

Role of Oxidative Stress and Neuroinflammation in the Etiology of Alzheimer’s Disease: Therapeutic Options

Abstract
1. Introduction
2. Synaptic Plasticity
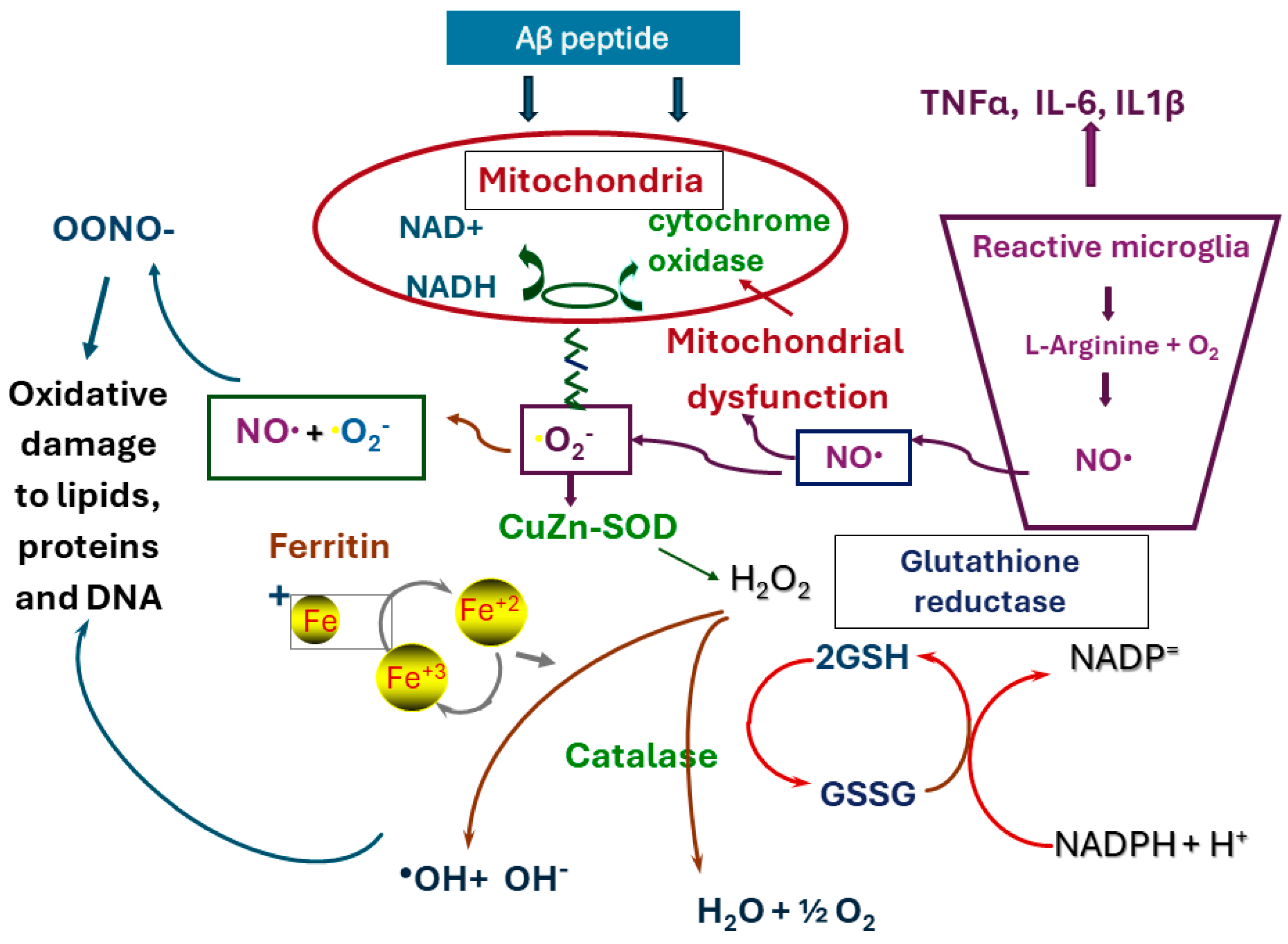
3. Mitochondrial Activity and Oxidative, Nitrative Stress
4. Role of Microglia in Neuroinflammation
5. Genes of the Immune System as Risk or Protective Factors in AD
6. Biomarkers for Aβ and Tau in AD
7. TREM2 in Combination with Aβ and Tau as Biomarkers for AD
8. Drugs That Reduce Oxidative Stress
8.1. Vitamin E
8.2. Curcumin
8.3. Resveratrol
8.4. Ginkgo Biloba
9. Anti-Inflammatory Drugs
9.1. Non-Steroidal Anti-Inflammatory Agents
9.2. Antibodies Against TNFα
10. Drug with Antioxidant and Anti-Inflammatory Activity
Ladostigil (SPE100)
11. Conclusions
Funding
Conflicts of Interest
References
- Alzheimer, A. A contribution concerning the pathological anatomy of mental disturbances in old age, 1899. Alzheimer Dis. Assoc. Disord. 1991, 5, 69–70. [Google Scholar] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Busciglio, J.; Gabuzda, D.H.; Matsudaira, P.; Yankner, B.A. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA 1993, 90, 2092–2096. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Lue, L.-F.; Kuo, Y.-M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- Rodrigue, K.; Kennedy, K.; Devous, M.D., Sr.; Rieck, J.; Hebrank, A.; Diaz-Arrastia, R.; Mathews, D.; Park, D. β-Amyloid burden in healthy aging: Regional distribution and cognitive consequences. Neurology 2012, 78, 387–395. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A.; Zheng, N.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R.; et al. p38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 1999, 72, 2053–2058. [Google Scholar] [CrossRef]
- Kelleher, I.; Garwood, C.; Hanger, D.P.; Anderton, B.H.; Noble, W. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 2007, 103, 2256–2267. [Google Scholar] [CrossRef]
- Lloret, A.; Badía, M.-C.; Mora, N.J.; Ortega, A.; Pallardó, F.V.; Alonso, M.-D.; Atamna, H.; Viña, J. Gender and age-dependent differences in the mitochondrial apoptogenic pathway in Alzheimer’s disease. Free Radic. Biol. Med. 2008, 44, 2019–2025. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Nery, E.S.M.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Van Dyk, D.; Llewellyn, R.L.; Gordon, P.C.; Brink, J.G. Sylvia Annigje Magdalena Heijke (1955–2022). S. Afr. Med. J. 2023, 113, 4. [Google Scholar]
- Bilousova, T.; Miller, C.A.; Poon, W.; Vinters, H.V.; Corrada, M.; Kawas, C.; Hayden, E.; Teplow, D.B.; Glabe, C.; Albay, R., 3rd; et al. Synaptic Amyloid-β Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. Am. J. Pathol. 2016, 186, 185–198. [Google Scholar] [CrossRef]
- Jeremic, D.; Navarro-López, J.D.; Jiménez-Díaz, L. Efficacy and safety of anti-amyloid-β monoclonal antibodies in current Alzheimer’s disease phase III clinical trials: A systematic review and interactive web app-based meta-analysis. Ageing Res. Rev. 2023, 90, 102012. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.; Grant, S.G. Synapse pathology in Alzheimer’s disease. Semin. Cell Dev. Biol. 2023, 139, 13–23. [Google Scholar] [CrossRef]
- Gonatas, N.K.; Anderson, W.; Evangelista, I. The contribution of altered synapses in the senile plaque: An electron microscopic study in Alzheimer’s dementia. J. Neuropathol. Exp. Neurol. 1967, 26, 25–39. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 2004, 256, 183–194. [Google Scholar] [CrossRef]
