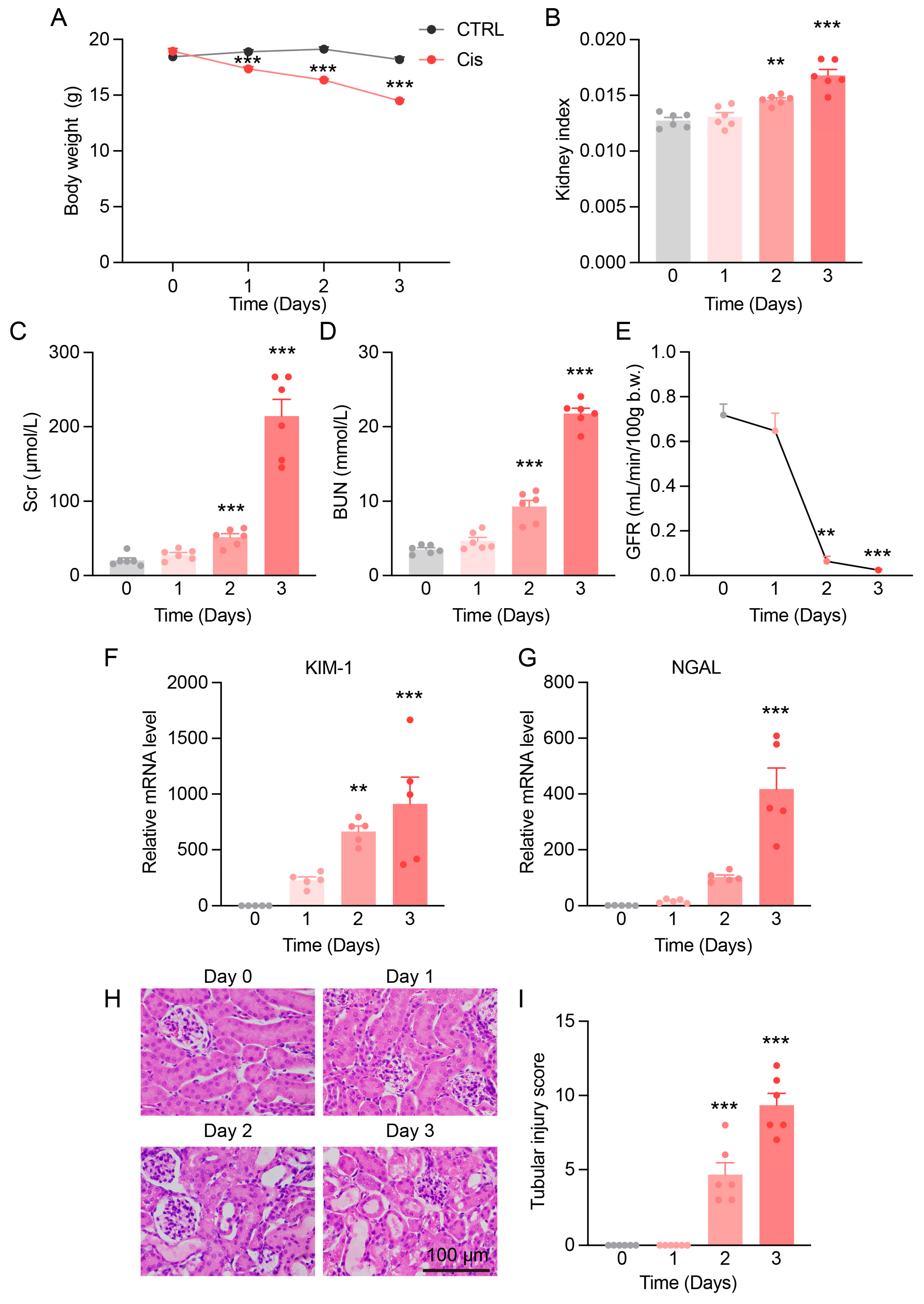
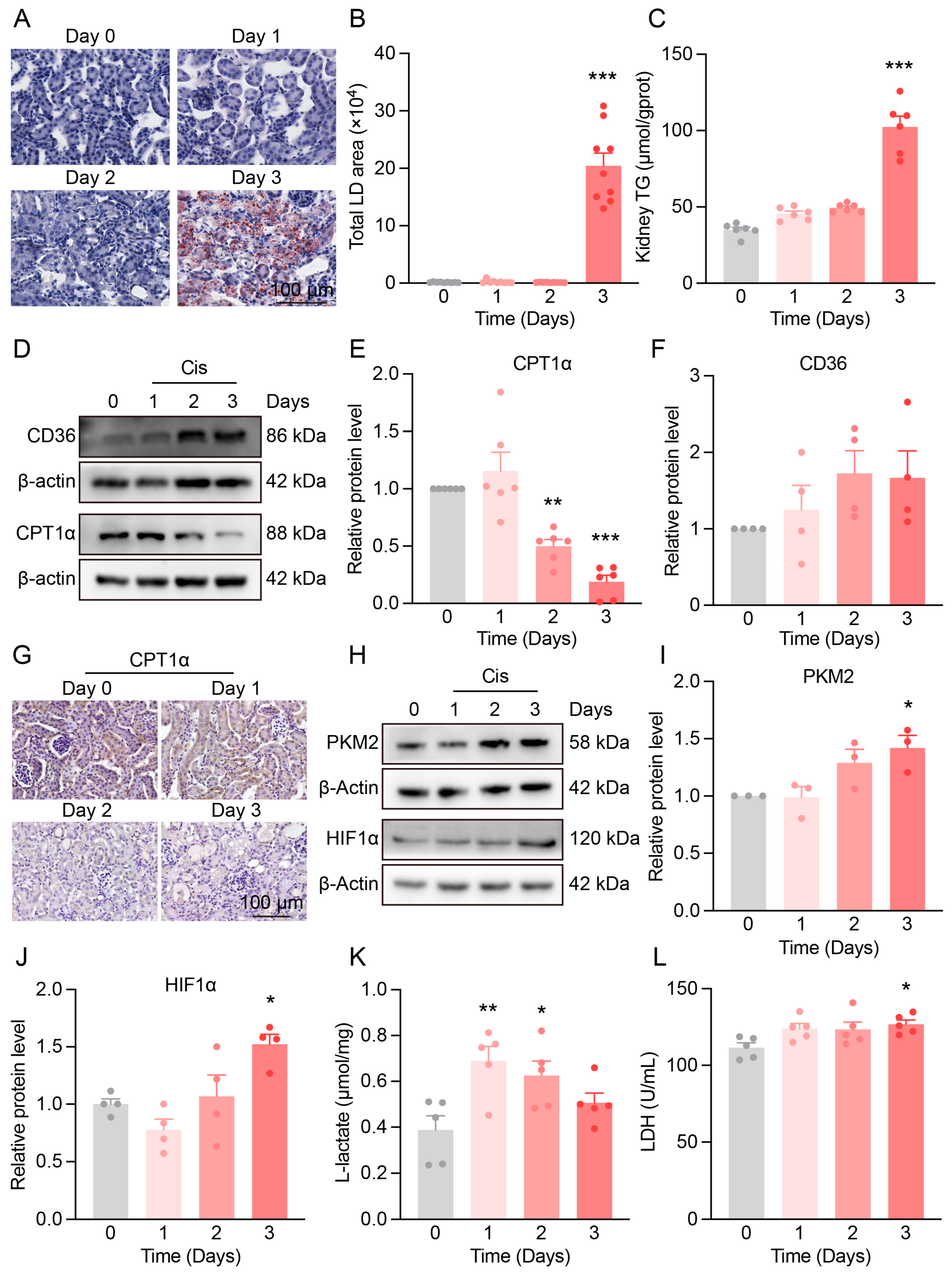
3.1. Progressive Kidney Function and Structural Damage in Cis-AKI Mice
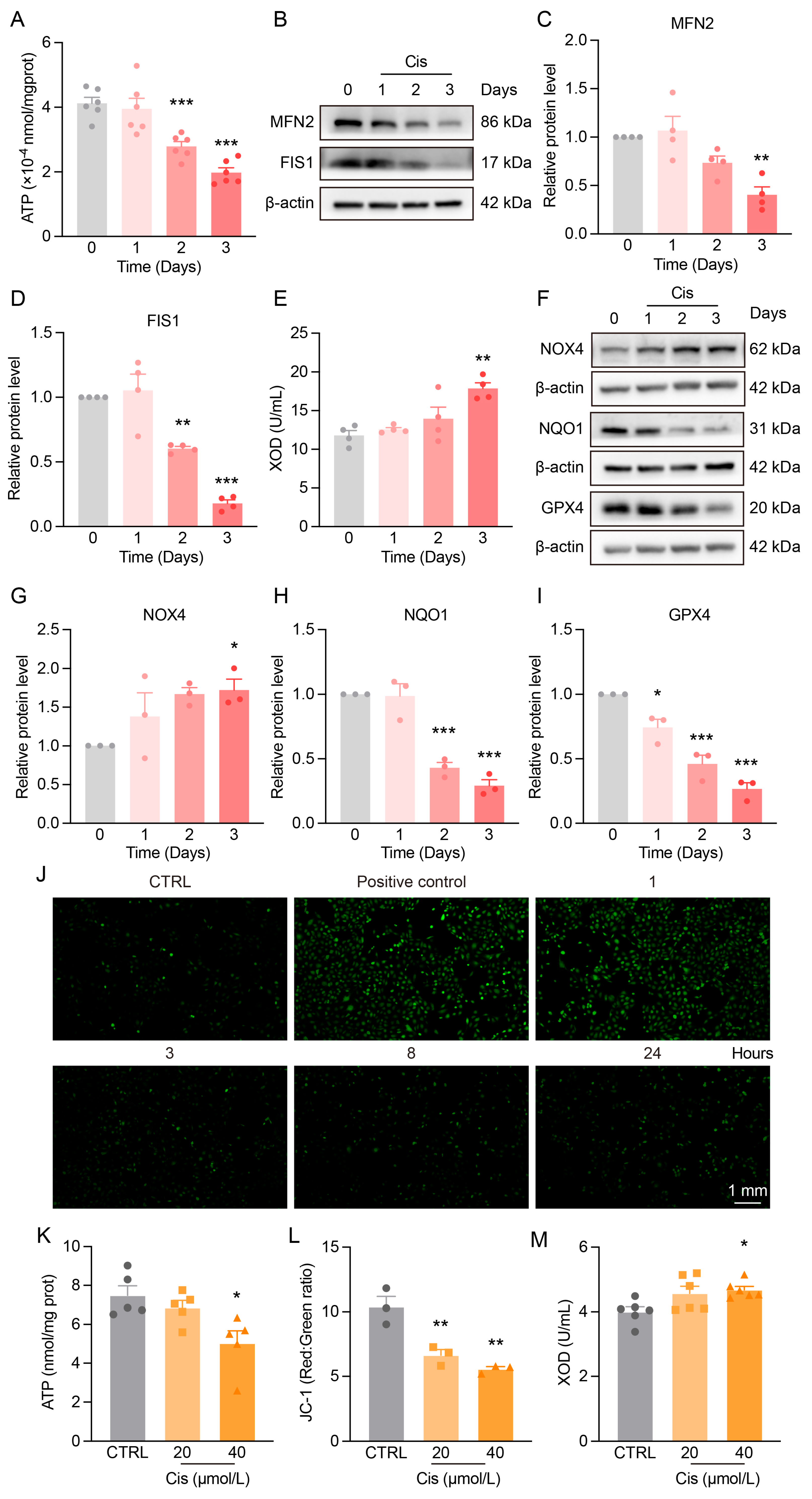
3.2. Mitochondrial Dysfunction and Redox Imbalance in the Kidneys of Cis-AKI Mice
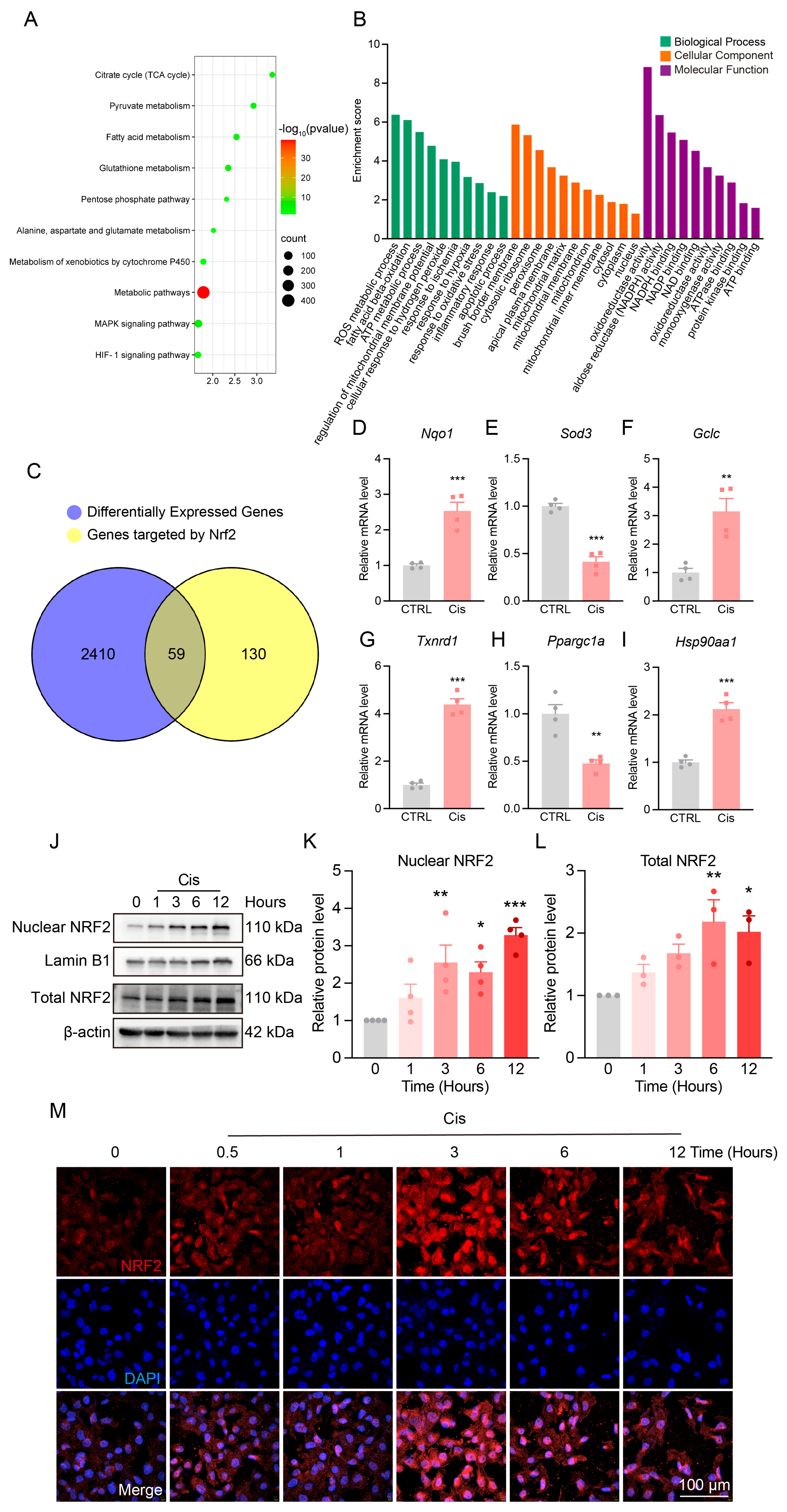
3.3. Metabolic Reprogramming in the Kidneys of Cis-AKI Mice
3.4. Changes in NRF2 Expression and Localization After Cisplatin Treatment
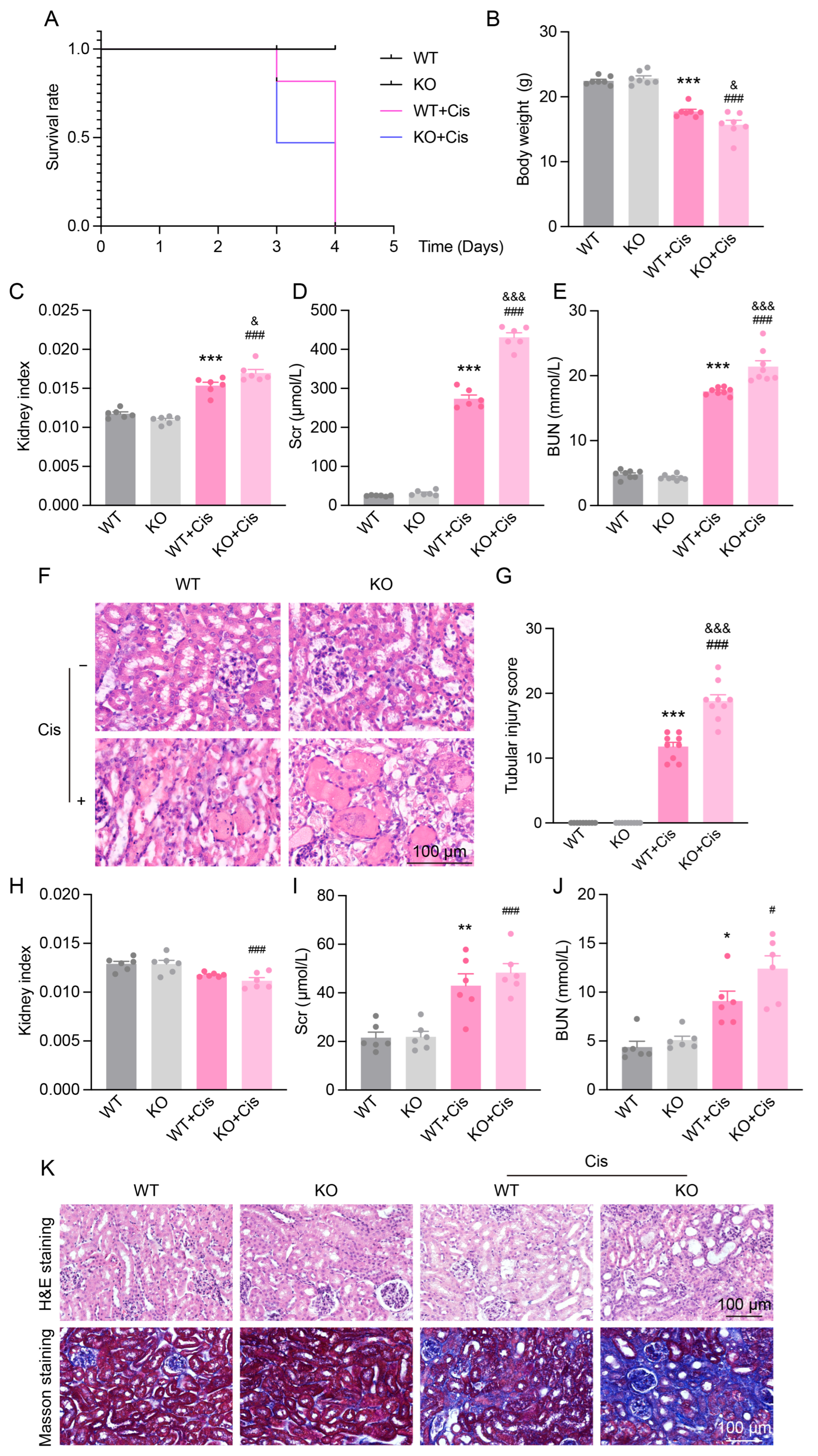
3.5. Loss of NRF2 Aggravates Cisplatin-Induced Renal Dysfunction and Structural Damage
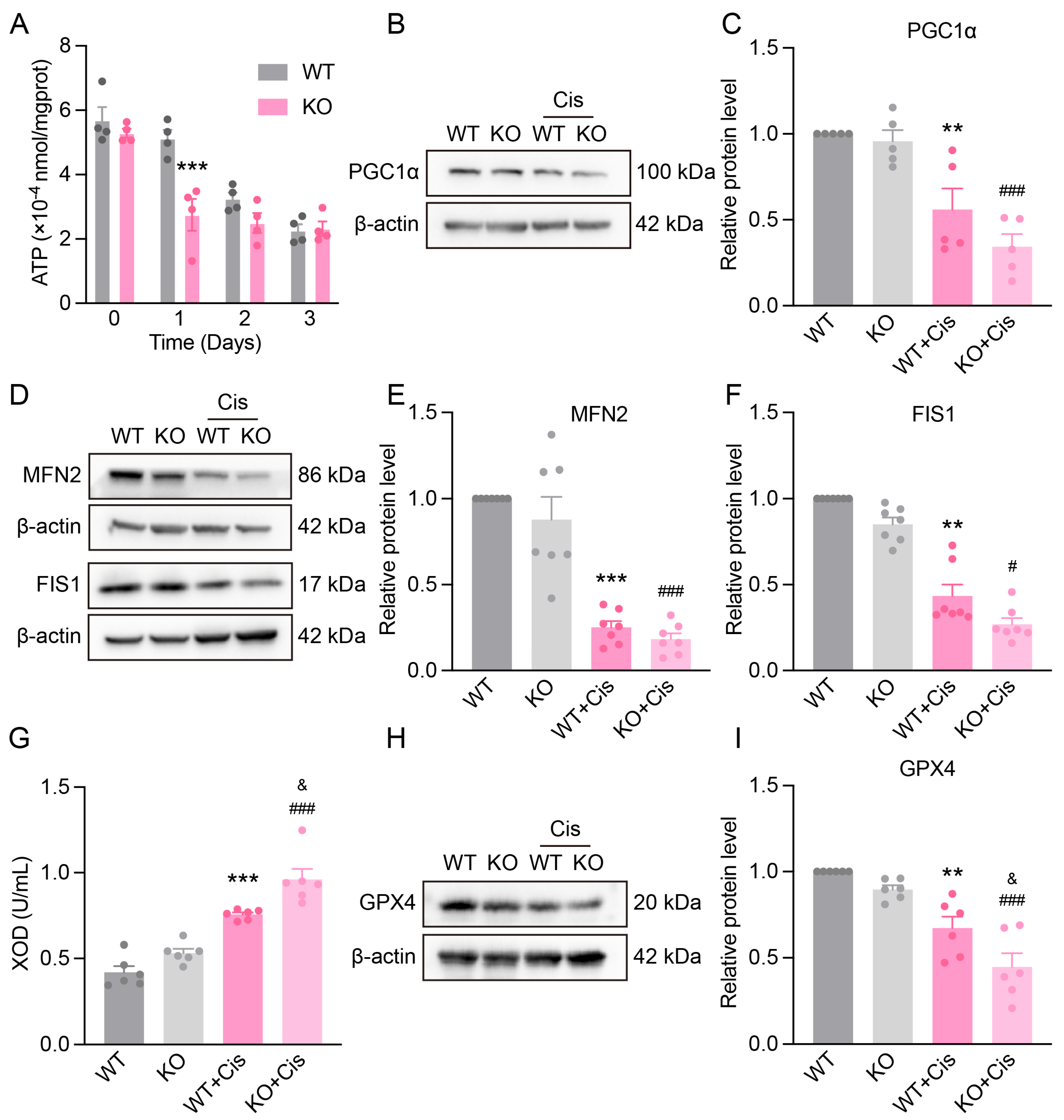
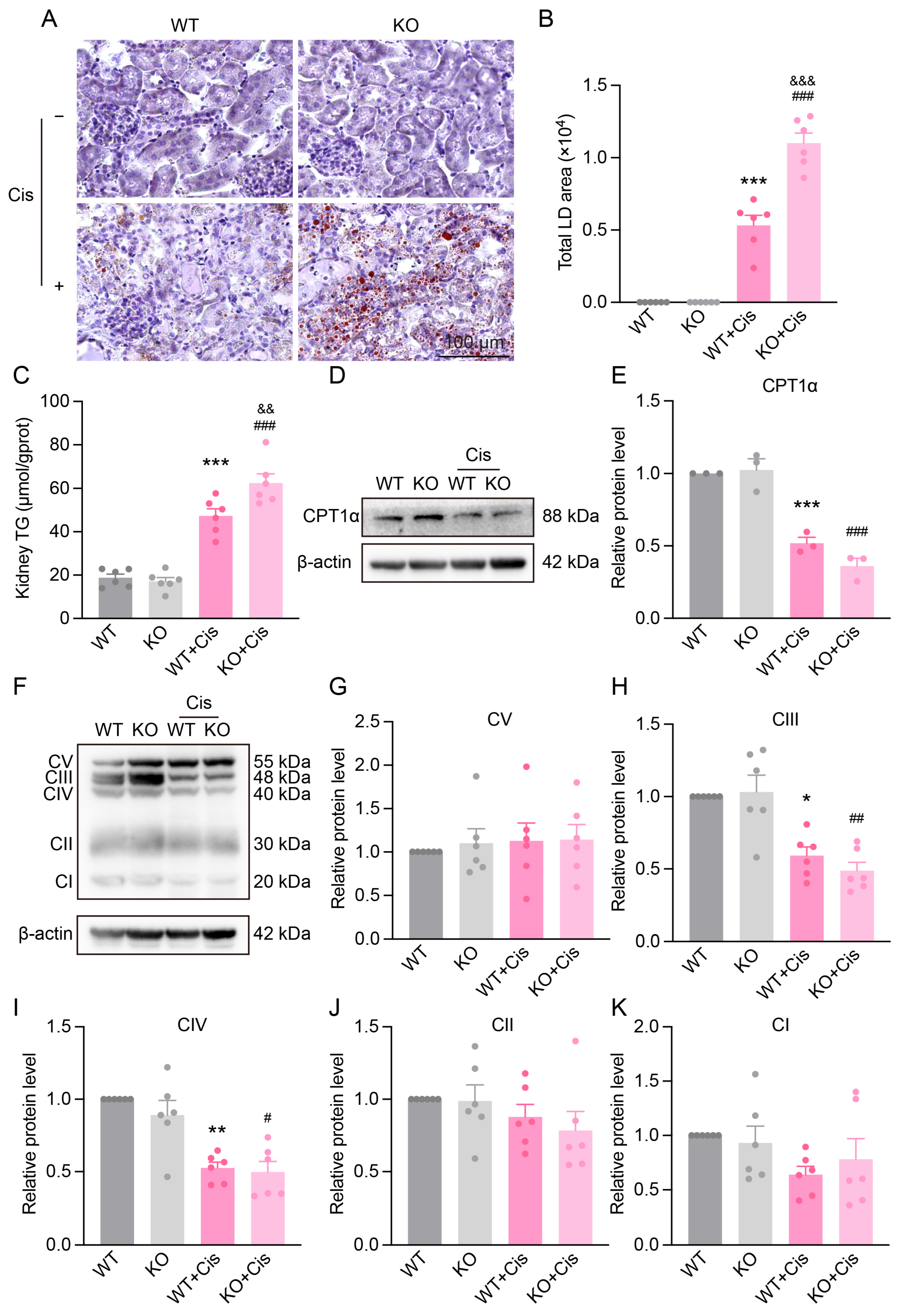
3.6. Loss of NRF2 Aggravates Cisplatin-Induced Renal Mitochondrial Dysfunction
3.7. Loss of NRF2 Aggravates Cisplatin-Induced Metabolism Disruption
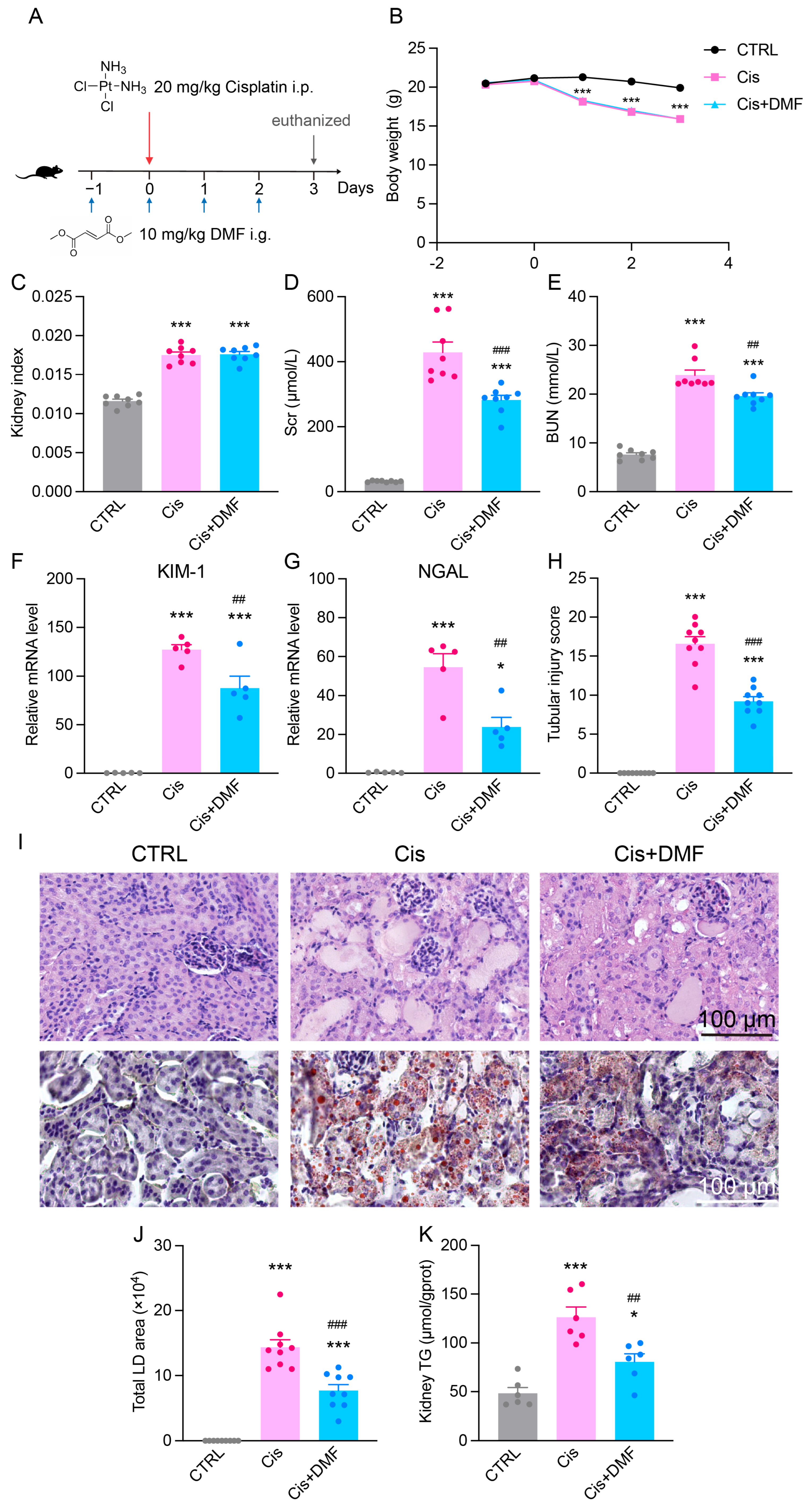
3.8. Pharmacological Activation of NRF2 Alleviates Cisplatin-Induced Renal Dysfunction and Lipid Accumulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKI | Acute Kidney Injury |
| ATP | Adenosine Triphosphate |
| BUN | Blood Urea Nitrogen |
| CD36 | Cluster of Differentiation 36 |
| CKD | Chronic Kidney Disease |
| CK-MB | Creatine Kinase-MB Isoenzyme |
| Cis | Cisplatin |
| Cis-AKI | Cisplatin-Induced Acute Kidney Injury |
| CPT1α | Carnitine Palmitoyltransferase 1α |
| DMF | Dimethyl Fumarate |
| FIS1 | Mitochondrial Fission 1 Protein |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| GPX4 | Glutathione Peroxidase 4 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KIM-1 | Kidney Injury Molecule-1 |
| LDH | Lactate Dehydrogenase |
| MFN2 | Mitofusin 2 |
| MS | Multiple Sclerosis |
| mtDNA | Mitochondrial DNA |
| nDNA | Nuclear DNA |
| NGAL | Neutrophil Gelatinase-Associated Lipocalin |
| NOX4 | NADPH Oxidase 4 |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| NQO1 | NAD(P)H Quinone Dehydrogenase 1 |
| PGC1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha |
| PKM2 | Pyruvate Kinase M2 |
| PTECs | Proximal Tubular Epithelial Cells |
| ROS | Reactive Oxygen Species |
| Scr | Serum Creatinine |
| XOD | Xanthine Oxidase |
| PKM2 | Pyruvate Kinase M2 |
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequences (5′-3′) |
|---|---|
| Havcr1 (mouse) NM_001166631 | F: ACATATCGTGGAATCACAACGAC R: ACTGCTCTTCTGATAGGTGACA |
| Lcn2 (mouse) NM_008491 | F: GAAATATGCACAGGTATCCTC R: GTAATTTTGAAGTATTGCTTGTTT |
| Gapdh (mouse) NM_008084 | F: TGTGTCCGTCGTGGATCTGA R: TTGCTGTTGAAGTCGCAGGAG |
| HAVCR1 (human) BC013325 | F: TCACATCCATGTGCTGGAAT R: CGTGTGTCCTTCCGATAGGT |
| LCN2 (human) NM_005564 | F: GTGAGCACCAACTACAACCAGC R: GTTCCGAAGTCAGCTCCTTGGT |
| ACTB (human) NM_001101 | F: CACCATTGGCAATGAGCGGTTC R: AGGTCTTTGCGGATGTCCACGT |
| Name | Catalog Number | Company | WB Dilution Ratio | IHC/IF Dilution Ratio |
|---|---|---|---|---|
| NQO1 | 67240-1-Ig | Proteintech | 1:10,000 | / |
| GPX4 | A11243 | ABclonal | 1:5000 | / |
| NOX4 | 14347-1-AP | Proteintech | 1:5000 | / |
| MFN2 | A12771 | ABclonal | 1:1000 | / |
| FIS1 | 10956-1-AP | Proteintech | 1:5000 | / |
| CD36 | A14714 | ABclonal | 1:1000 | / |
| CD36 | BD-PT-5585 | Biodragon | 1:2000 | / |
| CPT1A | A5307 | ABclonal | 1:2500 | 1:200 |
| PKM2 | A18799 | ABclonal | 1:1000 | / |
| HIF-1α | A11945 | ABclonal | 1:1000 | / |
| NRF2 | 80593-1-RR | Proteintech | 1:2500 | / |
| NRF2 | 16396-1-AP | Proteintech | / | 1:250 |
| PGC1α | A12348 | ABclonal | 1:1000 | / |
| OxPhos Rodent | 45-8099 | Invitrogen | 1:250 | / |
| Beta Actin | 81115-1-RR | Proteintech | 1:10,000 | / |
| Lamin B1 | 66095-1-Ig | Proteintech | 1:10,000 | / |
| HRP-conjugated Goat Anti-Mouse IgG (H + L) | SA00001-1 | Proteintech | 1:10,000 | / |
| HRP-conjugated Goat Anti-Rabbit IgG (H + L) | SA00001-2 | Proteintech | 1:10,000 | / |
| Cy3-conjugated Goat Anti-Rabbit IgG (H + L) | SA00009-2 | Proteintech | / | 1:100 |
| HRP-conjugated Goat Anti-Rabbit IgG (H&L) | BE0101 | EASYBIO | / | 1:200 |
References
- Fuertes, M.A.; Alonso, C.; Pérez, J.M. Biochemical modulation of Cisplatin mechanisms of action: Enhancement of antitumor activity and circumvention of drug resistance. Chem. Rev. 2003, 103, 645–662. [Google Scholar] [CrossRef]
- César, B.N.; Durão Júnior, M.S. Acute kidney injury in cancer patients. Rev. Assoc. Medica Bras. 2020, 66 (Suppl. 1), s25–s30. [Google Scholar] [CrossRef] [PubMed]