- Clark, D. An annotated bibliography of the publications of Cicely Saunders--2: 1968-77. Palliat. Med. 1999, 13, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 2004, 490, 115–125. [Google Scholar] [CrossRef]
- de Leon, M.J.; Convit, A.; Wolf, O.T.; Tarshish, C.Y.; DeSanti, S.; Rusinek, H.; Tsui, W.; Kandil, E.; Scherer, A.J.; Roche, A.; et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. USA 2001, 98, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Pupi, A.; de Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 2006, 27, 1564–1576. [Google Scholar] [CrossRef]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef]
- Montine, K.S.; Quinn, J.F.; Zhang, J.; Fessel, J.P.; Roberts, L.J., 2nd; Morrow, J.D.; Montine, T.J. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem. Phys. Lipids 2004, 128, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Kish, S.J. Mitochondria in Alzheimer’s disease. Int. Rev. Neurobiol. 2002, 53, 341–385. [Google Scholar] [CrossRef]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef]
- Parihar, M.S.; Brewer, G.J. Amyloid-β as a modulator of synaptic plasticity. J. Alzheimer’s Dis. 2010, 22, 741–763. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Schramm, E.; Waisman, A. Microglia as Central Protagonists in the Chronic Stress Response. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e200023. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Eyo, U.B.; Wu, L.-J. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. 2013, 2013, 456857. [Google Scholar] [CrossRef]
- Hanlon, L.; Raghupathi, R.; Huh, J. Differential effects of minocycline on microglial activation and neurodegeneration following closed head injury in the neonate rat. Exp. Neurol. 2017, 290, 1–14. [Google Scholar] [CrossRef]
- Illes, P.; Burnstock, G.; Tang, Y. Astroglia-Derived ATP Modulates CNS Neuronal Circuits. Trends Neurosci. 2019, 42, 885–898. [Google Scholar] [CrossRef]
- Savage, J.C.; Carrier, M.; Tremblay, M.E. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019, 2034, 13–26. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C.; Kufer, T.A. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in Alzheimer’s Disease: Risk Factors and Inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Orr, A.G.; Orr, A.L.; Li, X.-J.; Gross, R.E.; Traynelis, S.F. Adenosine A(2A) receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef]
- Gao, Z. Adenosine A(2A) receptor and glia. Int. Rev. Neurobiol. 2023, 170, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A(2A) receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef] [PubMed]
- Green, T.R.F.; Rowe, R.K. Quantifying microglial morphology: An insight into function. Clin. Exp. Immunol. 2024, 216, 221–229. [Google Scholar] [CrossRef]
- Perry, V.H.; Matyszak, M.K.; Fearn, S. Altered antigen expression of microglia in the aged rodent CNS. Glia 1993, 7, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef]
- Kelly, Á.; Vereker, E.; Nolan, Y.; Brady, M.; Barry, C.; Loscher, C.E.; Mills, K.H.G.; Lynch, M.A. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1β on long term potentiation in rat dentate gyrus. J. Biol. Chem. 2003, 278, 19453–19462. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Michetti, F.; Di Sante, G.; Clementi, M.E.; Sampaolese, B.; Casalbore, P.; Volonté, C.; Spica, V.R.; Parnigotto, P.P.; Di Liddo, R.; Amadio, S.; et al. Growing role of S100B protein as a putative therapeutic target for neurological- and nonneurological-disorders. Neurosci. Biobehav. Rev. 2021, 127, 446–458. [Google Scholar] [CrossRef]