- Sears, S.M.; Orwick, A.; Siskind, L.J. Modeling Cisplatin-Induced Kidney Injury to Increase Translational Potential. Nephron 2023, 147, 13–16. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Safirstein, R.; Dong, Z. Cisplatin nephrotoxicity: New insights and therapeutic implications. Nat. Rev. Nephrol. 2023, 19, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.W.; Tew, K.D.; Townsend, D.M. Cisplatin chemotherapy and renal function. Adv. Cancer Res. 2021, 152, 305–327. [Google Scholar] [CrossRef]
- Volovat, S.; Apetrii, M.; Stefan, A.; Vlad, C.; Voroneanu, L.; Hogas, M.; Haisan, A.; Volovat, C.; Hogas, S. Cisplatin and AKI: An ongoing battle with new perspectives—A narrative review. Int. Urol. Nephrol. 2023, 55, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef]
- Motwani, S.S.; Kaur, S.S.; Kitchlu, A. Cisplatin Nephrotoxicity: Novel Insights into Mechanisms and Preventative Strategies. Semin. Nephrol. 2022, 42, 151341. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e1266. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Aminuddin, A.; Ng, P.Y.; Leong, C.O.; Chua, E.W. Mitochondrial DNA alterations may influence the cisplatin responsiveness of oral squamous cell carcinoma. Sci. Rep. 2020, 10, 7885. [Google Scholar] [CrossRef] [PubMed]
- Cyran, A.M.; Zhitkovich, A. HIF1, HSF1, and NRF2: Oxidant-Responsive Trio Raising Cellular Defenses and Engaging Immune System. Chem. Res. Toxicol. 2022, 35, 1690–1700. [Google Scholar] [CrossRef]
- Zhu, Z.; Hu, J.; Chen, Z.; Feng, J.; Yang, X.; Liang, W.; Ding, G. Transition of acute kidney injury to chronic kidney disease: Role of metabolic reprogramming. Metabolism 2022, 131, 155194. [Google Scholar] [CrossRef]
- Xu, C.; Hong, Q.; Zhuang, K.; Ren, X.; Cui, S.; Dong, Z.; Wang, Q.; Bai, X.; Chen, X. Regulation of pericyte metabolic reprogramming restricts the AKI to CKD transition. Metabolism 2023, 145, 155592. [Google Scholar] [CrossRef]
- Ren, L.; Cui, H.; Wang, Y.; Ju, F.; Cai, Y.; Gang, X.; Wang, G. The role of lipotoxicity in kidney disease: From molecular mechanisms to therapeutic prospects. Biomed. Pharmacother. 2023, 161, 114465. [Google Scholar] [CrossRef]
- Bondi, C.D.; Hartman, H.L.; Tan, R.J. NRF2 in kidney physiology and disease. Physiol. Rep. 2024, 12, e15961. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.J.; Kim, J.E.; Bae, S.Y.; Cho, E.; Ahn, S.Y.; Kwon, Y.J.; Ko, G.J. Impaired NRF2 Inhibits Recovery from Ischemic Reperfusion Injury in the Aging Kidney. Antioxidants 2023, 12, 1440. [Google Scholar] [CrossRef]