- Petzold, A.; Jenkins, R.; Watt, H.; Green, A.; Thompson, E.; Keir, G.; Fox, N.; Rossor, M. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci. Lett. 2003, 336, 167–170. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Kleinberger, G.; Caballero, M.Á.A.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM 2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Peterson, B.S.; Bomze, H.M.; Hirschey, M.D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 2015, 26, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef]
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Bhole, R.P.; Chikhale, R.V.; Rathi, K.M. Current biomarkers and treatment strategies in Alzheimer disease: An overview and future perspectives. IBRO Neurosci. Rep. 2024, 16, 8–42. [Google Scholar] [CrossRef]
- Skillbäck, T.; Farahmand, B.Y.; Rosén, C.; Mattsson, N.; Nägga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; Schott, J.M.; et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 2015, 138, 2716–2731. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Lee, E.-H.; Kim, H.-J.; Jo, S.; Lee, S.; Seo, S.W.; Park, H.-H.; Koh, S.-H.; Lee, J.-H. The relationship of soluble TREM2 to other biomarkers of sporadic Alzheimer’s disease. Sci. Rep. 2021, 11, 13050. [Google Scholar] [CrossRef]
- Rauchmann, B.-S.; Sadlon, A.; Perneczky, R. Alzheimer’s Disease Neuroimaging Initiative. Soluble TREM2 and Inflammatory Proteins in Alzheimer’s Disease Cerebrospinal Fluid. J. Alzheimer’s Dis. 2020, 73, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. The Role of Natural Antioxidants in the Prevention of Dementia—Where Do We Stand and Future Perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 10, 59. [Google Scholar] [CrossRef]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Koppal, T.; Subramaniam, R.; Yatin, S. Vitamin Ε as an antioxidant/free radical scavenger against amyloid β-peptide-induced oxidative stress in neocortical synaptosomal membranes and hippocampal neurons in culture: Insights into Alzheimer’s disease. Rev. Neurosci. 1999, 10, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Grundman, M. Vitamin E and Alzheimer disease: The basis for additional clinical trials. Am. J. Clin. Nutr. 2000, 71, 630S–636S. [Google Scholar] [CrossRef]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s disease: What do we know so far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef]
- Ano, M.A.S.; Ernesto, C.H.; Homas, R.O.G.T.; Klauber, M.R.; Chafer, K.I.S.; Rundman, M.I.G.; Oodbury, J.O.W.; Growdon, J.; Otman, C.A.W.C.; Feiffer, E.R.P.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef]
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112. [Google Scholar] [CrossRef]
- Ammon, H.P.T.; Wahl, M.A. Pharmacology of Curcuma longa. Planta Med. 1991, 57, 1–7. [Google Scholar] [CrossRef]
- Ganguli, M.; Chandra, V.; Kamboh, M.I.; Johnston, J.M.; Dodge, H.H.; Thelma, B.K.; Juyal, R.C.; Pandav, R.; Belle, S.H.; DeKosky, S.T. Apolipoprotein E polymorphism and Alzheimer disease: The Indo-US Cross-National Dementia Study. Arch. Neurol. 2000, 57, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Bai, D.; Wei, Z.; Zhang, Y.; Huang, Y.; Deng, H.; Huang, X.; Mukhopadhyay, P. Curcumin attenuates oxidative stress in RAW264.7 cells by increasing the activity of antioxidant enzymes and activating the Nrf2-Keap1 pathway. PLoS ONE 2019, 14, e0216711. [Google Scholar] [CrossRef] [PubMed]
- Frautschy, S.A.; Hu, W.; Kim, P.; Miller, S.A.; Chu, T.; Harris-White, M.E.; Cole, G. M Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol. Aging 2001, 22, 993–1005. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Zhu, Q.; Zhang, S.; OuYang, D.; Lu, J.-H. Resveratrol in experimental Alzheimer’s disease models: A systematic review of preclinical studies. Pharmacol. Res. 2019, 150, 104476. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.-L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-S.; Li, Y.; Deora, G.S.; Ruan, B.-F. Derivatives and Analogues of Resveratrol: Recent Advances in Structural Modification. Mini Rev. Med. Chem. 2019, 19, 809–825. [Google Scholar] [CrossRef]
- Lizard, G.; Latruffe, N.; Vervandier-Fasseur, D. Aza- and Azo-Stilbenes: Bio-Isosteric Analogs of Resveratrol. Molecules 2020, 25, 605. [Google Scholar] [CrossRef]
- Griggs, B. Green Pharmacy: The History and Evolution of Western Herbal Medicine; Viking Press: New York, NY, USA, 1981; p. 326. [Google Scholar]
- Schötz, K.; Waimer, F.; Koch, E. Principal component analysis of production batches of the Ginkgo biloba extract EGb 761 over a period of 10 years on the basis of routine quality control parameters. Planta Med. 2015, 81, 16. [Google Scholar] [CrossRef]
- Oyama, Y.; Chikahisa, L.; Ueha, T.; Kanemaru, K.; Noda, K. Ginkgo biloba extract protects brain neurons against oxidative stress induced by hydrogen peroxide. Brain Res. 1996, 712, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Millan, A.; de la Asuncion, J.G.; Pla, R.; Juan, G.; Pallardo, F.V.; O’connor, E.; Martin, J.A.; Droy-Lefaix, M.-T.; Viña, J. A ginkgo biloba extract (EGb 761) prevents mitochondrial aging by protecting against oxidative stress. Free Radic. Biol. Med. 1998, 24, 298–304. [Google Scholar] [CrossRef]
- Wei, T.; NI, Y.; Hou, J.; Chen, C.; Zhao, B.; Xin, W. Hydrogen peroxide-induced oxidative damage and apoptosis in cerebellar granule cells: Protection by Ginkgo biloba extract. Pharmacol. Res. 2000, 41, 427–433. [Google Scholar] [CrossRef]
- Stoll, S.; Scheuer, K.; Pohl, O.; Müller, W. Ginkgo biloba extract (EGb 761) independently improves changes in passive avoidance learning and brain membrane fluidity in the aging mouse. Pharmacopsychiatry 1996, 29, 144–149. [Google Scholar] [CrossRef]
- Herrschaft, H.; Nacu, A.; Likhachev, S.; Sholomov, I.; Hoerr, R.; Schlaefke, S. Ginkgo biloba extract EGb 761® in dementia with neuropsychiatric features: A randomised, placebo-controlled trial to confirm the efficacy and safety of a daily dose of 240 mg. J. Psychiatr. Res. 2012, 46, 716–723. [Google Scholar] [CrossRef]
- Kandiah, N.; Ong, P.A.; Yuda, T.; Ng, L.; Mamun, K.; Merchant, R.A.; Chen, C.; Dominguez, J.; Marasigan, S.; Ampil, E.; et al. Treatment of dementia and mild cognitive impairment with or without cerebrovascular disease: Expert consensus on the use of Ginkgo biloba extract, EGb 761®. CNS Neurosci. Ther. 2019, 25, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Ihl, R.; Bachinskaya, N.; Korczyn, A.D.; Vakhapova, V.; Tribanek, M.; Hoerr, R.; Napryeyenko, O.; GOTADAY Study Group. Efficacy and safety of a once-daily formulation of Ginkgo biloba extract EGb 761 in dementia with neuropsychiatric features: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2011, 26, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Bohlken, J.; Peters, O.; Kostev, K. Association Between Ginkgo Biloba Extract Prescriptions and Dementia Incidence in Outpatients with Mild Cognitive Impairment in Germany: A Retrospective Cohort Study. J. Alzheimer’s Dis. 2022, 86, 703–709. [Google Scholar] [CrossRef]