- Tan, R.J.; Chartoumpekis, D.V.; Rush, B.M.; Zhou, D.; Fu, H.; Kensler, T.W.; Liu, Y. Keap1 hypomorphism protects against ischemic and obstructive kidney disease. Sci. Rep. 2016, 6, 36185. [Google Scholar] [CrossRef]
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, H.; Huang, L.; Zhu, X.; Sha, J.; Li, G.; Ma, G.; Zhang, W.; Gu, M.; Guo, Y. Nrf2 signaling attenuates epithelial-to-mesenchymal transition and renal interstitial fibrosis via PI3K/Akt signaling pathways. Exp. Mol. Pathol. 2019, 111, 104296. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Amador-Martínez, I.; Medina-Campos, O.N.; Garcia-Garcia, M.; Bernabe-Yepes, B.; León-Contreras, J.C.; Hernández-Pando, R.; Aparicio-Trejo, O.E.; Sánchez-Lozada, L.G.; et al. Sulforaphane protects from kidney damage during the release of unilateral ureteral obstruction (RUUO) by activating nuclear factor erythroid 2-related factor 2 (Nrf2): Role of antioxidant, anti-inflammatory, and antiapoptotic mechanisms. Free. Radic. Biol. Med. 2024, 212, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Sun, Y.; Wang, Y.; Lin, S.; An, Y.; Wang, L.; Liu, J.; Huang, Y.; Yang, B.; Zhou, H. Activation of NRF2 Signaling Pathway Delays the Progression of Hyperuricemic Nephropathy by Reducing Oxidative Stress. Antioxidants 2023, 12, 1022. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Takama, H.; Ichikawa, T.; Mukai, K.; Kojima, M.; Suzuki, Y.; Watada, H.; Wada, T.; Ueki, K.; Narita, I.; et al. Randomized, double-blind, placebo-controlled phase 3 study of bardoxolone methyl in patients with diabetic kidney disease: Design and baseline characteristics of the AYAME study. Nephrol. Dial. Transplant. 2023, 38, 1204–1216. [Google Scholar] [CrossRef]
- Rasmussen, M.; Hansen, K.H.; Scholze, A. Nrf2 Protein Serum Concentration in Human CKD Shows a Biphasic Behavior. Antioxidants 2023, 12, 932. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef]
- Paller, M.S.; Hoidal, J.R.; Ferris, T.F. Oxygen free radicals in ischemic acute renal failure in the rat. J. Clin. Investig. 1984, 74, 1156–1164. [Google Scholar] [CrossRef]