- Vellas, B.; Coley, N.; Ousset, P.-J.; Berrut, G.; Dartigues, J.-F.; Dubois, B.; Grandjean, H.; Pasquier, F.; Piette, F.; Robert, P.; et al. Long-term use of standardised Ginkgo biloba extract for the prevention of Alzheimer’s disease (GuidAge): A randomised placebo-controlled trial. Lancet Neurol. 2012, 11, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, R.V.; Malcher, N.S.; Amado, L.L.; Coleman, M.D.; Dos Santos, D.C.; Borges, R.S.; Valente, S.A.S.; Valente, V.C.; Monteiro, M.C.; Pant, A.B. In Vitro Protective Effect and Antioxidant Mechanism of Resveratrol Induced by Dapsone Hydroxylamine in Human Cells. PLoS ONE 2015, 10, e0134768. [Google Scholar] [CrossRef]
- Shi, C.; Zhao, L.; Zhu, B.; Li, Q.; Yew, D.T.; Yao, Z.; Xu, J. Protective effects of Ginkgo biloba extract (EGb761) and its constituents quercetin and ginkgolide B against β-amyloid peptide-induced toxicity in SH-SY5Y cells. Chem.-Biol. Interact. 2009, 181, 115–123. [Google Scholar] [CrossRef]
- Weinreb, O.; Bar-Am, O.; Amit, T.; Drigues, N.; Sagi, Y.; Youdim, M.B. The neuroprotective effect of ladostigil against hydrogen peroxide-mediated cytotoxicity. Chem.-Biol. Interact. 2008, 175, 318–326. [Google Scholar] [CrossRef]
- Shoham, S.; Linial, M.; Weinstock, M. Age-Induced Spatial Memory Deficits in Rats Are Correlated with Specific Brain Region Alterations in Microglial Morphology and Gene Expression. J. Neuroimmune Pharmacol. 2019, 14, 251–262. [Google Scholar] [CrossRef]
- Breitner, J.C.; Welsh, K.A.; Helms, M.J.; Gaskell, P.C.; Gau, B.A.; Roses, A.D.; Pericak-Vance, M.A.; Saunders, A.M. Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol. Aging 1995, 16, 523–530. [Google Scholar] [CrossRef]
- Landi, F.; Cesari, M.; Onder, G.; Russo, A.; Torre, S.; Bernabei, R. Non-steroidal anti-inflammatory drug (NSAID) use and Alzheimer disease in community-dwelling elderly patients. Am. J. Geriatr. Psychiatry 2003, 11, 179–185. [Google Scholar] [CrossRef]
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.; Zandi, P. Non-steroidal anti-inflammatory drugs and Alzheimers disease: The epidemiological evidence. CNS Neurol. Disord. Drug Targets 2010, 9, 132–139. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Mather, A.E.; Peters, R.; Lawrence, C.B.; Brough, D. Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun. 2020, 2, fcaa109. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-alpha modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed 2006, 8, 25. [Google Scholar] [PubMed]
- Cheng, X.; Shen, Y.; Li, R. Targeting TNF: A therapeutic strategy for Alzheimer’s disease. Drug Discov. Today 2014, 19, 1822–1827. [Google Scholar] [CrossRef]
- Allez, M.; Karmiris, K.; Louis, E.; Van Assche, G.; Ben-Horin, S.; Klein, A.; Van der Woude, J.; Baert, F.; Eliakim, R.; Katsanos, K.; et al. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: Definitions, frequency and pharmacological aspects. J. Crohn’s Colitis 2010, 4, 355–366. [Google Scholar] [CrossRef]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef]
- Scheinfeld, N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J. Dermatol. Treat. 2004, 15, 280–294. [Google Scholar] [CrossRef]
- Weinstock, M.; Gorodetsky, E.; Poltyrev, T.; Gross, A.; Sagi, Y.; Youdim, M. A novel cholinesterase and brain-selective monoamine oxidase inhibitor for the treatment of dementia comorbid with depression and Parkinson’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 555–561. [Google Scholar] [CrossRef]
- Maruyama, W.; Weinstock, M.; Youdim, M.B.; Nagai, M.; Naoi, M. Anti-apoptotic action of anti-Alzheimer drug, TV3326 [(N-propargyl)-(3R)-aminoindan-5-yl]-ethyl methyl carbamate, a novel cholinesterase-monoamine oxidase inhibitor. Neurosci. Lett. 2003, 341, 233–236. [Google Scholar] [CrossRef]
- Reichert, F.; Zohar, K.; Lezmi, E.; Eliyahu, T.; Rotshenker, S.; Linial, M.; Weinstock, M. Ladostigil Reduces the Adenoside Triphosphate/Lipopolysaccharide-Induced Secretion of Pro-Inflammatory Cytokines from Microglia and Modulate-Immune Regulators, TNFAIP3, and EGR1. Biomolecules 2024, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- García-García, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-kB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- Ravetti, M.G.; Rosso, O.A.; Berretta, R.; Moscato, P.; Wölfl, S. Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS ONE 2010, 5, e10153. [Google Scholar] [CrossRef]
- Abbasi, A.; Forsberg, K.; Bischof, F. The role of the ubiquitin-editing enzyme A20 in diseases of the central nervous system and other pathological processes. Front. Mol. Neurosci. 2015, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.N.; Wallace, J.L.; Hartzell, A.L.; Nematollahi, S.; Plange, K.; Barnes, C.A. Age-associated deficits in pattern separation functions of the perirhinal cortex: A cross-species consensus. Behav. Neurosci. 2011, 125, 836–847. [Google Scholar] [CrossRef]
- Reger, M.L.; Hovda, D.A.; Giza, C.C. Ontogeny of Rat Recognition Memory measured by the novel object recognition task. Dev. Psychobiol. 2009, 51, 672–678. [Google Scholar] [CrossRef]
- Linial, M.; Stern, A.; Weinstock, M. Effect of ladostigil treatment of aging rats on gene expression in four brain areas associated with regulation of memory. Neuropharmacology 2020, 177, 108229. [Google Scholar] [CrossRef]
- Schneider, L.S.; Geffen, Y.; Rabinowitz, J.; Thomas, R.G.; Schmidt, R.; Ropele, S.; Weinstock, M.; on behalf of the Ladostigil Study Group. Low-dose ladostigil for mild cognitive impairment: A phase 2 placebo-controlled clinical trial. Neurology 2019, 93, e1474–e1484. [Google Scholar] [CrossRef]
- Davatzikos, C.; Bhatt, P.; Shaw, L.M.; Batmanghelich, K.N.; Trojanowski, J.Q. Prediction of MCI to AD conversion, via MRI, CSF biomarkers, and pattern classification. Neurobiol. Aging 2011, 32, 2322.e19–2322.e27. [Google Scholar] [CrossRef]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer’s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Paglieri, C.; Bisbocci, D.; Caserta, M.; Rabbia, F.; Bertello, C.; Canadè, A.; Veglio, F. Hypertension and cognitive function. Clin. Exp. Hypertens. 2008, 30, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Alves, F.; Kallinowski, P.; Ayton, S. Accelerated Brain Volume Loss Caused by Anti–β-Amyloid Drugs: A Systematic Review and Meta-analysis. Neurology 2023, 100, e2114–e2124. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M. Therapeutic agents for Alzheimer’s disease: A critical appraisal. Front. Aging Neurosci. 2024, 16, 1484615. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Compound | Activity in Cells | Activity in Animal Models | Clinical Trials |