- Doke, T.; Mukherjee, S.; Mukhi, D.; Dhillon, P.; Abedini, A.; Davis, J.G.; Chellappa, K.; Chen, B.; Baur, J.A.; Susztak, K. NAD+ precursor supplementation prevents mtRNA/RIG-I-dependent inflammation during kidney injury. Nat. Metab. 2023, 5, 414–430. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Chen, Q.M. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free. Radic. Biol. Med. 2022, 179, 133–143. [Google Scholar] [CrossRef]
- Dong, W.; Sun, Y.; Cheng, H.; Yang, B.; Wang, L.; Jiang, Z.; Li, B.; Wen, S.; Guo, X.; Guan, D.; et al. Dynamic cell type-specific expression of Nrf2 after traumatic brain injury in mice. Eur. J. Neurosci. 2019, 50, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Ushimoto, C.; Sugiki, S.; Kunii, K.; Inoue, S.; Kuroda, E.; Akai, R.; Iwawaki, T.; Miyazawa, K. Dynamic change and preventive role of stress response via Keap1-Nrf2 during renal crystal formation. Free. Radic. Biol. Med. 2023, 207, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Kirita, Y.; Wu, H.; Uchimura, K.; Wilson, P.C.; Humphreys, B.D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. USA 2020, 117, 15874–15883. [Google Scholar] [CrossRef]
- Li, H.; Dixon, E.E.; Wu, H.; Humphreys, B.D. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. 2022, 34, 1977–1998.e9. [Google Scholar] [CrossRef]
- Hejazian, S.M.; Hosseiniyan Khatibi, S.M.; Barzegari, A.; Pavon-Djavid, G.; Razi Soofiyani, S.; Hassannejhad, S.; Ahmadian, E.; Ardalan, M.; Zununi Vahed, S. Nrf-2 as a therapeutic target in acute kidney injury. Life Sci. 2021, 264, 118581. [Google Scholar] [CrossRef]
- Liang, N.N.; Guo, Y.Y.; Zhang, X.Y.; Ren, Y.H.; He, Y.Z.; Liu, Z.B.; Xu, D.X.; Xu, S. Mitochondrial Dysfunction-Evoked DHODH Acetylation is Involved in Renal Cell Ferroptosis during Cisplatin-Induced Acute Kidney Injury. Adv. Sci. 2024, 11, e2404753. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.I.; Jo, E.R.; Song, H. Mitophagy Impairment Aggravates Cisplatin-Induced Ototoxicity. BioMed Res. Int. 2021, 2021, 5590973. [Google Scholar] [CrossRef]
- Granath-Panelo, M.; Kajimura, S. Mitochondrial heterogeneity and adaptations to cellular needs. Nat. Cell Biol. 2024, 26, 674–686. [Google Scholar] [CrossRef]
- Galichon, P.; Lannoy, M.; Li, L.; Serre, J.; Vandermeersch, S.; Legouis, D.; Valerius, M.T.; Hadchouel, J.; Bonventre, J.V. Energy depletion by cell proliferation sensitizes the kidney epithelial cells to injury. Am. J. Physiol.-Ren. Physiol. 2024, 326, F326–F337. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Kato, M.; Abdollahi, M.; Tunduguru, R.; Tsark, W.; Chen, Z.; Wu, X.; Wang, J.; Chen, Z.B.; Lin, F.M.; Lanting, L.; et al. miR-379 deletion ameliorates features of diabetic kidney disease by enhancing adaptive mitophagy via FIS1. Commun. Biol. 2021, 4, 30. [Google Scholar] [CrossRef]
- Zhang, S.; Qian, S.; Liu, H.; Xu, D.; Xia, W.; Duan, H.; Wang, C.; Yu, S.; Chen, Y.; Ji, P.; et al. LRRK2 aggravates kidney injury through promoting MFN2 degradation and abnormal mitochondrial integrity. Redox Biol. 2023, 66, 102860. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Han, Y.K.; Kong, M.J.; Park, K.M. Short-term control of diet affects cisplatin-induced acute kidney injury through modulation of mitochondrial dynamics and mitochondrial GSH. Physiol. Rep. 2022, 10, e15348. [Google Scholar] [CrossRef]
- Duan, H.; Yang, X.; Cai, S.; Zhang, L.; Qiu, Z.; Wang, J.; Wang, S.; Li, Z.; Li, X. Nrf2 mitigates sepsis-associated encephalopathy-induced hippocampus ferroptosis via modulating mitochondrial dynamic homeostasis. Int. Immunopharmacol. 2024, 143, 113331. [Google Scholar] [CrossRef]
- Katanić Stanković, J.S.; Selaković, D.; Rosić, G. Oxidative Damage as a Fundament of Systemic Toxicities Induced by Cisplatin—The Crucial Limitation or Potential Therapeutic Target? Int. J. Mol. Sci. 2023, 24, 14574. [Google Scholar] [CrossRef]
- García-Caballero, C.; Guerrero-Hue, M.; Vallejo-Mudarra, M.; Palomino Antolin, A.; Decouty-Pérez, C.; Sánchez-Mendoza, L.M.; Villalba, J.M.; González-Reyes, J.A.; Opazo-Rios, L.; Vázquez-Carballo, C.; et al. Nox4 is involved in acute kidney injury associated to intravascular hemolysis. Free. Radic. Biol. Med. 2024, 225, 430–444. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Wang, B.; Zhang, Z.; Jiang, L.; Qin, Z.; Zhao, Y.; Su, B. NOX4 is a potential therapeutic target in septic acute kidney injury by inhibiting mitochondrial dysfunction and inflammation. Theranostics 2023, 13, 2863–2878. [Google Scholar] [CrossRef]