|---|---|---|---|
| Vitamin E (α tocopherol) | Vit E reduced cell death induced by H2O2 in hippocampal neuronal cells and by Aβ in rat hippocampal cell cultures [77] | Vit E improved cognitive performance in aged animals; prevented oxidative damage in animal models of AD [77] | Vit E (2000 IU/qd) given for 3 years slowed progression of memory loss in subjects with AD [80]. In patients with MCI, giving 2000 IU/qd produced no difference from placebo in rate of conversion to AD [81] |
| Curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) (active principle of turmeric) | Restored levels of SOD, catalase and glutathione peroxidase in macrophages exposed to H2O2 [85,86] | Curcumin (5.43 μmol/g) given in diet improved spatial memory of aging rats infused with Aβ40 and Aβ42 [87] | Curcumin, 2–4 gm qd given for 24 weeks to patients with mild to moderate AD, showed no difference from placebo in measures of cognitive function [88] |
| Resveratrol (3,5,4-trihydroxystilbene) | Resveratrol, 10–100 µM, reduced oxidative stress in human erythrocytes [106] | Resveratrol (300 mg/kg) reduced oxidative stress and number of plaques in Tg19959 transgenic mouse model [107] | Resveratrol (0.5–1 gm qd) had no effect on cognitive function, hippocampal volume or Aβ42, CSF Aβ42, CSF tau, CSF phospho-tau181 in patients with mild to moderate AD [91] |
| Ginkgo biloba extract (EGb761) | EGb761 (100 µg/mL) protected SH-SY5Y against H2O2-induced cell death and fall in mitochondrial function induced by Aβ (1–42) [97,98] | EGb761 (100 mg/kg) improved short-term memory and membrane fluidity avoidance learning in aged female mice [99] | EGb761 (240 qd) given for 5 years to subjects with MCI showed no significant difference from controls in % of converters to AD [105]. Subjects with dementia given EGb761 (240 qd) for 24 weeks showed a greater improvement in cognition than those on placebo [102] |
| Ladostigil (6-(N- ethyl, N- methyl carbamyloxy)-N propargyl-1(R)-aminoindan hemitartrate | Ladostigil prevented oxidative stress in SH-SY5Y neuroblastoma cells induced by H2O2 [108] and restored normal gene expression of NADPH and catalase | Ladostigil 1 mg/kg/day) restored object recognition and spatial memory in aging rats to those of young adults [109] | Ladostigil (1 mg/day) significantly slowed decline in hippocampal volume in patients with MCI and reduced number of subjects without ApoE4 gene who converted to AD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinstock, M. Role of Oxidative Stress and Neuroinflammation in the Etiology of Alzheimer’s Disease: Therapeutic Options. Antioxidants 2025, 14, 769. https://doi.org/10.3390/antiox14070769
Weinstock M. Role of Oxidative Stress and Neuroinflammation in the Etiology of Alzheimer’s Disease: Therapeutic Options. Antioxidants. 2025; 14(7):769. https://doi.org/10.3390/antiox14070769
Chicago/Turabian StyleWeinstock, Marta. 2025. "Role of Oxidative Stress and Neuroinflammation in the Etiology of Alzheimer’s Disease: Therapeutic Options" Antioxidants 14, no. 7: 769. https://doi.org/10.3390/antiox14070769
APA StyleWeinstock, M. (2025). Role of Oxidative Stress and Neuroinflammation in the Etiology of Alzheimer’s Disease: Therapeutic Options. Antioxidants, 14(7), 769. https://doi.org/10.3390/antiox14070769