- Schmidt, H.M.; Kelley, E.E.; Straub, A.C. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019, 21, 101072. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Xuan, C.; Li, X.; Tan, Y.; Yang, M.; Cao, M.; Chen, C.; Huang, X.; Hu, R. DPP9 regulates NQO1 and ROS to promote resistance to chemotherapy in liver cancer cells. Redox Biol. 2024, 75, 103292. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in cell death, autophagy, and disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Vomhof-Dekrey, E.E.; Picklo, M.J., Sr. The Nrf2-antioxidant response element pathway: A target for regulating energy metabolism. J. Nutr. Biochem. 2012, 23, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef]
- Li, Y.; Huang, X.; Yang, G.; Xu, K.; Yin, Y.; Brecchia, G.; Yin, J. CD36 favours fat sensing and transport to govern lipid metabolism. Prog. Lipid Res. 2022, 88, 101193. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, L.; Zhang, Z.; Yang, P.; Chen, Q.; Zeng, X.; Tan, F.; Wang, C.; Ruan, X.; Liao, X. CD36 promotes tubular ferroptosis by regulating the ubiquitination of FSP1 in acute kidney injury. Genes Dis. 2024, 11, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Ren, X.; Tan, E.; Wan, X.; Wang, Y.; Shi, H.; Hou, Y.; Wang, L. CD36 deletion ameliorates diabetic kidney disease by restoring fatty acid oxidation and improving mitochondrial function. Ren. Fail. 2023, 45, 2292753. [Google Scholar] [CrossRef] [PubMed]
- Liang, K. Mitochondrial CPT1A: Insights into structure, function, and basis for drug development. Front. Pharmacol. 2023, 14, 1160440. [Google Scholar] [CrossRef]
- Miguel, V.; Tituaña, J.; Herrero, J.I.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Expósito, L.; Ruiz-Ortega, M.; et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 2021, 131, e140695. [Google Scholar] [CrossRef]
- Deja, S.; Fletcher, J.A.; Kim, C.W.; Kucejova, B.; Fu, X.; Mizerska, M.; Villegas, M.; Pudelko-Malik, N.; Browder, N.; Inigo-Vollmer, M.; et al. Hepatic malonyl-CoA synthesis restrains gluconeogenesis by suppressing fat oxidation, pyruvate carboxylation, and amino acid availability. Cell Metab. 2024, 36, 1088–1104.e12. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Huang, H.; Zhao, Y. Interplay between metabolic reprogramming and post-translational modifications: From glycolysis to lactylation. Front. Immunol. 2023, 14, 1211221. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Cai, X.; Wang, T.; Ye, C.; Xu, G.; Xie, L. Relationship between lactate dehydrogenase and albuminuria in Chinese hypertensive patients. J. Clin. Hypertens. 2021, 23, 128–136. [Google Scholar] [CrossRef]
- Azushima, K.; Kovalik, J.P.; Yamaji, T.; Ching, J.; Chng, T.W.; Guo, J.; Liu, J.J.; Nguyen, M.; Sakban, R.B.; George, S.E.; et al. Abnormal lactate metabolism is linked to albuminuria and kidney injury in diabetic nephropathy. Kidney Int. 2023, 104, 1135–1149. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; Xiong, J. The Role of Hypoxia-Inducible Factor-1 Alpha in Renal Disease. Molecules 2022, 27, 7318. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Takagaki, Y.; Kumagai, A.; Kanasaki, K.; Koya, D. The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J. Diabetes Investig. 2021, 12, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, Y.; Qiao, P.; Ying, Y.; Lin, S.; Lu, F.; Gao, C.; Li, M.; Yang, B.; Zhou, H. Mechanisms of Cisplatin-Induced Acute Kidney Injury: The Role of NRF2 in Mitochondrial Dysfunction and Metabolic Reprogramming. Antioxidants 2025, 14, 775. https://doi.org/10.3390/antiox14070775
Liu J, Wang Y, Qiao P, Ying Y, Lin S, Lu F, Gao C, Li M, Yang B, Zhou H. Mechanisms of Cisplatin-Induced Acute Kidney Injury: The Role of NRF2 in Mitochondrial Dysfunction and Metabolic Reprogramming. Antioxidants. 2025; 14(7):775. https://doi.org/10.3390/antiox14070775
Chicago/Turabian StyleLiu, Jihan, Yiming Wang, Panshuang Qiao, Yi Ying, Simei Lin, Feng Lu, Cai Gao, Min Li, Baoxue Yang, and Hong Zhou. 2025. "Mechanisms of Cisplatin-Induced Acute Kidney Injury: The Role of NRF2 in Mitochondrial Dysfunction and Metabolic Reprogramming" Antioxidants 14, no. 7: 775. https://doi.org/10.3390/antiox14070775
APA StyleLiu, J., Wang, Y., Qiao, P., Ying, Y., Lin, S., Lu, F., Gao, C., Li, M., Yang, B., & Zhou, H. (2025). Mechanisms of Cisplatin-Induced Acute Kidney Injury: The Role of NRF2 in Mitochondrial Dysfunction and Metabolic Reprogramming. Antioxidants, 14(7), 775. https://doi.org/10.3390/antiox14070775